

Abstract
Random cationic copolymer brushes composed of 2-(dimethylamino)ethyl methacrylate (DMAEMA) and N-isopropylacrylamide (NIPAAm) were synthesized using the atom transfer radical polymerization (ATRP) method. The effects of varying the monomer feed ratios (30:70 and 70:30 DMAEMA:NIPAAm) and polymerization times on the film height, morphology and stimuli response to pH of the brush were evaluated. While the polymerization time was found to have little influence on the properties of the brushes, the monomer feed ratios had a great impact. The 70 % DMAEMA polymer brush had similar height as the 30 % DMAEMA brush after 45 min; however, it had a greater response to pH and morphological change compared to the 30 % DMAEMA. The 70 % DMAEMA brush was used to demonstrate an efficient approach to alleviate the ion suppression effect in MALDI analysis of complex mixtures by effectively fractionating a binary mixture of peptides prior to MALDI-MS analysis.
Keywords: Polymer Brush, Peptide Fractionation, Stimuli Response
Introduction
Polymers end grafted to the surface of inorganic substrate have become important in many fields of polymer science. Given a high enough grafting density, the steric hindrance of the polymer chains will force them to stretch away from the surface forming a “so called” polymer brush.1 For many polymer brushes, the microscopic properties, which are derived from the polymer structure and environment, determine the efficacy of their particular functions. For example, polymer brushes with different functional units have different micro- and sometimes macroscopic structures depending on their environment. In a good solvent system or in response to stimulation (pH, temp., press. etc), they can reorganize their conformation to adjust to their environmental stimulus. These polymer brushes are termed “responsive polymer brushes” or “smart materials” and they are of value in polymer science because of their wide range of technological applications. For instance, polymer brushes have been used in various applications such as protein fractionation,2 anti-fouling,3 environmentally responsive polymers,2b,3b,4 bio- and chemical sensing,5,6,7 cell adhesion and wetting,3,4,8,9 microfluidics,10 microfabrication,3a,6b molecular recognition,11 and optics.12
Stimuli responsive polymers have attracted interest in many fields; especially materials for biomedical science and technology.13 Specifically those containing the acrylate functional group have been exploited for their dual response to temperature and pH. Several combinations of polyacrylic anions/PNIPAAm copolymers have been prepared, such as polymethacrylic acid (PMAA),2d polyacrylic acid (PAAc),14,15 polyhydroxyethyl methacrylate (HEMA),16 and poly(2-aminoethylmethacrylate) (PAEMA).2c One recurrent combination of thermal/pH polymer consists of 2-(dimethylamino)ethyl methacrylate (DMAEMA) and N-isopropylacrylamide (NIPAAm). For example, Liu et al. has reported the use of a pH-dependent thermo-responsive amphiphilic star block copolymer comprised of a hydrophobic poly(methyl methacrylate) (PMMA) block and hydrophilic poly(NIPAAm-co-DMAEMA) tri-arm block for drug delivery,17 and Hinrichs et al. explored the use of various ratios of DMAEMA and NIPAAm copolymers to deliver DNA.18 Owing to the importance of these stimuli responsive polymer films, it is important to continue studying their micro and macroscopic features under different conditions and using different techniques in order to improve their physical properties and expand their applications.
In previous studies, we demonstrated the efficiency of polyelectrolyte copolymer brushes of 70% PNIPAAm and 30% PMAA,2d and also 70% PAEMA and 30% PNIPA Am2c in the fractionation of peptides and small proteins. Those brushes were synthesized by the photoinitiation free radical polymerization method; however it is well known that free radical polymerizations suffers from poor control over the macromolecular structure including molecular weights, polydispersity, chain architecture and composition. This lack of control over the structure of the polymer chains typically results in inconsistent structures and, thus, surfaces. Although the brushes were effective in the separation process, it is necessary to have reproducible surfaces that can be altered in a controlled manner in order to produce a more robust system. In this paper, we explore the use of surface initiated – atom transfer radical polymerization (SI-ATRP) as a controlled polymerization method to synthesize random copolymers of NIPAAm and DMAEMA with different monomer feed rations (Scheme 1), which to our knowledge has not been explored before. Previous reports of NIPAAm/DMAEMA copolymers consisted of diblock copolymers prepared using a controlled polymerization technique such as ATRP,19,20 or random polymers prepared by free radical polymerization methods that are not controlled.21 We investigated the effects of the monomer feed ratio and polymerization time on the swelling properties and surface morphology of the polymers. Finally, the copolymers were used as a substrate to fractionate a dual peptide mixture.
