

Abstract
Lentiviral vectors have become mainstream gene transfer vehicles for their ability to delivery and integrate into host cells. In RNA interference (RNAi) applications, lentiviral constructs constitutively express dsRNA molecules usually as short hairpin RNA (shRNA) enabling long-term gene silencing and when pseudotyped with a broad host glycoprotein envelope; allows a multitude of cell types to be transduced. Their successful use ultimately relies on the production of lentiviral particles in high-titer and uniformity. Typical methods require the transfection of three or more plasmids in which essential viral elements have been encoded separated so as to remain replication deficient. These transfection procedures are of critical importance; however, methods often vary among laboratories making it difficult to assess the overall efficiency of lentiviral particle production. In this report, we focused exclusively on this step and compared the overall impact of the commercial transfection reagent FuGENE 6 to FuGENE HD. We found that FuGENE HD resulted in at least 5-fold improvement in viral particle titer as assessed by the p24 standard ELISA assay. We present the complete optimized workflow and demonstrate this utility in which a single modification of this transfection step improved the lentiviral particle production.
Keywords: shRNA, FuGENE 6, FuGENE HD, RNAi, lentiviral particles, viral titer, plasmid, DNA
INTRODUCTION
Lentiviral vectors are widely used gene delivery vehicles in RNAi applications able to transduce different cell types including stem cell-derived, non-dividing, and slow-growing lines (1,2). Lentiviral based systems as opposed to other retroviruses, adeno-associated, and adenoviruses allow for the stable integration of RNAi expression cassette yielding long term expression and targeted knockdown of genes in the host. Typically, lentiviral vectors are derived from human immunodeficiency virus type 1 (HIV-1) and commonly pseudotyped with glycoprotein G from vesicular stomatitis virus (VSV-G) allowing for the transduction of virtually any cell type with high efficiency. These viruses have been modified such that factors required for replication are removed or separated from the HIV-1 genome to provide for safety and protection. In the current generation lentiviral vectors, these major viral elements are encoded into several plasmids to prevent possible recombination into replication competent viruses (3).
In screening applications, lentiviral systems offer the opportunity to study cellular functions up to a genome-wide scale and has largely evolved around two different approaches: pooled versus arrayed formats. RNAi libraries are currently available through a select number of commercial vendors in different shRNA platforms; a majority of which are pooled as opposed to arrayed formats. In pooled formats, libraries are either simple stem loop or mi-R30-based lentiviral vectors available from the following vendors Thermo Fisher Scientific, Cellecta, and Sigma-Aldrich, and are used as a bulk load of thousands of lentiviral particles to transduce a single population of cells in a one step transaction. Afterwards, deconvolution studies are required for hit selection and to eliminate off-targeting effects as the observed phenotype may be a result of multiple integration events in a single cell (4). For arrayed screening, the RNAi Consortium (TRC) libraries have been made available through Sigma-Aldrich and utilize lentiviral particles targeting one gene in a single transduction reaction (5). In contrast, deconvolution studies are not required as phenotypes can be individually scored for each shRNA systematically. Nevertheless, the successful application of these technologies in screening mainly depends on the ease of lentiviral production with sufficient yield and in high titer.
For lentiviral particle production, these engineered systems commonly utilize a three plasmid-based method in which all major viral elements are encoded separately in the packaging vector, envelope vector, and expression vector (6). The packaging vector contains the minimal genes required for HIV-1 structural proteins encoding such elements as gag, rev, and pol. The envelope vector encodes the VSV-G coat needed for pseudo-typing. The expression vector also known as transfer vector is the lentiviral backbone containing the simple stem loop or miR-30 based shRNA cassette driven by a promoter H1 or U6, fluorescent protein or antibiotic selection markers, and necessary viral packaging elements. Production methods rely on a packaging cell line, typically HEK293T cell line for transfection of the required plasmids to produce functional virus. This process is often demanding as hundreds to thousands of unique constructs must be made uniformly in sufficient titer to achieve lentiviral particles capable of infecting a large amount of cells. Packaging and envelope plasmids are readily available for purchasing and the plasmid purification systems for the expression plasmids are of high-quality; however, a wide variety of protocols such as calcium phosphate precipitation and transfection reagents can be applied in this system and lentiviral production seems to rely on the efficiency of these methods.
