

Abstract
Inflammation is a hallmark of inflammatory bowel disease (IBD) that involves macrophages. Given the inverse link between selenium (Se) status and IBD-induced inflammation, our objective was to demonstrate that selenoproteins in macrophages were essential to suppress pro-inflammatory mediators, in part, by the modulation of arachidonic acid metabolism. Acute colitis was induced using 4% DSS in wild type mice maintained on Se-deficient (<0.01 ppm Se), Se-adequate (0.1 ppm; sodium selenite), and two supraphysiological levels in the form of Se-supplemented (0.4 ppm; sodium selenite) and high Se (1.0 ppm; sodium selenite) diets. Transfer RNASec (tRNA[sec]) knockout mice (Trspfl/flLysMCre) were used to examine the role of selenoproteins in macrophages on disease progression and severity using histopathological evaluation, expression of pro-inflammatory and anti-inflammatory genes, and modulation of prostaglandin (PG) metabolites in urine and plasma. While Se-deficient and Se-adequate mice showed increased colitis and exhibited poor survival, Se supplementation at 0.4 and 1.0 ppm increased survival of mice and decreased colitis-associated inflammation with an up-regulation of expression of pro-inflammatory and anti-inflammatory genes. Metabolomic profiling of urine suggested increased oxidation of PGE2 at supraphysiological levels of Se that also correlated well with Se-dependent upregulation of 15-hydroxy-PG dehydrogenase (15-PGDH) in macrophages. Pharmacological inhibition of 15-PGDH, lack of selenoprotein expression in macrophages, and depletion of infiltrating macrophages indicated that macrophage-specific selenoproteins and upregulation of 15-PGDH expression were key for Se-dependent anti-inflammatory and pro-resolving effects. Selenoproteins in macrophages protect mice from DSS-colitis by enhancing 15-PGDH-dependent oxidation of PGE2 to alleviate inflammation, suggesting a therapeutic role for Se in IBD.
Keywords: Selenium, macrophage activation, PGE2, IBD, 15-PGDH
INTRODUCTION
Micronutrient trace element selenium (Se) is incorporated as selenocysteine (Sec) in selenoproteins that play an important role in shaping the immune response during inflammation (1). Particularly in inflammatory bowel disease (IBD), decreased levels of Se are seen in IBD patients of ulcerative colitis (UC) or Crohn’s disease (2–4). Recent studies using a two-stage carcinogenesis model in mice demonstrated Se deficiency to exacerbate disease severity and colon cancer risk by enhanced epithelial injury (5). Previous studies from our laboratory have shown that Se through its incorporation into selenoproteins suppressed inflammation and decreased the production of tumor necrosis factor-α (TNFα) and prostaglandin E2 (PGE2) (6), which have also been shown to be elevated in the plasma of UC patients (7).
Selenium at supranutritional levels (as inorganic selenite) differentially modulated transcription factors, nuclear factor-κB (NFκB) and peroxisome proliferator activated receptor-γ (PPARγ) in macrophages by skewing the cyclooxygenase (COX)-dependent PG pathways (6). As a result, metabolism of arachidonic acid in LPS-activated macrophages was shunted towards PGD2 and its cyclopentenone PGJ2 (CyPG) metabolites, Δ12-PGJ2 and 15d-Δ12,14-PGJ2, rather than the production of PGE2 and TXA2 (6). The production of CyPGs was effected by the differential expression of downstream PG synthases that were tightly controlled by selenoprotein-mediated changes in cellular redox status (6). PGD2-derived CyPG metabolites exert significant anti-inflammatory effects (8, 9), whereas elevated levels of PGE2 and TNF-α have been linked to the promotion of inflammation, ulceration, edema, and pain (10, 11). Thus, inhibition of PGE2 could be a double-edged sword because of its pro-angiogenic role in the maintenance of mucosal integrity in experimental colitis (12) and its link to the activation of resolution by stimulating the eicosanoid class switching to anti-inflammatory and pro-resolving mediators in human leukocytes (13). The mice deficient in PGE2 receptor (EP) subtype EP4 (12) were found to develop experimental colitis and treatment with an EP4 agonist ameliorated colitis in wild-type mice (12). These studies suggest a pleiotropic role for PGE2, where the control of PGE2 production and catabolism may have a significant bearing on the pathophysiology of UC. Little is known about the impact of selenoprotein expression on PGE2 catabolism and subsequent effect on pathways of inflammation and resolution. In fact the role of 15-hydroxy prostaglandin dehydrogenase (15-PGDH), a NAD+-dependent dehydrogenase that oxidizes many eicosanoid metabolites, including PGE2, in IBD is well known (14). 15-PGDH is abundantly expressed in the gut and is involved in the catabolism of PGs and other pro-resolving lipid mediators, including resolvin E1 (RvE1), which effect resolution pathways in colitis suggesting a dual role for this enzyme (15–17). However, 15-PGDH has been reported to preferentially bind to PGE2 leading to the production of downstream 15-keto PGE2 metabolites (18, 19). It appears that such a metabolic event in response to selenoprotein expression may serve as a key determinant in the pathology of UC to modulate resolution of inflammation to alleviate colitis.
Increasing dietary Se intake from adequate levels to supranutritional levels that are well within the non-toxic dose range could potentially facilitate the resolution of UC-associated inflammation by regulating the metabolism of key lipid metabolites, such as PGD2. A recent study reported elevated levels of PGD2 in colitis patients in remission (20). In the current study, we demonstrate the ability of Se supplementation to upregulate the expression of 15-PGDH leading to the increased catabolism of PGE2 that alleviates inflammation and activating pathways of resolution in the DSS-model of experimental colitis. Macrophage-specific selenoprotein knockout mice (Trspfl/flLysMCre) that do not express the selenoproteome due to the loss of both tRNA[Sec] alleles reproduced the effects of whole body Se deficiency on colitis susceptibility towards an inflammatory phenotype. Thus, nutritional intervention with Se supplementation may alleviate GI inflammation and promote resolution through the expression of macrophage selenoproteins.
METHODS
Mice
Wild type male C57BL/6 mice (3 week old; Taconic) were divided into five groups (n=5–10) and weaned on torula yeast-based diets (from Harlan Teklad, WI, USA) containing <0.01 ppm Se. To the basal diet either no Se was added (Se -deficient; Se-D), or sodium selenite was added at adequate levels of 0.08 ppm (Se-adequate; Se-A), or supplemented with supranutritional doses of selenite at 0.4 ppm Se (Se-supplemented; Se-S) or 1.0 ppm Se (Se-high; Se-H) for at least 10–12 weeks. Trspfl/fl mice were prepared as described earlier (21) and crossed with LysMCre mice to produce Trspfl/flLysMCre mice that lack the expression of selenoproteins in monocyte and macrophages (and to a minor extent in granulocytes) (22). These mice were maintained on Se-D or Se-S diets. In experiments using Trspfl/flLysMCre mice, corresponding sex, age, and diet group matched WT littermates were used for comparison. All procedures were reviewed and preapproved by the Institutional Animal Care and Use Committee at the Pennsylvania State University.
Induction of colitis
Mice were administered 4.0 % DSS (MW=40 kDa; ICN Biomedicals, Aurora, OH) dissolved in Milli-Q water ad libitum for 5 days, after which the mice were switched to Milli-Q water for the remainder of the experiment. Animals were weighed daily and monitored clinically for rectal bleeding, diarrhea, and general signs of morbidity. Moribund mice or mice that had lost more than 25% of their body weight were sacrificed and listed as dead following induction of colitis. Day −1 represented one day before the initiation of DSS.