Scheme 1.
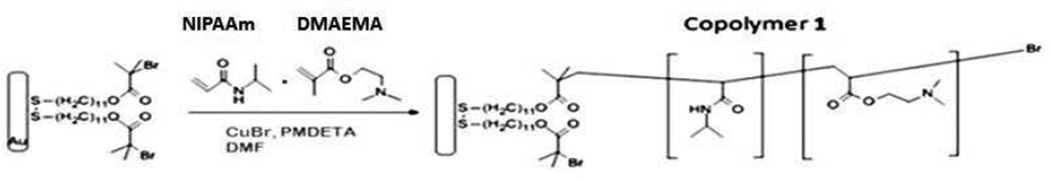
Representative synthesis of PDMAEMA/PNIPAAM random copolymer brush by ATRP
Materials and Methods
All supplies were purchased from Acros, Aldrich, or Fisher Scientific and used without further purification unless otherwise stated. The initiator (BrC(CH3)2COO(CH2)11S)2 (SAM-Br) that was used in the ATRP synthesis was prepared as previously described.22 Molecular weight measurements were performed by Waters Alliance 2690 GPC using a miniDAWN® multi-angle light scattering detector and a refractive index detector Waters 2410. AFM film thickness was carried out with VeecoNanoman in a tapping mode using NSG30 tip by NT-MDT. SAM thickness of 1.34 ±0.05nm was adjusted for in the final film height. For ozone cleaning of gold substrates, a Jelight UVO Cleaner Model 42 was used.
Brush synthesis
SAM Formation
A 2 × 2.5 cm gold coated Si wafer was cleaned in Piranha solution for 60 minutes, after which it was rinsed with Milipore-filtered water, followed by HPLC grade dimethylformamide (DMF) solution. The wafer was then blown dry with liquid nitrogen boil off and then placed in the ozone cleaner for 45 minutes. Next, the wafer was placed in a vial containing a 5 mM of ATRP initiator (SAM-Br) in ethanol for 18 hours. Once removed, the wafer was rinsed with DMF three times and dried with nitrogen. A Nicolet 670 FTIR spectrometer with a nitrogen cooled MCT-B detector and PIKE grazing angle accessory at 80° grazing angle was used to collect IR spectra of the modified wafers. Self -assembly of a monolayer was confirmed by reflection absorption infrared spectroscopy (RAIRS) and the presence of the initiator was confirmed by observing carbonyl moiety at 1735 cm−1, and C-O stretch at 1300 cm−1.
ATRP
A desired concentration of DMAEMA, NIPAAm and 1,1,4,7,7-pentamethyldiethylenetriamine (PMDETA) (0.14 µL) totaling 33 mmol were dissolved in 5 mL of DMF, followed by three freeze-thaw cycles inside of a Schlenk tube. The Schlenk tube was then placed inside of a glove box and copper (I) bromide (10 mg) was added to the solution. Once the copper (I) bromide was dissolved, the gold wafer containing the SAM initiator was placed into the solution and the polymerization was allowed to take place. After the desired polymerization time, the reaction was terminated by removing the gold wafers from the solution and immersing them in DMF. The substrates were then rinsed with copious amount of DMF and dried with nitrogen blow-off. The RAIRS spectra of the brushes were recorded, and the NIPAAm carbonyl amide (I) band was found at 1662 cm−1, and amide (II) N-H band at 1528 cm−1. The DMAEMA was confirmed with the carbonyl band at 1721 cm−1.