Current protocols for lentiviral production can vary significantly from different laboratories and therefore difficult to compare their effectiveness as there are no uniform practices. In this report, we focused solely on optimizing the transfection stage during cell line packaging and assessed the overall impact of transfection reagent on lentiviral particle production. First, we performed a direct comparison of commonly used FuGENE 6 to FuGENE HD reagent in HEK293T as the packaging cell line and only varied transfection reagents while maintaining constant ratio of plasmid to packaging mix. We measured the efficiency of FuGENE 6 in comparison to FuGENE HD by determining the overall titer using an ELISA assay for the HIV-1 p24 antigen and found a minimum 5-fold improvement in lentiviral particle production using FuGENE HD. Next, we demonstrate the utility of this application in a custom focused library comprised of 2,407 shRNA clones covering 461 genes supplied in an arrayed format. Here, we present our complete optimized workflow for lentiviral particle production starting from bacterial glycerol stocks through purification of the pLKO.1 expression plasmid containing a simple stem loop shRNA cassette for each clone and finally transfection of the three plasmid-based system into HEK293T cells to demonstrate its enhancement in overall titer with FuGENE HD.
MATERIALS AND METHODS
Cell culture and materials
HEK293T cells (ATCC, Manassas, VA) were grown at 37°C and 5% CO2-95% air in cell growth media containing DMEM, high glucose with L-glutamine, and sodium pyruvate supplemented with 10% heat inactivated FBS. Cell culture supplies were from Life Technologies (Carlsbad, CA) and Sigma-Aldrich (St. Louis, MO).
Liquid handling and dispensing
Several liquid handling and dispensing devices were used throughout this study. Transfection complexes and lentiviral supernatants were transferred using a 96 stainless steel head with disposable low-volume polypropylene tips on a PP-384-M Personal Pipettor (Apricot Designs, Monrovia, CA). The addition of cell suspensions and growth media was performed using the Multidrop 384 (Thermo Scientific, Waltham, MA).
Bacterial cultures and plasmid purification
A custom focused library comprised of 2,407 clones covering 461 genes was plated in 26 96-well plates as bacterial glycerol stocks; the plasmids contain the pLKO.1 expression vector carrying a cloning site between AgeI and EcoRI for the shRNA sequences with puromycin resistance marker for selection. The bacterial glycerol stocks were inoculated using a 96 head pin-tool into 1 mL of TB media containing carbencillin (Thermo Scientific) and cultured in 96 deep well plates (Corning 3961, Corning Life Sciences, Lowell, MA) for 14 h at 37°C on an orbital shaker (Excella E24, Eppendorf, Hauppauge, NY). The bacterial cultures were checked for growth by measuring optical density at 600 nm (OD600). Next, the 96 deep well plates were centrifuged at 3,000 rpm (Sorvall Legend T, Thermo Scientific) to form bacterial pellets and culture media was removed by gentle aspiration. The plates were processed using vacuum pump and filtration procedure following manufacturer's specifications with the Wizard SV 9600 Plasmid DNA Purification System (Promega, Madison, WI) and the concentration of purified pLKO.1 expression plasmid was determined by measuring absorbance at 260 nm (A260) (SpectraMax Plus384, Molecular Devices, Sunnyvale, CA). To check for contaminants, the absorbance was measured at 280 nm (A280) for presence of protein and all samples had a ratio of A260/A280 greater than 1.8 indicative of good overall quality.