Pathological examination and scoring of colitis
The entire colon (from cecum to anus) was removed and the length was measured and reported as colonic length as described (23). Distal colon was removed, fixed in 10% formalin, sectioned and stained with H & E for histopathology (at the Pennsylvania State University Animal Diagnostic Laboratory, University Park, PA). Histological analysis was performed blindly and the slides were scored as follows: severity of inflammation (0–4: none, slight, moderate, severe), extent of injury (0–4: none, mucosal, mucosal and submucosal/transmural), and crypt damage (0–4: none, basal 1/3 damaged, basal 2/3 damaged, only surface epithelium intact with a loss of entire crypt) (24). Sections were scored for a total of 12 points and divided by three main criterion scores to fit within the 0–4 scale.
Cell culture and Se treatments
Murine RAW264.7 immortalized macrophage-like cell line and primary bone marrow-derived macrophages (BMDMs) isolated from mice were cultured in DMEM media supplemented with 5% defined FBS (ATCC) with low Se (~ 6 nM by atomic absorption spectroscopy) and sterile filtered 10 % (v/v) L929 murine fibroblast culture media supernatant. These Se-deficient cells were conditioned with LPS (100 ng/ml) and then cultured with sodium selenite (0.1–0.5 μM). To address if bioavailable Se was critical to effect the expression of 15-PGDH, the source of Se was changed from inorganic sodium selenite to organo-Se compounds such as MSA (methylseleninic acid), SeMet (L-selenomethionine) or p-XSC (1,4-phenylenebis(methylene)selenocyanate) at the concentrations indicated. Cells were allowed to grow for at least three days with daily media changes before being used for experiments. None of these treatments affected cell viability or growth rates (data not shown).
Gene and protein expression analysis
Changes in various classical inflammatory markers and 15-PGDH expression were studied in the macrophages and the colonic tissues extracted from mice as described below.
Cells
Cells cultured in different Se concentrations (as described above) were harvested and used for isolation of RNA and protein. Expression of 15-PGDH, TNF-α, IFN-γ, and IL-1β was analyzed by quantitative realtime PCR (qPCR) using an ABI 7300 real-time system (Applied Biosystems). Data was analyzed according to the method of Livak et al (25, 26) and results were expressed as 2−ΔΔCT, which is the expression of target gene relative to the housekeeping gene (GAPDH) that is normalized to the negative control in the Se-D group. To study protein expression, lysates were prepared using the mammalian protein extraction reagent (M-PER; ThermoFisher-Scientific, IL) and immunoblots were developed using specific primary and appropriate secondary antibodies for 15-PGDH (Cayman Chemicals, MI), GPX-1 (Abcam, MA), and GAPDH (Fitzgerald Industries, MA). The bands were visualized by using an enhanced chemiluminescence assay kit.
Mice
Distal colons from mice were collected and washed in PBS containing penicillin (100 U/ml) and streptomycin (100 μg/ml). RNA was isolated from the colon with Trizol reagent and used for qPCR analysis with Taqman probes (Applied Biosystems).
Western immunoblotting
Tissue homogenates were prepared in ice-cold RIPA buffer and centrifuged at 13,000 rpm for 5 min at 4 °C. Protein concentrations were determined in the supernatants using BCA method (Thermo Scientific, IL, USA) and subjected to Western immunoblots as described above.
Biochemical Analyses
The activity of 15-PGDH was measured by recording the changes in absorbance due to formation of NADH from NAD+ by 15-PGDH (27). Myeloperoxidase (MPO) activity assay in the colonic tissue was performed as described (28).
Metabolomics and ELISA
Urine samples were diluted 1:10 in 50% acetonitrile:water containing 5 μM chlorpropamide (internal standard). The samples were vortexed and centrifuged at maximum speed for 20 min at 4 °C. The supernatants were subjected to mass spectrophotometry and the spectral data was deconvoluted using MarkerLynx software program (Waters). Characteristics such as mass-to-charge ratio (m/z), retention time, peak area of each data point were used for analysis. PCA (principal components analysis) and PLS-DA (supervised projection to latent structures-discriminant analysis) were conducted using SIMCA P12+ software (Umetrics). Candidate biomarkers were searched using METLIN (Metabolite and Tandem MS Database), database from the Scripps Center for Metabolomics. Levels of PGE2, and its downstream metabolites (PGEM) 13, 14-dihydro-15-keto PGE2 and 13,14-dihyro-15-keto-PGA2 were measured by respective ELISA kits (as per manufacturer’s protocol) in the plasma isolated from mice maintained on different Se diets upon treatment with DSS.
Depletion of macrophages
Macrophages were depleted using clodronate-containing liposomes (ClodronateLiposomes.org, Amsterdam, The Netherlands) as described (29). Briefly, mice were anesthetized and 200 μl of clodronate-loaded liposomes (CL) were injected via retro-orbital sinus into mice on days −1, 1, 3, and 5 post DSS treatment. PBS-encapsulated liposomes were not used as a negative control, since the uptake of these liposomes by colonic macrophages was shown to result in partial reduction of macrophages (28).
Synthesis of 2-hydroxy-5-(4-ethoxycarbonylphenylazo)benzeneacetic acid (CAY10397)
Ethyl p-aminobenzoate (Compound 1; 0.33 g, 2 mmol; Fig. S1A) was dissolved with cooling to 0°C in HCl (6 N, 4 ml). A solution of sodium nitrite (0.21 g, 3 mmol) in water (2 ml) was added and the reaction mixture was stirred at 0°C for 25 min. The diazonium salt solution was added to 2-hydroxy-benzeneaccetic acid (0.33 g, 2.2 mmol) and sodium hydroxide (0.11 g, 2.8 mmol) in water (2 ml) at 4°C. The reaction mixture was adjusted to pH 9–10 and the mixture was stirred for 2 h and acidified by HCl. The precipitated product was filtered and washed with water, which on recrystallization from ethanol/water resulted in 0.38 g (57 %) of pure compound. Ethyl p-aminobenzoate was treated with NaNO2 under acidic conditions to form a diazonium salt (Compound 2; Fig. S1A), which was used in the next step without purification. Compound 2 was coupled with 2-hydroxybenzeneacetic acid under basic conditions to form Compound 3, which on recrystallization from aqueous ethanol resulted in pure 2-hydroxy-5-(4-ethoxycarbonylphenylazo)benzeneacetic acid (Compound 3; Fig. S1A) with a 57 % yield. The structure of Compound 3 was confirmed by 1H-NMR (Fig. S1B). Selected physical data for compound 3: 1H NMR (d6-DMSO) δ 1.35 (t, 3H, CH3), 3.61 (s, 2H, CH2), 4.35 (dd, 2H, OCH2), 7.01 (d, 1H, aromatic, J = 8.5 Hz), 7.81-7.70 (m, 2H, aromatic), 7.90 (d, 2H, aromatic, J = 8.5 Hz), 8.13 (d, 2H, aromatic, J = 9.0 Hz); 13C NMR: δ 14.63, 35.78, 61.47, 115.80, 122.65, 123.77, 125.37, 126.17, 130.90, 131.31, 145.55, 155.29, 160.43, 165.70, 172.81. Purity of Compound 3 was further confirmed by reverse phase HPLC using Vydac Analytical column. Solvent gradient of 0–100% methanol in 30 minutes with a flow rate of 1 ml/min was used. The purity of compound 3 (retention time = 25 min) was >97 % (data not shown).
Pharmacological inhibition of 15-PGDH activity
CAY10397, a selective inhibitor of 15-PGDH, was administered by oral gavage at a dose of 100 mg/kg in Se-S mice every alternate day starting at day −1 of DSS treatment. CAY10397 (100 mg/ml) was formulated in 20 mM glycine buffer (pH 10) containing 5 % (v/v) cell culture grade DMSO (Sigma). CAY10397 was synthesized as described in supplementary methods (Fig. S1A). 1H-NMR spectroscopic analysis (Fig. S1B) and enzyme inhibition of purified human recombinant 15-PGDH (Cayman Chemicals, Ann Arbor, MI) analysis indicated that the chemically synthesized compound was identical to the commercially available CAY10397 (from Cayman Chemicals). Based on the inhibition of 15-PGDH activity in kidney extracts of mice (Fig. S2), an in-vivo dose of 100 mg/kg of CAY10397 was used. No toxic effects were observed in mice at this dose (data not shown).