Swelling Studies: pH
The samples were incubated in 3 mL of a 0.100 M citrate pH 5 buffer solution for 5 hours. The samples were removed and rinsed with nanopure water then dried with nitrogen blow off.
Solvent
for the solvent studies, a similar procedure was followed where the samples were incubation in 3 mL of THF solution for 5 hours. The samples were removed and dried with nitrogen blow off.
Peptide Procedure (β-casomorphin and bradykinin)
The polymer brush (70:30 DMAEMA/NIPAAM copolymer) was initially placed in 100 µL of a 0.100 µM ammonium acetate buffer solution (pH 7) and allowed to stand for 1 hour. Next, a 1 µL aliquot of a peptide mixture (10 pmol β-casomorphin: 1 pmol bradykinin) was placed directly onto a conventional MALDI target (control sample) and onto the polymer-modified surface. After 3 minutes, the residual solution on the polymer-modified surface was removed with a pipet and deposited onto a separate location on a conventional MALDI target. Subsequently, three 1 µL aliquots of acetate buffer solution were deposited onto the peptide coated polymer-modified surface, removed immediately by pipet, and then combined with the originally removed droplet on the MALDI target. Next, 1 µL aliquots of 6 mg/mL α-cyano-4-hydroxycinnamic acid (CHCA) in 0.1% trifluoroacetic acid (TFA) were added to the control sample and to the polymer-fractionated combined droplets on the MALDI target and MALDI mass spectra were acquired from these samples. The peptides that remained bound to the polymer brush were analyzed directly on the polymer brush. Direct analysis was accomplished by placing 1 µL of 6 mg/mL CHCA in 0.1% TFA solution directly on the peptide coated polymer brush. MALDI mass spectra were then acquired directly from the brush polymer surface.
Results and Discussion
The cationic copolymer brushes consisting of either 30:70 DMAEMA/NIPAAm or 70:30 DMAEMA/NIPAAm monomer feed ratio were polymerized for different periods of time (Figure 1). It is assumed here that the heights of the films, which were measured using AFM, reached a maximum at about 45 min before leveling off. Interestingly, the brush heights for the 70:30 DMAEMA/NIPAAm were about 5 nm lower than the 30:70 DMAEMA/NIPAAm in the initial stage of the polymerization; however, an increase in the polymerization rate occurred after 30 min and the final heights of the two brushes were essentially the same at 12 h. It should be noted that the assumption is made that the height of the films correlate proportionally with the length of the polymer and the ratio of the monomer units in the final polymer is proportional to the monomer feed ratio as was reported by Dyer and co-workers.2e The lower film heights in the early stage of polymerization at the higher cationic monomer ratio suggest that DMAEMA has slower kinetics compared to NIPAAm for this process. Zhang et al. synthesized homopolymer brushes of PNIPAAm and PDMAEMA grafted from a nylon membrane by ATRP and showed that the rate of polymerization was slower for DMAEMA than for NIPAAm.20 This report corroborates our finding that the polymerization rate was slower for the brush containing higher ratio of DMAEMA.
Figure 1.

AFM height data for 30:70 and 70:30 DMAEMA:NIPAAm polymer brush at different polymerization times
The swelling behavior, or stimuli response, of the films is an important property that affects the performance of the brush; therefore, monitoring this process is essential. The swelling heights and conformational changes of the brushes are directly related to the functional groups and their ability to interact with their environment. The stimuli response to pH is of particular interest to us, since our application is oriented towards proteomics, in which we use pH to drive the binding of peptides and small protein to the ionic brushes. The swelling response of the brushes as a function of pH was monitored by both AFM and contact angle. The response of the polymer brushes to pH is a result of the ionization of the DMAEMA group at low or acidic pH (<7); as the pH is adjusted to increase the number of charges in the brush, there is an increase in the electrostatic repulsion causing the polymer chains to stretch out into the solution.23,24 The swelling heights of polyelectrolytes brushes can also be affected by high salt concentration due to screening by the charges, which results in a decrease in the brush heights.25 However it was shown that the height of weak polyelectrolyte brushes increases at high salt concentration (0.10 M buffer solution) of monovalent ions such as sodium nitrate. The high dissociation constant for sodium carboxylate causes an increase in the charges species, which resulted in an increase in the osmotic pressure, causing swelling in the brush.26 This swelling behavior causes a change in the height of the brush film as well as the surface charge. The brush height is monitored by AFM while the surface charge is monitored by contact angle. Since the number of charged segments is higher in the 70 % DMAEMA brushes, it is expected that they will respond more significantly to pH as compared to the 30 % DMAEMA brushes. In this case, the NIPAAm groups are not expected to have significant impact in the swelling response of the brushes and are only integrated into the brush to control the hydrophobic/hydrophilic nature of the brush.15 The stimuli response experiments were carried out below the LCST of NIPAAm and therefore temperature should not play a role in the response.