Transfection reagent assessment on lentiviral particle production
HEK293T cell suspensions at 20,000 cells per well were dispensed into 96-well microtiter plates (Corning 3603) in 100 μL of media and incubated overnight at 37°C. The Lentiviral Packaging Mix (Sigma-Aldrich) containing the packaging plasmid and envelope plasmid were thawed from −20°C storage at room temperature in preparation for transfection. A master mix containing 16.5 μL of Opti-Mem (Life Technologies), 0.3 μL or 0.6 μL of FuGENE 6 and HD transfection reagent (Promega) diluted to 0.75 μL, and 1 μL of Lentiviral Packaging Mix was dispensed into an intermediate 96-well polypropylene storage plate (Corning 3357) containing 0.1 μg of expression plasmid and incubated for 15 minutes to promote plasmid-transfection reagent complex formation. Next, 20 μL was transferred into the cell plates and incubated for 48 h at 37°C. For the first harvest, 85 μL of lentiviral supernatant was transferred into an intermediate 96-well storage plate and stored at 4°C. Cell plates were replenished with 85 μL of media and incubated for an additional 24 h at 37°C. For the second harvest, 95 μL of lentiviral supernatant was transferred into the corresponding intermediate storage plate already yielding a total harvest of lentiviral particles in 180 μL. Plates were briefly centrifuged at 1,000 rpm to remove any cell debris and aliquots of lentiviral supernatants were stored at −80°C.
Lentiviral particle production of a custom focused library
HEK293T cell suspensions at 20,000 cells per well were dispensed into 26 96-well microtiter plates in 100 μL of media and incubated overnight at 37°C. To prepare for transfection, a master mix solution containing 16.5 μL of Opti-Mem, 0.6 μL of FuGENE HD transfection reagent diluted to 0.75 μL, and 1 μL of Lentiviral Packaging Mix was dispensed into an intermediate 96-well polypropylene storage plate containing 0.1 μg of expression plasmid followed by incubation for 15 minutes. Next, 20 μL of plasmid-transfection reagent complex was transferred into the cell plates and incubated for 48 h at 37°C. For the first harvest, 85 μL of lentiviral supernatant was transferred into an intermediate 96-well storage plate and stored at 4°C. Cell plates were replenished with 85 μL of media and incubated for an additional 24 h at 37°C to yield 95 μL of lentiviral supernatant for the second harvest. Plates containing a total harvest of 180 μL lentiviral particles were briefly centrifuged at 1,000 rpm to remove any cell debris and aliquots of lentiviral supernatants were stored at −80°C until use.
Lentiviral particle titer
Lentiviral particle titer was measured at 490 nm (A490) using the HIV-1 p24 Antigen ELISA assay (Perkin Elmer, Waltham, MA) according to manufacturer's specifications. A standard curve was prepared using the p24 controls and fitted using a linear regression model. Individual samples were measured in duplicate to calculate overall titer.
RESULTS and DISCUSSION
To assess the impact of transfection on lentiviral particle production, we focused exclusively on this step and evaluated the overall efficiency of two different commercially available transfection reagents. We selected the commonly used FuGENE 6 as a baseline in our approach and tested FuGENE HD in comparison. Previous work revealed FuGENE HD amongst a panel of 7 transfection reagents as the most optimal for DNA plasmids and we sought to adapt this for lentiviral particle production (7). The two transfection reagents were tested in parallel at two recommended concentrations while maintaining all other parameters constant and measured the resulting output for effectiveness.