Data analysis
All data are expressed as mean ± SEM. An unpaired, two-tailed t-test was used to compare the mean for each treatment group with the mean of the control group and one-way ANOVA (Tukey multiple comparison method) or two-way ANOVA was performed in order to compare various treatment groups within in vivo studies using GraphPad Prism 5.0 program (GraphPad Software Inc., San Diego, CA). p values ≤ 0.05 were considered as statistically significant.
RESULTS
Se supplementation alleviates DSS-induced colitis
Fecal occult blood was detected in the feces of Se-D and Se-A diet fed mice as early as day 1 post-DSS administration. Body weight changes, diarrhea, and bleeding were significantly more severe in Se-D and Se-A mice compared to mice on Se-S and Se-H diets. Se-D and Se-A had significantly lower body weights than Se-S and Se-H mice (Fig. 1A). The colons of Se-D and Se-A mice were significantly shorter and had higher blood scores compared to Se-S and Se-H mice (Fig. 1B). Colon sections from Se-D and Se-A mice showed edema and complete destruction of the mucosal surface with the loss of the epithelium and crypt damage. In addition, there was significant neutrophil infiltration and damage to colonic mucosa of Se-D and Se-A mice indicating higher levels of tissue inflammation and failure to resolve epithelial injury (Fig. 1C). On the contrary, colonic sections from DSS-treated Se-S and Se-H mice indicated negligible signs of inflammation and mucosal damage in addition to pronounced healing of the colon (Fig. 1C). Also, the classical molecular hallmarks of colitis such as TNFα, IFNγ, IL-1β, and COX-2 were significantly higher in Se-D and Se-A than in Se-S and Se-H mice groups (Fig 2A). On the other hand, Arg-1, Fizz-1 and Muc-2 were increased in Se-S and Se-H groups compared to Se-D and Se-A mice (Fig 2B). These studies suggest a critical role for Se in the control of inflammation in DSS-induced experimental colitis.
Figure 1. Selenium supplementation mitigates and resolves inflammation in DSS-treated mice.
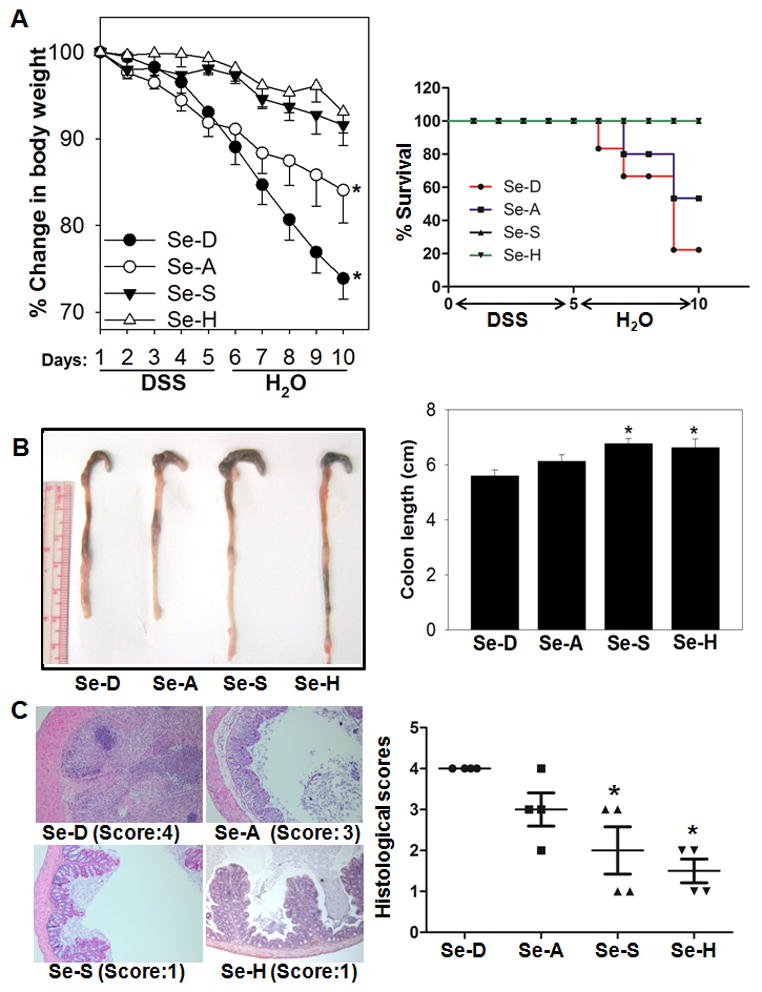
C57BL/6 wildtype mice on Se-D (Se-deficient), Se-A (Se-adequate; 0.08 ppm; as sodium selenite), Se-S (Se-supplemented; 0.4 ppm; as sodium selenite), and Se-H (Se-high; 1.0 ppm; as sodium selenite) diets for 12 weeks were treated with DSS (4% w/v in drinking water) for 5 days followed by water for the remainder of the experiment. (A) Loss in body weight (left panel) and Kaplan Meier curves showing the survival (right panel) as a function of dietary Se in DSS-treated mice. (B) Shrinkage of colons (left panel) and changes in colonic length (right panel) showing macroscopic inflammation in DSS-treated Se-D and Se-A mice compared to their Se-S and Se-H counterparts. Representative colons from n=10 per group shown. (C) H&E-stained colonic sections of mice treated with DSS (left panel) were histologically scored based on neutrophil infiltration, edema and cellular inflammation, and the extent of colitis on a scale of 0–4 (right panel) as described under “Methods”. Representative slides from each treatment along with their corresponding scores are shown. Values are represented as means ± SEM, * represents p<0.05 of n=10 (for H&E, n=4) compared to the Se-D group.
Figure 2. Selenium supplementation differentially regulates the colonic expression of proinflammatory and anti-inflammatory genes in DSS colitis mice.
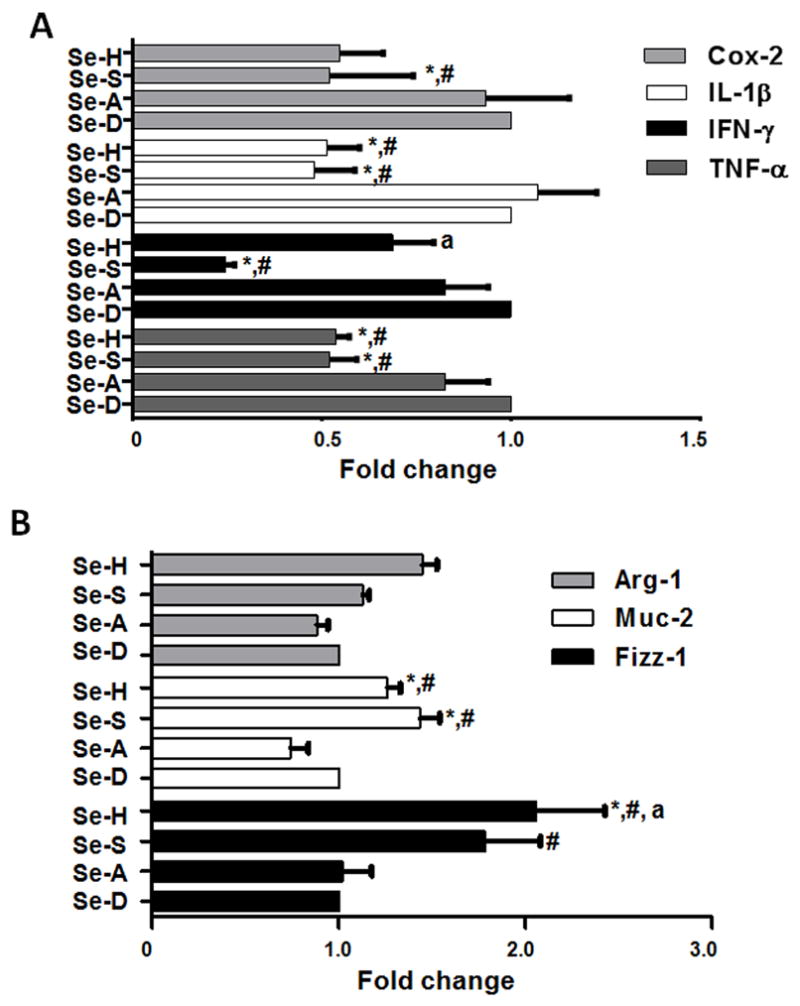
C57BL/6 wildtype mice on Se-D, Se-A, Se-S, and Se-H diets for 12 weeks were treated with DSS (4% w/v in drinking water) for 5 days followed by water for the remainder of the experiment. Total colonic RNA was isolated on day 10 and used for qPCR analysis. (A) Inhibition of colonic expression of COX-2, IL-1β, IFNγ, and TNFα with increasing levels of dietary Se. (B) Changes in colonic expression of alternatively activated macrophage (M2) markers Arg-1, Muc-2, and Fizz-1 as a function of increasing dietary Se on day 10 post DSS treatment. Values are represented as means ± SEM * represents p<0.05 of n=6 (for M2 markers n=3). ‘a’ and ‘#’ represent p<0.05 when compared to Se-S and Se-A groups, respectively.