The polymer brushes were stimulated by a pH 5 buffer solution and the swelling response of 30:70 and 70:30 DMAEMA/NIPAAm brush prepared at various polymerization times were monitored (Figure 2). As expected, the heights of the brushes increased upon changing from neutral conditions (pH 7) to the lower pH conditions (pH 5) due to the increased interaction of the charged portions with the aqueous environment. It is interesting to note that the changes in brush heights were consistent over the range of polymerization time, which attests to the controlled nature of the ATRP process. Specifically, the 30 % DMAEMA swollen brushes exhibited an average height increase of 2±0.14 nm over the neutral brush, while the average increase for the 70 % DMAEMA brushes was 3.63±0.06 nm (Figure 2A and 2B respectively). While the difference in the average height increase for the two brushes was expected since the amount of the charged species is higher in the 70 % DMAEMA brush, a larger increase was expected for the 70 % DMAEMA brush than was observed. At this point, the explanation for the small increase in the swelling height for the 70% DMAEMA brush is unclear. One possible explanation is that the brushes shrink as the water is removed in order to perform the AFM measurements; therefore, the true swelling heights of the brushes are not recorded. As the brushes swell under the low pH conditions, the surface charge also increases. This is confirmed by the decrease in the contact angle as the brushes go from the neutral conditions to the low pH conditions (Figure 2C and 2D). Due to the higher percentage of the charged monomer in the 70 % DMAEMA brushes, there was a greater change in the contact angle; the average decrease in the contact angle is 40.93 ± 3.12 and 25.14 ± 2.67 for 70 % DMAEMA and 30 % DMAEMA, respectively.
Figure 2.

Swelling response to pH of the PDMAEMA/PNIPAAm brushes at different polymerization time and monomer feed ratio. A and C represent the height and contact angle of the 30 % DMAEMA brush respectively. B and D represent the height and contact angle of the 70 % DMAEMA brush respectively.
The swelling effect on the topography of the polymer brushes were investigated by tapping mode AFM. ATRP is expected to produce uniform films due to the controlled polymerization step. Figure 3 shows the AFM topography of the two brushes at pH 7 and pH 5 along with the gold substrate as the control. The surfaces of the two brushes after polymerization (pH 7) show homogeneity with some small grooves.27 The topology of the 70 % DMAEMA film surface is a bit rougher (Figure 3A.) having rms z-deviations of 3.27 nm compared to the 30 % DMAEMA film surface (Figure 3C), which has rms z-deviations of 2.49 nm. Although the surfaces were not smooth, their homogeneity denotes the uniformity of the polymerization process. The polymer brushes are expected to undergo conformational changes in response to changes in pH; this response is expected to produce a topographical change in the AFM micrographs. When both films were exposed to a pH 5 buffer solution, there was a significant change in the surface morphology (Figure 3B and 3D). These changes were likely a result of the reorientation of the polymer strands as they interact with the aqueous environment. The surfaces reveal a smoother morphology, yet with less uniformity, with rms z-deviations of 2.33 nm for the 70 % DMAEMA films (Figure 3B), and 1.36 nm for the 30 % DMAMA films (Figure 3D). The lack of uniformity in the pH treated films could be a result of polymer coagulating at the surface due to differences in the monomer sequence and ratio of different polymer chains, as this was a random polymerization process. Nonetheless, the difference in the changes in the morphology for the two polymers is consistent with the monomer ratios present; i.e., more change in the morphology occurred for the 70 % ionic monomer compared to the film containing 30 % ionic monomer. It is unclear to us whether the “bumps” in the pH treated brushes are connected to the ‘bumps’ in the uncoated gold substrates, since is it possible that the polymer could delaminate under the acidic conditions. We are currently probing this process by preparing brushes from silicon surfaces instead of gold and also looking at anionic polymers that respond at higher pH on both gold and silicon surfaces. In any event, the pH treated brushes provide sufficient charged surfaces that allow us to probe the fractionation of peptides.