In the first step of optimization, HEK293T cells as the packaging line was seeded into a 96-well source plate followed by transient transfection of a three plasmid-based lentivirus packaging system that separates critical viral elements as to remain replication deficient (6). We varied FuGENE reagents at 0.3 μL to 0.6 μL per well and maintained a constant ratio of pLKO.1 expression plasmid at 0.1 μg to 1 μL of packaging mix per reaction. Brightfield imaging revealed that FuGENE 6 at 0.3 μL and 0.6 μL per well alone did not produce any noticeable toxicity to the cells as compared to untreated control as the cell monolayer remained intact (Figure 1). Similarly, FuGENE HD at 0.3 μL and 0.6 μL per well alone did not produce any noticeable cytotoxicity (Figure 1) consistent with previous results that transfection reagent alone does not produce any antiproliferative effects (7). However, FuGENE 6 and FuGENE HD transfection reagent at 0.3 μL and 0.6 μL per well in combination with the packaging mix alone resulted in toxicity. On the contrary, transfection with packaging mix and the expression vector attenuated these effects indicating all components are necessary for complex formation to minimize cytotoxicity in the packaging cell line.
Figure 1. Production of lentiviral particles using FuGENE 6 and FuGENE HD.

Brightfield images of HEK293T cells 48 h after FuGENE transfection at 0.3 μL and 0.6 μL per well.
After the transfection step, cells were incubated for 48 h and 180 μL of supernatant was harvested over a 24 h period from each of the samples. To determine the titer, we utilized an ELISA assay to detect the presence of the HIV-1 p24 antigen on the surface of lentiviral particles. A standard curve was generated using p24 antigen controls and lentiviral particle samples from FuGENE 6 and FuGENE HD were compared to assess the overall performance of each reagent at the concentrations tested (Figure 2A). For FuGENE 6, transfection of expression plasmid with 0.3 μL per well did not yield any lentiviral particles while 0.6 μL per well produced a titer of 1.96 x 106 TU/mL (Figure 2B). Similarly, FuGENE HD at 0.3 μL per well yielded lentiviral particles at 1.68 x 106 TU/mL. Using FuGENE HD at 0.6 μL per well, we increased titer to 8.56 x 106 TU/mL representing a nearly 5-fold enhancement in production.
Figure 2. Comparison of lentiviral particle titer.
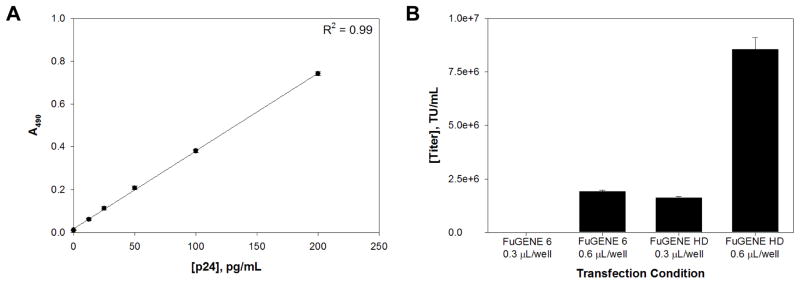
(A) Standard curve using controls in HIV-1 p24 Antigen ELISA assay. (B) Comparison of titers between FuGENE 6 and FuGENE HD at transfection reagent concentrations of 0.3 μl and 0.6 μL per well.
Following our optimized workflow, we demonstrated this utility for the production of a lentiviral particle library in an arrayed format. First, we assembled a custom focused library comprised of 461 genes contains approximately 5 shRNA clones per gene for a total of 2,407 clones across 26 96-well source plates as bacterial glycerol stocks. For ease, each of the source plates was inoculated into TB media using a 96 head pin-tool and grown overnight in deep well plates. The pLKO.1 expression plasmids were isolated from the bacterial glycerol stocks using a DNA purification system formatted for high-throughput applications. As a quality control, the plasmid concentration for each well across all source plates was calculated yielding an average of 20 μg/mL (Figure 3A) in 100 μL volume. Furthermore, all expression plasmids had a minimum A260/A280 ratio of 1.8 indicating high-quality extraction and at sufficient concentrations for multiple transfections. Next, we used FuGENE HD as our transfection reagent at 0.6 μL per well and continued with the lentiviral production process until all 26 96-well microtiter plates were completed. We measured a single lentiviral particle from each plate and obtained an average titer of 1.49 x 107 TU/mL across the entire procedure with lowest concentration at 6.66 x 106 TU/mL and highest at 3.39 x 107 TU/mL (Figure 3B). The entire process yielded 180 μL of supernatant for each of the 2,407 clones which enabled us to aliquot and store multiple copies of the library screening sets. With this current method, a single modification yielded approximately 3,000,000 lentiviral particles per clone to sufficiently transduce multiple cell lines at a given time.