Macrophage-specific selenoproteins are required for resolution of colitis associated inflammation
Since macrophages play a key role in the DSS-model of experimental colitis (30), and that selenoprotein expression in macrophages was earlier demonstrated to be key to mitigate inflammation (6), we utilized the Trspfl/flLysMCre mice. DSS treated Trspfl/flLysMCre mice lost weight despite being on Se-S diet (Fig. 3A). Se-D and Se-S Trspfl/flLysMCre mice displayed shorter colons (Fig. 3B) and increased histopathological scores than the Se-S WT mice (Fig. 3C). The symptoms of DSS colitis in the Se-S Trspfl/flLysMCre mice resembled those of the Se-D WT mice treated with DSS. Histological analysis of the colon of Se-S Trspfl/flLysMCre mice treated with DSS confirmed severe colitis and increased infiltration of neutrophils along with edema, cryptitis, and increased tissue inflammation (Fig. 3C). WT littermates maintained on Se-S diet showed no lesions following DSS treatment (Fig 3C). The expression of TNFα, IFNγ, IL-1β, and COX-2 were upregulated in the Se-S Trspfl/flLysMCre mice compared to the littermate Se-S controls (Fig 3D). These studies suggested that selenoprotein expression in macrophages was critical for the protective effects of Se in DSS colitis.
Figure 3. Macrophage-specific selenoprotein knockout (Trspfl/flLysMCre) mice fail to resolve colonic inflammation upon DSS treatment.

(A) Changes in body weight (left panel) and survival (right panel) in DSS treated Trspfl/flLysMCre and WT littermates maintained on Se-D or Se-S diets. (B) Shrinkage in colonic length upon DSS treatment on day 10 (left panel; representative of n= 5 per group shown) and quantitation of colon lengths (right) in DSS-treated Trspfl/flLysMCre and WT mice maintained on Se-D or Se-S diets. n= 5 per group. (C) Representative H&E-stained colonic sections (left panel) and histology scores from Trspfl/flLysMCre and WT littermate controls following DSS treatment on day 10 (right panel). Representative slides for each treatment along with their corresponding histology scores are shown. n= 7 per group. * p<0.05 compared between Se-S/WT and Se-S/Trspfl/flLysMCre groups. (D) Colonic expression of TNF-α, IL-1β, IFN-γ, and COX-2 by qPCR in DSS-treated WT and Trspfl/flLysMCre mice on Se-D and Se-S diets. Values are represented as means ± SEM, * represents p<0.05 of n=5 per group.
Selenium-dependent changes in the metabolome indicate oxidation of PGE2
A 24 h pooled urine was collected from Se-D, Se-A, and Se-S mice prior to treatment with DSS for metabolomic analysis. Results indicate differences in the metabolite pools in three diet groups (Fig. S4). Of the various metabolites that were differentially regulated by increasing Se in the diet, 13,14-dihydro-15-keto PGE2, a product of PGE2 oxidation, was produced at higher levels in the urine of Se-S mice compared to other groups (data not shown). Following this lead, a targeted LC-MS/MS analysis was performed on the urine and plasma of mice on day 5 post-DSS treatment. While urinary 13,14-dihydro-15-keto PGE2 was higher in the Se-S mice following DSS treatment, the differences were not statistically significant (data not shown). However, 15-keto-PGE2 in the plasma of Se-S mice post-DSS treatment was significantly higher than in Se-D mice (Fig 4A). The role of 15-PGDH in the oxidation of PGE2 to 15-keto-PGE2 that is further reduced to form 13,14-dihydro-15-keto-PGE2, which subsequently undergoes dehydration to form 13,14-dihydro-15-keto-PGA2, is well known (31). Plasma levels of PGE2, 15-keto-PGE2, and 13,14-dihydro-15-keto-PGE2 were estimated by ELISA from DSS treated mice on day 10. PGE2 levels decreased in the plasma as a function of dietary Se, while 15-keto metabolites of PGE2 increased as a function of dietary Se (Fig. 4B). Se-dependent increase in 15-PGDH expression of mRNA, protein, and enzymatic activity were seen in DSS treated Se-S and Se-H mice (Fig. 4C–E). Similarly, GPX2, a Se-dependent glutathione peroxidase predominantly expressed in the GI tract, was also increased in a dose-dependent manner in the diet groups with saturation in the Se-S and Se-H groups (Fig. 4D). Taken together, these results suggest that Se-dependent upregulation of 15-PGDH may play a critical role in alleviating inflammation with a subsequent increase in resolution of mucosal injury.
Figure 4. Selenium-dependent upregulation of 15-PGDH in the colon of mice treated with DSS.

(A) Plasma levels of 15-keto-PGE2, and 13,14-dihydro-15-keto-PGE2 by LC-MS/MS in Se-D and Se-S mice treated with DSS on day 10. n= 3 per group. (B) Serum levels of PGE2 and keto-metabolites (15-keto-PGE2 and 13,14-dihydro-15-keto-PGE2) by ELISA in Se-D, Se-A, Se-S, and Se-H mice on day 10 post-DSS treatment. n= 3 per group. (C) Se-dependent upregulation of 15-PGDH expression by qPCR in the colonic samples. (D) Western immunoblot and (E) enzymatic activity of 15-PGDH in the colonic lysates of DSS-treated Se-D, Se-A, Se-S, and Se-H mice. Values are represented as means ± SEM, *, # represent p<0.05 of n=3 when compared to Se-D and Se-A respectively.
Macrophage expression of 15-PGDH is regulated by selenoproteins
To examine the regulation of expression of 15-PGDH by selenoproteins, BMDMs isolated from mice maintained on the Se-D diet and murine RAW264.7 macrophage-like cells were cultured in the presence of graded levels of sodium selenite (0–500 nM Se). The expression (and activity) of 15-PGDH in RAW264.7 cells and BMDMs displayed Se dose response with a saturation at ~250 nM of Se (Fig. 5A, 5B). Western immunoblot analysis of GPX1 expression confirmed the ability of both cell types to readily incorporate Se into selenoproteins (data not shown). Interestingly, BMDMs and RAW264.7 cells treated with two organo-Se compounds, SeMet, or pXSC, which do not readily release Se for incorporation into selenoproteins, failed to increase the expression of 15-PGDH (Fig. 5C, D). However, treatment with MSA that releases Se for incorporation into selenoproteins increased the expression of 15-PGDH. To further provide conclusive evidence of the requirement of selenoproteins, we examined the colonic expression of 15-PGDH in DSS-treated Trspfl/flLysMCre mice. Analysis of the colonic extracts on day 10 post-DSS treatment clearly showed an increased expression and activity of 15-PGDH only in Se-S WT littermates, while significantly reduced levels of 15-PGDH was seen in Trspfl/flLysMCre mice on Se-S diet (Fig. 5E–F).