Figure 3.

AFM spectra showing morphological changes In the brush surface before and after treatment In pH 5 buffer solutions at 45 min polymerization time A and B represent 70:30 DMAEAM/NIPAAM at pH 7 and pH 5 respectively. C and D represent 30:70 DMAEAM/NIPAAM at pH 7 and pH 5 respectively. E represents uncoated gold surface. (Bar scale 10 nm).
It was clear from the contact angle and height changes for the swelling experiments that the 70:30 DMAEMA/NIPAAm polymer brush had more surface charge and volume in response to pH compared to the 30:70 DMAEMA/NIPAAm polymer brush; therefore, that polymer brush was chosen to test the peptide fractionation application. We have previously demonstrated the use of ionic polymer brushes to separate a binary peptide mixture for subsequent MALDI-MS analysis.2c,2d Here we apply this polymer brush to separate a mixture of β-casomorphin (pI = 5.52, MW = 579.6) and bradykinin (pI = 12, MW = 1059). Due to the low ionization efficiency of the β-casomorphin, a large excess was needed in the control experiment in order to obtain a detectable signal in the mass spectrum in the presence of bradykinin (Figure 4A). A mixture of the peptides was deposited on the brush and MALDI MS was performed on both the unbound and bound peptides (Figure 4B and Figure 4C respectively) after fractionation. From the MALDI mass spectra shown, it is evident that the basic bradykinin peptide did not bind to the polymer surface; while as expected, the β-casomorphin was adsorbed to the polymer surface during the fractionation process. The isolation of the acidic β-casomorphin peptide using this approach substantially overcomes the ion suppression effect seen in the control experiment. It is worth noting in this example that the MALDI analysis of the bound peptide was performed directly on the polymer surface which was used as the MALDI target. Careful examination of the unbound peptides in Figure 4B reveals that a small signal from β-casomorphin was detected in this fraction. This result is likely due to saturation of the polymer brush with β-casomorphin resulting in excess β- casomorphin appearing in the unbound peptide fraction. Regardless of the reason, the MALDI mass spectra clearly reveal that the polymer brush modified substrate can effectively fractionate the peptide mixture.
Figure 4.

(A) MALDI-MS spectra of a 6:1 ratio ²–casomorphin fragment. (B) shows the spectrum of the unbound peptide washed off the surface. (C) shows the unbound peptide washed off the surface. (C) shows the spectrum of the peptide that was bound to the surface.
Conclusion
Mixed cationic copolymer nanobrushes with different monomer feed ratios were synthesized on gold substrate by the ATRP method. The copolymer nanobrush response to changes in the pH of the solution was evaluated by contact angle and AFM measurements as a function of polymerization time. As expected, the film heights increased in response to the change in pH and as the polymerization times increased. The contact angle decreased as the polymer brush films became more ionic in character due to the protonation of the DMAEMA moiety. Furthermore, a striking difference in the topography of the films with change in the pH was recorded by AFM. Specifically, the films went from a rougher, yet homogeneous surface before treatment with a pH 5 buffer solution to a smoother, yet less uniform surface after pH treatment. The film height, contact angle and topography changes were more pronounced in the 70 % DMAEMA brushes compared to the 30 % DMAEMA brushes. Ultimately, application of the 70 % DMAEMA brush for the fractionation of a binary mixture of peptides prior to MALDI MS analysis was effective and this approach may serve as an efficient approach to alleviate the ion suppression effect in MALDI analysis of complex mixtures.