Figure 3. Quantification of the custom focused library.

(A) Quantification of expression plasmid obtained using the Wizard SV 9600 Plasmid DNA Purification System for the custom focused library consisting of 461 genes with 2,407 shRNA clones (approximately 5 clones per gene). Each well across the 26 source plates was measured at A260 for pLKO.1 expression plasmid concentration. (B) Quantification of titer for one sample well across the 26 source plates using the HIV-1 p24 ELISA antigen assay at A490.
In conclusion, our optimized workflow using FuGENE HD can dramatically enhance lentiviral particle production that result in high-titer preparations. Using FuGENE 6, our optimization experiment yielded an average titer 1.96 x 106 TU/mL while our method with FuGENE HD increased the titer to an average of 1.49 x 107 TU/mL; allowing for many more transduction experiments to be processed at any given time. Although FuGENE HD is more expensive, it is important to take into consideration all the costs needed with FuGENE 6 to generate similar titer amounts including materials such as tissue culture plates, packaging cell line, expression plasmid, and packaging plasmids. Overall, we demonstrate that this simple modification yielded a dramatic improvement allowing for the optimized preparation of high-titer lentiviral particles in RNAi applications.
Supplementary Material
Acknowledgments
The HTS Core Facility is partially supported by Mr. William H. Goodwin and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research, the Experimental Therapeutics Center of MSKCC, the William Randolph Hearst Fund in Experimental Therapeutics, the Lillian S Wells Foundation and by an NIH/NCI Cancer Center Support Grant 5 P30 CA008748-44.
Footnotes
AUTHOR CONTRIBUTIONS
D.S. designed the study, performed the experiments, interpreted the data, and wrote the manuscript. H.D. managed the overall study and wrote the manuscript.
COMPETING INTERESTS
The authors declare no competing interests.
References
- 1.Lambeth LS, Smith CA. Short hairpin RNA-mediated gene silencing. Methods Mol Biol. 2013;942:205–232. doi: 10.1007/978-1-62703-119-6_12. [DOI] [PubMed] [Google Scholar]
- 2.Alimperti S, Lei P, Tian J, et al. A novel lentivirus for quantitative assessment of gene knockdown in stem cell differentiation. Gene Ther. 2012;19:1123–1132. doi: 10.1038/gt.2011.208. [DOI] [PubMed] [Google Scholar]
- 3.Dull T, Zufferey R, Kelly M, et al. A third-generation lentivirus vector with a conditional packaging system. J Virol. 1998;72:8463–8471. doi: 10.1128/jvi.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bhinder B, Djaballah H. Systematic analysis of RNAi reports identifies dismal commonality at gene-level and reveals an unprecedent enrichment in pooled shRNA screens. Comb Chem High Throughput Screen. 2013;16:665–681. doi: 10.2174/13862073113169990045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Root DE, Hacohen N, Hahn WC, et al. Genome-scale loss-of-function screening with a lentiviral RNAi library. Nat Methods. 2006;3:715–719. doi: 10.1038/nmeth924. [DOI] [PubMed] [Google Scholar]
- 6.Zufferey R, Dull T, Mandel RJ, et al. Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J Virol. 1998;72:9873–9880. doi: 10.1128/jvi.72.12.9873-9880.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Antczak C, Mahida JP, Singh C, et al. A high content assay to assess cellular fitness. Comb Chem High Throughput Screen. 2014;17:12–24. doi: 10.2174/13862073113169990056. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.