Figure 5. Selenoprotein-dependent expression of 15-PGDH in-vitro and in-vivo.

RAW264.7 macrophage-like cells and BMDMs were treated with increasing concentrations of Se (as sodium selenite, 0–500 nM) for at least 3 days and analyzed for 15-PGDH expression. (A) Western immunoblot analysis of RAW264.7 cells (left panel) and BMDMs (right panel). (B) Enzymatic activity of 15-PGDH in RAW264.7 cells (left panel) and BMDMs (right panel). * p<0.05 compared to cells without any exogenously added Se. (C) RAW264.7 cells were cultured in SeD media and supplemented with indicated concentrations of Se in various forms; while BMDMs were isolated from untreated Se-D mice and cultured ex-vivo in the presence of selenite (NaSe; 100, 500 nM), methylseleninic acid (MSA; 100, 500 nM), Seleno-L-methionine (SeMet; 100, 500 nM), or 1,4-phenylenebis(methylene)selenocyanate (pXSC; 100, 500 nM) for at least 3 days. RAW264.7 cells (left panel) and BMDMs (right panel) and protein expression was examined by Western immunoblot of 15-PGDH expression. Representative blot of n= 3 shown. (D) Modulation of 15-PGDH expression by qPCR in RAW264.7 cells (left panel) and BMDMs (right panel) treated with above mentioned Se compounds. *p<0.05 compared to untreated cells. (E) Colonic extracts isolated from DSS-treated Trspfl/flLysMCre and WT littermate control mice (on day 10) maintained on Se-D and Se-S diets were used to examine the (E) expression of 15-PGDH mRNA by qPCR, (F) up-regulation of 15-PGDH expression by Western immunoblot, and (G) modulation of enzymatic activity of 15-PGDH. qPCR and enzymatic assay data for Trspfl/flLysMCre mice shown are mean ± SEM of n=3 per group, * represents p<0.05 Western blot data shown are representative of n=3 independent observations. ‘#’ and ‘a’ represent p<0.05 when compared Se-A and Se-S groups, respectively.
Macrophage depletion in Se supplemented mice blocks the protective effects during DSS treatment
To examine if macrophages were essential players in the Se-mediated protective effects in DSS-colitis, clodronate-liposomes (CL) were used to deplete infiltrating macrophages (29). Depletion of macrophages even under Se-S conditions exacerbated the severity of colitis compared to PBS control Se-S mice (Fig. 6A–E). CL treatment of Se-S mice resulted in symptoms of colitis such as weight loss and rectal bleeding that resembled DSS-treated Se-D mice (Fig 6A). As a control, we examined if Se-D mice treated with CL to address if Se-deficiency was a critical factor to support our results with the Trspfl/flLysMCre mice. As shown in Fig 6A–B, clodronate-treatment of Se-D mice also exhibited severe DSS-colitis as in Se-D mice. Clodronate treatment significantly affects on the onset of the disease in the Se-D mice, but was more or less similar to the PBS-treated Se-D mice subjected to DSS treatment. Taken together with the Trspfl/flLysMCre data, these results suggest that selenoprotein status and Se-dependent macrophage function are critical in DSS-colitis susceptibility (Fig 6E).
Figure 6. Macrophage depletion increases severity of DSS-induced colitis in Se-supplemented mice.

C57BL/6 wildtype mice maintained on Se-D and Se-S diets were injected with clodronate-encapsulated liposomes or PBS on days −1, 1, 3, and 5 of DSS-treatment and monitored for changes in body weight until day 10. DSS-treated Se-S and Se-D mice injected with PBS were used as controls. (A) Changes in body weight following clodronate (200 μL) or PBS treatment in Se-D and Se-S mice. (B) Kaplan Meier curves showing the survival of mice on the above mentioned treatments. (C) Changes in colon length of clodronate or PBS treated Se-S mice (left panel; representative of n=3 shown). Quantitation of colon length in DSS-treated Se-S mice with PBS or clodronate (right panel; * p<0.05 compared to Se-S PBS control group). (D) Expression of TNF-α, IL-1β, and 15-PGDH mRNA in the colonic mucosa of DSS-treated Se-S PBS and Se-S clodronate mice on day 10. Data are presented as mean ± SEM on n=3 per group. * represents p<0.05 compared to Se-S PBS control.
Pharmacological inhibition of 15-PGDH abrogates protective effects of Se
To examine the role of 15-PGDH in the Se-S mice, we used CAY10397, a selective inhibitor of 15-PGDH (32). Oral gavage of CAY10397 at 100 mg/kg significantly blocked the protective effects of Se supplementation and exacerbated symptoms of colitis (Fig. 7A, B, E). Although there was no effect on the expression of 15-PGDH mRNA per se (Fig. 7C), CAY10397 blocked the Se-mediated inhibition of colonic expression of TNF-α, IL-1β, IFN-γ and COX-2 (Fig. 7C and 7D). Furthermore, Se-mediated increase in Muc-2 (Fig. 7D) and colonic 15-PGDH activity were also significantly reduced in Se-S mice treated with DSS and CAY10397 (Fig. 7F). Taken together, these studies suggest a critical role for Se-dependent modulation of 15-PGDH expression in alleviating inflammation and promoting resolution of the gut epithelium.
Figure 7. Pharmacological inhibition of 15-PGDH activity blocks the protective effect of Se supplementation in DSS-colitis.

C57BL/6 wildtype mice maintained on a Se-S diet were treated with CAY10397, a 15-PGDH specific inhibitor at 100 mg/kg body weight, or vehicle (Veh; 20 mM glycine buffer, pH 10, containing 5 % (v/v) DMSO) by oral gavage every alternate day of DSS treatment starting at day −1. (A) Changes in the body weight upon treatment with DSS. (B) Changes in the colon length were measured on day 10. Inset shows a representative colons from Se-S+CAY10397 and Se-S+Veh control mice on day 10 post-treatment with DSS. (C) Expression of TNF-α, IL-1β, IFN-γ, and 15-PGDH by qPCR in the colonic tissue of Se-S mice treated with CAY10397 or vehicle on day 10. (D) Expression of COX-2 and Muc-2 by qPCR in the colonic tissue of Se-S+CAY10397 and Se-S+vehicle treated mice on day 10 post-DSS treatment. (E) Representative H&E-stained colonic sections from DSS- treated mice in the presence of Se-S+CAY10397 or Se-S+Veh controls. Representative slides for each treatment along with their corresponding histology scores are shown. Inset: Histological scores of colonic sections from the above treatments. (F) Enzymatic activity of 15-PGDH in colonic samples of CAY10397 treated Se-S mice and Se-S vehicle control on day 10 post DSS treatment. All data shown are mean ± SEM of n=3 per group.
DISCUSSION
Elevated levels of inflammatory mediators, such as PGE2 and cytokines, are a hallmark in IBD patients (33). Dysregulated COX-dependent metabolism of arachidonic acid has been implicated in UC (34–36). However, emerging evidence suggests that the role of the COX pathway may be more complex given both the protective and harmful effects of COX-derived eicosanoids (37, 38). Therefore, pharmacological inhibition of COXs may have unintended consequences. In fact, COX-1−/− and COX-2−/− mice are more susceptible to DSS colitis (39) and NSAIDs exacerbate experimental models of IBD (40). Thus, targeting PG metabolic pathways downstream of COX could lead to more effective pharmacological strategies to treat colitis.