Acknowledgments
We thank the National Science Foundation (CHE-0719426) and the National Institutes of Health (NIGMSR15GM083325) for financial support. The authors also thank Southern Illinois University for partial support of this research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jordan R. Advances in Polymer Science: Surface Initiated Polymerization I. 197th ed. Vol. 1. Springer; 2006. [Google Scholar]
- 2.(a) Halperin A, Kröger M. Macromolecules. 2011;44:6986–7005. [Google Scholar]; (b) Morisada S, Namazuda K, Suzuki S, Kikuchi N, Kanda H, Hirokawa Y, Nakano Y. Ind Eng Chem Res. 2011;50:12358–12365. [Google Scholar]; (c) Mitrovic B, Eastwood S, VenNey W, Dyer D, Kinsel G, Scott C. Langmuir. 2012;29:696–700. doi: 10.1021/la3033995. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Wong VN, Fernando G, Wagner AR, Zhang J, Kinsel GR, Zaucher S, Dyer DJ. Langmuir. 2009;25:1459–1465. doi: 10.1021/la802723r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Kaholek M, Lee W-K, Feng J, LaMattina B, Dyer DJ, Zauscher S. Chem. Mater. 2006;18:3660–3664. [Google Scholar]
- 3.(a) Gunkel G, Weinhart M, Becherer T, Haag R, Huck WTS. Biomacromolecules. 2011;12:4169–4172. doi: 10.1021/bm200943m. [DOI] [PubMed] [Google Scholar]; (b) Sileika TS, Kim H-D, Maniak P, Messersmith PB. ACS Appl Mater Interfaces. 2011;3:4602–4610. doi: 10.1021/am200978h. [DOI] [PubMed] [Google Scholar]; (c) Gon S, Santore MM. Langmuir. 2011;27:15083–15091. doi: 10.1021/la203293k. [DOI] [PubMed] [Google Scholar]
- 4.Lee H-S, Eckmann DM, Lee D, Hickok NJ, Composto RJ. Langmuir. 2011;27:12458–12465. doi: 10.1021/la202616u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Sheth SR, Leckband D. Proc. Natl. Acad. Sci. 1997;94:8399–8404. doi: 10.1073/pnas.94.16.8399. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Becker AL, Welsch N, Schneider C, Ballauff M. Biomacromolecules. 2011;12:3936–3944. doi: 10.1021/bm200954j. [DOI] [PubMed] [Google Scholar]; (c) Löbbicke R, Chanana M, Schlaad H, Pilz-Allen C, Günter C, Möhwald H, Taubert A. Biomacromolecules. 2011;12:3753–3760. doi: 10.1021/bm200991b. [DOI] [PubMed] [Google Scholar]
- 6.(a) Kricka L. Clin. Chim. Acta. 2001;307:219–223. doi: 10.1016/s0009-8981(01)00451-x. [DOI] [PubMed] [Google Scholar]; (b) Krishnan M, Namasivayam V, Lin R, Pal R, Burns MA. Curr. Opin. Biotech. 2001;12:92–98. doi: 10.1016/s0958-1669(00)00166-x. [DOI] [PubMed] [Google Scholar]
- 7.Rouhi AM. Chem. Eng. News. 1997;75:41–45. [Google Scholar]
- 8.(a) Huang N-P, De Paul SM, Textor M. Biomacromolecules. 2011;12:4213–4220. doi: 10.1021/bm2009872. [DOI] [PubMed] [Google Scholar]; (b) Gao G, Yu K, Kindrachuk J, Brooks DE, Hancock REW, Kizhakkedathu JN. Biomacromolecules. 2011;12:3715–3727. doi: 10.1021/bm2009697. [DOI] [PubMed] [Google Scholar]