Prostaglandins are widely reported to have a dual role in inflammation and anti-inflammation. While PGD2 and its downstream metabolites, Δ12-PGJ2 and 15d-PGJ2, exert significant anti-inflammatory effects (8, 9), PGE2 has been linked with the promotion of inflammation and ulceration (11, 39). A prominent role for PGE2 in UC was suggested by studies where increased incidences of active UC (36, 41, 42) was associated with increasing PGE2 concentrations leading to the activation of the PGE2-TNFα axis (43, 44). However, PGE2 is also essential in the suppression of colitis, as shown in the EP4−/− mice, where increased sensitivity towards experimental colitis was seen (12). Thus, Se status appears be a key regulator of the levels of PGE2 by 15-PGDH-dependent metabolism to 15-keto-PGE2 and 13,14-dihydro-15-keto-PGA2 to alleviate inflammation as well as promote resolution. In addition, Se-dependent lowering of pro-inflammatory PGE2 could be further complemented by the ability of the downstream PGEM to activate PPARγ that together effect an active resolution program in addition to their already known tumor suppressor functions (45). These data support an inverse causal relationship between Se status and PGE2 production, which establishes Se as a key dietary factor in colitis. Clinical studies have shown supplementation with Se in UC patients leads to symptomatic relief from active colitis (2–4). Furthermore, our studies demonstrating the need for selenoproteins to alleviate DSS-colitis are in agreement with a recent study demonstrating that decreased levels of dietary Se exacerbate DSS-colitis (5). More importantly, our studies implicate macrophage selenoproteins to be critical in resolution of injury.
Increased susceptibility of Trspfl/flLysMCre mice and the ability of clodronate to block the protective effects of Se strongly suggest that selenoprotein expression in macrophages are key components for the successful protection by Se. Absence of these selenoproteins renders the body susceptible to oxidative damage, as seen in the form of increased myeloperoxidase (MPO), lipid peroxidation, and 8-isoprostane in the colonic extracts (Fig. S3) that are associated with inflammation in DSS-treated Se-D and Se-A mice compared to the Se-S and Se-H mice. These studies suggest that macrophages infiltrate in response to a myriad of inflammatory signals to alleviate inflammation and aid in resolution. Along these lines, studies from our laboratory have demonstrated that Se status has a significant impact on the polarization of macrophages towards an M1 (classically-activated) or M2 (alternatively-activated)-like phenotype that are endowed with pro-inflammatory or anti-inflammatory properties, respectively (46). In agreement with these observations, colonic expression of Arg1, a prototypical M2 marker was significantly increased along with Fizz1 in Se-S mice treated with DSS, while the M1 markers (Tnfα, Ifnγ, Il1β) were significantly decreased. In addition, Muc2, predominantly expressed by goblet cells that help in maintaining gut homeostasis and protection from pathogenic microbes by secreting mucin, was downregulated in Se-D mice rendering them susceptible to colitis as reported in Muc2−/− mice (47). These results suggest that macrophages may impact other cells to alleviate inflammation and facilitate resolution responses. It remains to be seen if the expression of specific selenoproteins in macrophages changes the microenvironment in the gut to facilitate resolution over continued inflammation.
The current study extends the paradigm that cellular metabolism is sensitive to selenoprotein expression as seen in form of global changes in the metabolome. More specifically, the metabolism of PGE2 being sensitive to the expression of the selenoproteome suggests the importance of redox-modulated metabolic pathways in alleviating inflammation. These studies also suggest that maintenance of inflammatory PGs and cytokines at homeostatic levels are important in the protection against colitis. In fact, while low concentrations of PGE2 are essential for angiogenesis and mucus production (48), high levels of PGE2 at the site of inflammation is associated with increased colitis activity (36) therefore making PGE2 as one of the major drivers of pathology in IBD (49, 50). Thus, increased oxidative catabolism of PGE2 by Se through the upregulation of 15-PGDH is a critical event in reducing excess PGE2 that effectively alleviates inflammation. Although it is possible that 15-PGDH can oxidize other eicosanoids (e.g., resolvins) that aid in anti-inflammation and resolution, 15-PGDH has been reported to preferentially oxidize PGE2, particularly in inflamed tissues (18, 19). It remains to be seen whether 15-PGDH expression is spatio-temporally regulated during inflammation to limit the metabolism of other anti-inflammatory eicosanoids to initiate pathways of resolution.
Use of various forms of Se along with macrophages that lack selenoproteins clearly demonstrates that bioavailable Se is crucial for the upregulation of proteins, such as 15-PGDH, outside the selenoproteome. Preliminary studies suggest that 15-PGDH expression is regulated by the nuclear receptor, PPARγ in the colonic tissue, which is in agreement with that reported by the Lu laboratory (45). Given that PPARγ activation is seen in Se supplemented macrophages via the generation of endogenous ligands in the form of PGD2 metabolites (6) and the ability of PGEM to activate PPARγ (45), it is very likely that a feed-forward loop may be initiated leading to the upregulation of 15-PGDH and H-PGDS to effect anti-inflammatory and pro-resolution pathways. The ability of PPARγ to activate 15-PGDH in cancer cell types is reminiscent of an anti-inflammatory cell type that is programmed to effectively accelerate wound healing while alleviating inflammation (45). It remains to be seen if pharmacological agonists of PPARγ also enhance PGE2 catabolism to the extent that they can be used as a therapy for colitis. Based on our studies, it appears that 15-PGDH expression and activity may be associated with alternatively-activated macrophages that are endowed with anti-inflammatory and wound healing properties (51).
In summary, our studies demonstrate the anti-inflammatory role of selenoproteins to impinge on the modulation of PG metabolic pathways to protect against experimental colitis (Fig. 8). Our data suggest that macrophages are key players that mediate the protective effects of selenoproteins where metabolic inactivation of PGE2 appears to be the major regulator of disease pathogenesis. Based on our data, it is clear that the metabolic inactivation of PGE2 increases with increased dietary Se, suggesting that higher than adequate levels of Se may be beneficial in IBD. Currently, little is known about the molecular basis of these therapies in IBD. Therefore, a better understanding of these redox-dependent processes may help develop more efficient regimens to effectively resolve inflammation and restore the gut epithelium in IBD.
Figure 8. Proposed mechanism of Se-dependent modulation of inflammation and resolution in macrophages.

Selenoprotein-dependent upregulation of PGEM and PGD2-derived CyPGs and differential modulation of pro-inflammatory and anti-inflammatory genes in macrophages that may play a key role in resolving inflammation in DSS-induced colitis.
Supplementary Material
Acknowledgments
Grant Support: This work was supported, in part, by PHS grants DK077152 (KSP) and ES022186 (ADP) from the National Institutes of Health
We thank the Histology Core of the Animal Diagnostic Laboratory, Penn State University, and Dr. Ramesh Ramachandran for help with microscopy.
Abbreviations used
- Se
selenium
- CyPGs
cyclopentenone prostaglandins
- 15-PGDH
15-hydroxy-prostaglandin dehydrogenase
- IBD
inflammatory bowel disease
- UC
ulcerative colitis
- COX
cyclooxygenase
- PGEM
PGE2 metabolites
- Sec
selenocysteine
- CL
clodronate
- Trsp
tRNASec
- NSAIDs
non-steroidal anti-inflammatory drugs
- PGEM
PGE2 metabolites
Footnotes
Disclosures: No competing financial interests
Author Contributions: N.K. designed and carried out experiments, analyzed data, and contributed to manuscript and figure preparation; A.D.P., A.K.K., C.C. carried out LC-MS, analyzed data, and contributed figures; M.J.K. performed histological evaluation; D. D. and S. A. performed synthesis; B.A.C. provided mice and expertise in the use of mice and contributed to the manuscript preparation; M.T.C. assisted in experimental design and data interpretation; K.S.P. carried out overall experimental design, conceived the research plan, analyzed data, and contributed to manuscript and figure preparations.
References
- 1.Hoffmann PR, Berry MJ. The influence of selenium on immune responses. Mol Nutr Food Res. 2008;52:1273–1280. doi: 10.1002/mnfr.200700330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andoh A, Hirashima M, Maeda H, Hata K, Inatomi O, Tsujikawa T, Sasaki M, Takahashi K, Fujiyama Y. Serum selenoprotein-P levels in patients with inflammatory bowel disease. Nutrition. 2005;21:574–579. doi: 10.1016/j.nut.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 3.Geerling BJ, Badart-Smook A, Stockbrugger RW, Brummer RJ. Comprehensive nutritional status in recently diagnosed patients with inflammatory bowel disease compared with population controls. Eur J Clin Nutr. 2000;54:514–521. doi: 10.1038/sj.ejcn.1601049. [DOI] [PubMed] [Google Scholar]
- 4.Ringstad J, Kildebo S, Thomassen Y. Serum selenium, copper, and zinc concentrations in Crohn’s disease and ulcerative colitis. Scand J Gastroenterol. 1993;28:605–608. doi: 10.3109/00365529309096096. [DOI] [PubMed] [Google Scholar]
- 5.Barrett CW, Singh K, Motley AK, Lintel MK, Matafonova E, Bradley AM, Ning W, Poindexter SV, Parang B, Reddy VK, Chaturvedi R, Fingleton BM, Washington MK, Wilson KT, Davies SS, Hill KE, Burk RF, Williams CS. Dietary selenium deficiency exacerbates DSS-induced epithelial injury and AOM/DSS-induced tumorigenesis. PLoS One. 2013;8:e67845. doi: 10.1371/journal.pone.0067845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gandhi UH, Kaushal N, Ravindra KC, Hegde S, Nelson SM, Narayan V, Vunta H, Paulson RF, Prabhu KS. Selenoprotein-dependent up-regulation of hematopoietic prostaglandin D2 synthase in macrophages is mediated through the activation of peroxisome proliferator-activated receptor (PPAR) gamma. J Biol Chem. 2011;286:27471–27482. doi: 10.1074/jbc.M111.260547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wiercinska-Drapalo A, Jaroszewicz J, Tarasow E, Flisiak R, Prokopowicz D. Transforming growth factor beta1 and prostaglandin E2 concentrations are associated with bone formation markers in ulcerative colitis patients. Prostaglandins Other Lipid Mediat. 2005;78:160–168. doi: 10.1016/j.prostaglandins.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 8.Ajuebor MN, Singh A, Wallace JL. Cyclooxygenase-2-derived prostaglandin D(2) is an early anti-inflammatory signal in experimental colitis. Am J Physiol Gastrointest Liver Physiol. 2000;279:G238–244. doi: 10.1152/ajpgi.2000.279.1.G238. [DOI] [PubMed] [Google Scholar]
- 9.Rajakariar R, Hilliard M, Lawrence T, Trivedi S, Colville-Nash P, Bellingan G, Fitzgerald D, Yaqoob MM, Gilroy DW. Hematopoietic prostaglandin D2 synthase controls the onset and resolution of acute inflammation through PGD2 and 15-deoxyDelta12 14 PGJ2. Proc Natl Acad Sci U S A. 2007;104:20979–20984. doi: 10.1073/pnas.0707394104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Louis E. The immuno-inflammatory reaction in Crohn’s disease and ulcerative colitis: characterisation, genetics and clinical application. Focus on TNF alpha. Acta Gastroenterol Belg. 2001;64:1–5. [PubMed] [Google Scholar]
- 11.Murakami M, Kudo I. Prostaglandin E synthase: a novel drug target for inflammation and cancer. Curr Pharm Des. 2006;12:943–954. doi: 10.2174/138161206776055912. [DOI] [PubMed] [Google Scholar]
- 12.Kabashima K, Saji T, Murata T, Nagamachi M, Matsuoka T, Segi E, Tsuboi K, Sugimoto Y, Kobayashi T, Miyachi Y, Ichikawa A, Narumiya S. The prostaglandin receptor EP4 suppresses colitis, mucosal damage and CD4 cell activation in the gut. J Clin Invest. 2002;109:883–893. doi: 10.1172/JCI14459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN. Lipid mediator class switching during acute inflammation: signals in resolution. Nat Immunol. 2001;2:612–619. doi: 10.1038/89759. [DOI] [PubMed] [Google Scholar]
- 14.Otani T, Yamaguchi K, Scherl E, Du B, Tai HH, Greifer M, Petrovic L, Daikoku T, Dey SK, Subbaramaiah K, Dannenberg AJ. Levels of NAD(+)-dependent 15-hydroxyprostaglandin dehydrogenase are reduced in inflammatory bowel disease: evidence for involvement of TNF-alpha. Am J Physiol Gastrointest Liver Physiol. 2006;290:G361–368. doi: 10.1152/ajpgi.00348.2005. [DOI] [PubMed] [Google Scholar]
- 15.Backlund MG, Mann JR, Holla VR, Buchanan FG, Tai HH, Musiek ES, Milne GL, Katkuri S, DuBois RN. 15-Hydroxyprostaglandin dehydrogenase is down-regulated in colorectal cancer. J Biol Chem. 2005;280:3217–3223. doi: 10.1074/jbc.M411221200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tai HH, Ensor CM, Tong M, Zhou H, Yan F. Prostaglandin catabolizing enzymes. Prostaglandins Other Lipid Mediat. 2002;68–69:483–493. doi: 10.1016/s0090-6980(02)00050-3. [DOI] [PubMed] [Google Scholar]
- 17.Ishida T, Yoshida M, Arita M, Nishitani Y, Nishiumi S, Masuda A, Mizuno S, Takagawa T, Morita Y, Kutsumi H, Inokuchi H, Serhan CN, Blumberg RS, Azuma T. Resolvin E1, an endogenous lipid mediator derived from eicosapentaenoic acid, prevents dextran sulfate sodium-induced colitis. Inflamm Bowel Dis. 2010;16:87–95. doi: 10.1002/ibd.21029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ruckrich MF, Schlegel W, Jung A. Prostaglandin endoperoxide analogues and prostaglandid D2 as substrates of human placental 15-hydroxy prostaglandin dehydrogenase. FEBS Lett. 1976;68:59–62. doi: 10.1016/0014-5793(76)80404-8. [DOI] [PubMed] [Google Scholar]
- 19.Sun FF, Armour SB, Bockstanz VR, McGuire JC. Studies on 15-hydroxyprostaglandin dehydrogenase from monkey lung. Adv Prostaglandin Thromboxane Res. 1976;1:163–169. [PubMed] [Google Scholar]
- 20.Vong L, Ferraz JG, Panaccione R, Beck PL, Wallace JL. A pro-resolution mediator, prostaglandin D(2), is specifically up-regulated in individuals in long-term remission from ulcerative colitis. Proc Natl Acad Sci U S A. 2010;107:12023–12027. doi: 10.1073/pnas.1004982107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kumaraswamy E, Carlson BA, Morgan F, Miyoshi K, Robinson GW, Su D, Wang S, Southon E, Tessarollo L, Lee BJ, Gladyshev VN, Hennighausen L, Hatfield DL. Selective removal of the selenocysteine tRNA [Ser]Sec gene (Trsp) in mouse mammary epithelium. Molecular and cellular biology. 2003;23:1477–1488. doi: 10.1128/MCB.23.5.1477-1488.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carlson BA, Yoo MH, Sano Y, Sengupta A, Kim JY, Irons R, Gladyshev VN, Hatfield DL, Park JM. Selenoproteins regulate macrophage invasiveness and extracellular matrix-related gene expression. BMC Immunol. 2009;10:57. doi: 10.1186/1471-2172-10-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Okayasu I, Hatakeyama S, Yamada M, Ohkusa T, Inagaki Y, Nakaya R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology. 1990;98:694–702. doi: 10.1016/0016-5085(90)90290-h. [DOI] [PubMed] [Google Scholar]