- 9.(a) Lenz P. Adv. Mater. 1999;11:1531–1534. [Google Scholar]; (b) Xia Y, Qin DYY. Curr. Opin. Colloid Inter. Sci. 2001;6:54–64. [Google Scholar]
- 10.Kataoka DE, Trolan SM. Nature. 1999;402:794–797. [Google Scholar]
- 11.Lahiri J, Isaacs L, Grzybowski B, Carbeck JD, Whitesides GM. Langmuir. 1999;15:7186–7198. [Google Scholar]
- 12.Lee CH, Lim HS, Kim J, Cho JH. ACS Nano. 2011;5:7397–7403. doi: 10.1021/nn202328y. [DOI] [PubMed] [Google Scholar]
- 13.(a) Alarcon CdlH, Pennadam S, Alexander C. Chem. Soc. Rev. 2005;34:276–285. doi: 10.1039/b406727d. [DOI] [PubMed] [Google Scholar]; (b) Kumar A, Srivastava A, Galaev IY, Mattiasson B. Progress in Polymer Science. 2007;32:1205–1237. [Google Scholar]; (c) Galaev IY, Mattiasson B. Trends in Biotechnology. 1999;17:335–340. doi: 10.1016/s0167-7799(99)01345-1. [DOI] [PubMed] [Google Scholar]
- 14.Han CK, Bae YH. Polymer. 1998;39:2809–2812. [Google Scholar]
- 15.Xia F, Feng L, Wang S, Sun T, Song W, Jiang W, Jiang L. Adv. Mater. 2006;18:432–436. [Google Scholar]
- 16.Quynh TR, Yoneyamab M, Maki Y, Dobashi T. J Appl Polym Sci. 2012;123:2368–2376. [Google Scholar]
- 17.Liu Y, Cao X, Luo M, Le Z, Xu W. J. Colloid Interface Sci. 2009;329:244–252. doi: 10.1016/j.jcis.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 18.Hinrichs LJ, Schuurmans-Nieuwenbroek NME, van de Wetering P, Hennink WE. J. Controlled Release. 1999;60:249–259. doi: 10.1016/s0168-3659(99)00075-9. [DOI] [PubMed] [Google Scholar]
- 19.Zhang B-Y, He W-D, Li W-T, Li L-Y, Zhang K-R, Zhang H. Polymer. 2010;51:3039–3046. [Google Scholar]
- 20.Zhang ZB, Zhub XL, Xua FJ, Neoha KG, Kanga ET. J. Membr. Sci. 2009;342:300–306. [Google Scholar]
- 21.Zha L, Hu J, Wang C, Fu S, Elaissari A, Zhang Y. Colloid. Polym. Sci. 2002:280 1–280 6. [Google Scholar]
- 22.Plunkett KN, Zhu X, Moore JS, Leckband DE. Langmuir. 2006;22:4259–4266. doi: 10.1021/la0531502. [DOI] [PubMed] [Google Scholar]
- 23.Ross RS, Pincus P. Macromolecules. 1992;25:2177–2183. [Google Scholar]
- 24.Rühe J, Ballauff M, Biesalski M, Dziezok P, Gröhn F, Johannsmann D, Houbenov N, Hugenberg N, Konradi R, Minko S, Motornov M, Netz RR, Schmidt M, Seidel C, Stamm M, Stephan T, Usov D, Zhang H. Adv. Polym. Sci. 2004;165:79–150. [Google Scholar]
- 25.Gelbert M, Biesalski M, Rühe J, Johannsmann D. Langmuir. 2000;16:5774. [Google Scholar]
- 26.Guo X, Ballauff M. Phys Rev E. 2001;64 doi: 10.1103/PhysRevE.64.051406. 051406. [DOI] [PubMed] [Google Scholar]
- 27.(a) Munirasu S, Karunakaran RG, Rühe J, Dhamodharan R. Langmuir. 2011;27:13284–13292. doi: 10.1021/la202855u. [DOI] [PubMed] [Google Scholar]; (b) Bates FS, Fredrickson GH. Phys Today. 1999:32. [Google Scholar]; (c) Zhao B, Brittain W. J. Prog. Polym. Sci. 2000;25:677. [Google Scholar]