- 24.Froicu M, Cantorna MT. Vitamin D and the vitamin D receptor are critical for control of the innate immune response to colonic injury. BMC Immunol. 2007;8:5. doi: 10.1186/1471-2172-8-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Livak KJ, Flood SJ, Marmaro J, Giusti W, Deetz K. Oligonucleotides with fluorescent dyes at opposite ends provide a quenched probe system useful for detecting PCR product and nucleic acid hybridization. PCR Methods Appl. 1995;4:357–362. doi: 10.1101/gr.4.6.357. [DOI] [PubMed] [Google Scholar]
- 26.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 27.Alix E, Schmitt C, Strazielle N, Ghersi-Egea JF. Prostaglandin E2 metabolism in rat brain: Role of the blood-brain interfaces. Cerebrospinal Fluid Res. 2008;5:5. doi: 10.1186/1743-8454-5-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bradley PP, Priebat DA, Christensen RD, Rothstein G. Measurement of cutaneous inflammation: estimation of neutrophil content with an enzyme marker. J Invest Dermatol. 1982;78:206–209. doi: 10.1111/1523-1747.ep12506462. [DOI] [PubMed] [Google Scholar]
- 29.Qualls JE, Kaplan AM, van Rooijen N, Cohen DA. Suppression of experimental colitis by intestinal mononuclear phagocytes. J Leukoc Biol. 2006;80:802–815. doi: 10.1189/jlb.1205734. [DOI] [PubMed] [Google Scholar]
- 30.Weisser SB, Brugger HK, Voglmaier NS, McLarren KW, van Rooijen N, Sly LM. SHIP-deficient, alternatively activated macrophages protect mice during DSS-induced colitis. J Leukoc Biol. 2011;90:483–492. doi: 10.1189/jlb.0311124. [DOI] [PubMed] [Google Scholar]
- 31.Tai HH. Prostaglandin catabolic enzymes as tumor suppressors. Cancer Metastasis Rev. 2011;30:409–417. doi: 10.1007/s10555-011-9314-z. [DOI] [PubMed] [Google Scholar]
- 32.Berry CN, Hoult JR, Peers SH, Agback H. Inhibition of prostaglandin 15-hydroxydehydrogenase by sulphasalazine and a novel series of potent analogues. Biochem Pharmacol. 1983;32:2863–2871. doi: 10.1016/0006-2952(83)90390-8. [DOI] [PubMed] [Google Scholar]
- 33.Shanahan F, Targan S. Medical treatment of inflammatory bowel disease. Annu Rev Med. 1992;43:125–133. doi: 10.1146/annurev.me.43.020192.001013. [DOI] [PubMed] [Google Scholar]
- 34.de Silva PS, Olsen A, Christensen J, Schmidt EB, Overvaad K, Tjonneland A, Hart AR. An association between dietary arachidonic acid, measured in adipose tissue, and ulcerative colitis. Gastroenterology. 2010;139:1912–1917. doi: 10.1053/j.gastro.2010.07.065. [DOI] [PubMed] [Google Scholar]
- 35.Nishida T, Miwa H, Shigematsu A, Yamamoto M, Iida M, Fujishima M. Increased arachidonic acid composition of phospholipids in colonic mucosa from patients with active ulcerative colitis. Gut. 1987;28:1002–1007. doi: 10.1136/gut.28.8.1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sharon P, Ligumsky M, Rachmilewitz D, Zor U. Role of prostaglandins in ulcerative colitis. Enhanced production during active disease and inhibition by sulfasalazine. Gastroenterology. 1978;75:638–640. [PubMed] [Google Scholar]
- 37.Chen C. COX-2’s new role in inflammation. Nat Chem Biol. 2010;6:401–402. doi: 10.1038/nchembio.375. [DOI] [PubMed] [Google Scholar]
- 38.Wallace JL. COX-2: a pivotal enzyme in mucosal protection and resolution of inflammation. Scientific World Journal. 2006;6:577–588. doi: 10.1100/tsw.2006.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morteau O, Morham SG, Sellon R, Dieleman LA, Langenbach R, Smithies O, Sartor RB. Impaired mucosal defense to acute colonic injury in mice lacking cyclooxygenase-1 or cyclooxygenase-2. J Clin Invest. 2000;105:469–478. doi: 10.1172/JCI6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bjarnason I, Hayllar J, MacPherson AJ, Russell AS. Side effects of nonsteroidal anti-inflammatory drugs on the small and large intestine in humans. Gastroenterology. 1993;104:1832–1847. doi: 10.1016/0016-5085(93)90667-2. [DOI] [PubMed] [Google Scholar]
- 41.Lauritsen K, Hansen J, Bytzer P, Bukhave K, Rask-Madsen J. Effects of sulphasalazine and disodium azodisalicylate on colonic PGE2 concentrations determined by equilibrium in vivo dialysis of faeces in patients with ulcerative colitis and healthy controls. Gut. 1984;25:1271–1278. doi: 10.1136/gut.25.11.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wiercinska-Drapalo A, Flisiak R, Prokopowicz D. Plasma and mucosal prostaglandin E2 as a surrogate marker of ulcerative colitis activity. Rocz Akad Med Bialymst. 2001;46:60–68. [PubMed] [Google Scholar]
- 43.Khan I, Al-Awadi FM, Thomas N, Haridas S, Anim JT. Cyclooxygenase-2 inhibition and experimental colitis: beneficial effects of phosphorothioated antisense oligonucleotide and meloxicam. Scand J Gastroenterol. 2002;37:1428–1436. doi: 10.1080/003655202762671314. [DOI] [PubMed] [Google Scholar]
- 44.Szumilas D, Krysiak R, Okopien B. The role of TLR4 receptor in development of inflammation and carcinogenesis in ulcerative colitis and pharmacotherapy of this disorder. Wiad Lek. 2013;66:3–9. [PubMed] [Google Scholar]
- 45.Lu D, Han C, Wu T. 15-hydroxyprostaglandin dehydrogenase-derived 15-keto-prostaglandin E2 inhibits cholangiocarcinoma cell growth through interaction with peroxisome proliferator-activated receptor-gamma, SMAD2/3, and TAP63 proteins. J Biol Chem. 2013;288:19484–19502. doi: 10.1074/jbc.M113.453886. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 46.Nelson SM, Lei X, Prabhu KS. Selenium levels affect the IL-4-induced expression of alternative activation markers in murine macrophages. J Nutr. 2011;141:1754–1761. doi: 10.3945/jn.111.141176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bergstrom KS, Kissoon-Singh V, Gibson DL, Ma C, Montero M, Sham HP, Ryz N, Huang T, Velcich A, Finlay BB, Chadee K, Vallance BA. Muc2 protects against lethal infectious colitis by disassociating pathogenic and commensal bacteria from the colonic mucosa. PLoS Pathog. 2010;6:e1000902. doi: 10.1371/journal.ppat.1000902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dey I, Lejeune M, Chadee K. Prostaglandin E2 receptor distribution and function in the gastrointestinal tract. Br J Pharmacol. 2006;149:611–623. doi: 10.1038/sj.bjp.0706923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hernandez Y, Sotolongo J, Breglio K, Conduah D, Chen A, Xu R, Hsu D, Ungaro R, Hayes LA, Pastorini C, Abreu MT, Fukata M. The role of prostaglandin E2 (PGE 2) in toll-like receptor 4 (TLR4)-mediated colitis-associated neoplasia. BMC Gastroenterol. 2010;10:82. doi: 10.1186/1471-230X-10-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sheibanie AF, Yen JH, Khayrullina T, Emig F, Zhang M, Tuma R, Ganea D. The proinflammatory effect of prostaglandin E2 in experimental inflammatory bowel disease is mediated through the IL-23-->IL-17 axis. J Immunol. 2007;178:8138–8147. doi: 10.4049/jimmunol.178.12.8138. [DOI] [PubMed] [Google Scholar]
- 51.Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496:445–455. doi: 10.1038/nature12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.