

Abstract
The noninvasive imaging of σ-1 receptors (S1Rs) could provide insight into their role in different diseases and lead to novel diagnostic/treatment strategies. The main objective of this study was to assess the S1R radiotracer 18F-FTC-146 in rats. Preliminary squirrel monkey imaging and human serum/liver microsome studies were performed to gain information about the potential of 18F-FTC-146 for eventual clinical translation.
Methods
The distribution and stability of 18F-FTC-146 in rats were assessed via PET/CT, autoradiography, γ counting, and high-performance liquid chromatography (HPLC). Preliminary PET/MRI of squirrel monkey brain was conducted along with HPLC assessment of 18F-FTC-146 stability in monkey plasma and human serum.
Results
Biodistribution studies showed that 18F-FTC-146 accumulated in S1R-rich rat organs, including the lungs, pancreas, spleen, and brain. Pretreatment with known S1R compounds, haloperidol, or BD1047, before radioligand administration, significantly attenuated 18F-FTC-146 accumulation in all rat brain regions by approximately 85% (P < 0.001), suggesting radiotracer specificity for S1Rs. Similarly, PET/CT and autoradiography results demonstrated accumulation of 18F-FTC-146 in rat brain regions known to contain S1Rs and that this uptake could be blocked by BD1047 pretreatment. Ex vivo analysis of 18F-FTC-146 in the brain showed that only intact radiotracer was present at 15, 30, and 60 min, whereas rapid metabolism of residual 18F-FTC-146 was observed in rat plasma. Preliminary monkey PET/MRI studies demonstrated specific accumulation of 18F-FTC-146 in the brain (mainly in cortical structures, cerebellum, and vermis) that could be attenuated by pretreatment with haloperidol. HPLC of monkey plasma suggested radioligand metabolism, whereas 18F-FTC-146 appeared to be stable in human serum. Finally, liver microsome studies revealed that 18F-FTC-146 has a longer half-life in human microsomes, compared with rodents.
Conclusion
Together, these results indicate that 18F-FTC-146 is a promising tool for visualizing S1Rs in pre-clinical studies and that it has potential for mapping these sites in the human brain.
Keywords: σ-1 receptor, PET, 18F
Over the past few decades, the enigmatic σ receptor (SR) has been studied extensively (1–3). We now understand that SRs are ligand-regulated molecular chaperones (4) and that there are at least 2 subtypes—that is, σ-1 receptor (S1R) and σ-2 receptor (S2R) (5,6).
S1Rs are 223 amino acid, interorganelle signaling modulators (7) located predominately at the mitochondria-associated endoplasmic reticulum (ER) membrane (MAM). Upon ligand binding or extreme ER stress, S1Rs can translocate to other parts of the cell, including the extended ER network and the plasma membrane. At the MAM, S1Rs modulate the level of reactive oxygen species (8) and also chaperone inositol 1,4,5-triphosphate receptors to regulate Ca2+ signaling (4), whereas, at the plasma membrane, S1Rs can interact with a large variety of receptors, ion channels, and protein kinases (7).
There is increasing evidence linking the dysregulation of S1Rs to various neurologic conditions (9), including Alzheimer disease (10,11), neuropathic pain (12), and addiction (13); however, the exact role of S1Rs in these conditions remains unknown. Numerous S1R agonists and antagonists are currently being evaluated for different clinical applications. For example, the S1R agonist ANAVEX 2–73 is being assessed in clinical trials for its use in attenuating mitochondrial/oxidative stress in Alzheimer disease, whereas S1R antagonists BD1047 and S1RA are showing promise as potential treatments for neuropathic pain (14,15).
Imaging S1Rs via PET could serve as a useful means for detecting S1R-related diseases and monitoring their treatment noninvasively. Additionally, S1R PET could help us investigate the in vivo role of S1Rs in different diseases and enhance our understanding of the pathology/progression of such conditions.
Several PET radioligands have been developed for S1Rs, including but not limited to 11C-SA4503 (16), 18F-FM-SA4503 (17), 18F-FPS (18), 18F-SFE (19), and 18F-fluspidine (20). Most of these radioligands possess high affinity for S1Rs, and although some have progressed to human studies, each has its disadvantages, whether it be nonspecific binding or suboptimal selectivity. A radioligand with high affinity and high specificity and selectivity for S1Rs is required to accurately image and quantify these sites in a living subject.
Previously, we reported the synthesis, radiosynthesis, and initial evaluation of a new S1R radioligand 18F-FTC-146 (21). Results from in vitro binding studies revealed that 18F-FTC-146 has the highest affinity and selectivity of any reported radioligand for S1R over S2R to date. Furthermore, our reported mouse PET imaging data indicated specific binding of 18F-FTC-146 to S1Rs in vivo, thus highlighting its promise as an imaging tool for studying these orphan receptors. There was, however, some evidence of radiotracer metabolism in mice and subsequent PET signal in bone (21). Although only intact 18F-FTC-146 was present in the brain (i.e., radiolabeled metabolites were unable to penetrate the blood–brain barrier), metabolism leading to bone uptake is not desirable and can make quantitation of brain signal difficult because of the close proximity of skull to brain and the partial-volume effect of PET. That is, the radioactive signal in the skull can spill over into parts of the brain, thus affecting the accuracy of determining S1R levels.
Because the metabolic profile of a radioligand in mice does not necessarily reflect its stability in other species (22), we set out to evaluate the biodistribution, kinetics, and stability of 18F-FTC-146 in rats and nonhuman primates. The aim of this study was to assess 18F-FTC-146 in rats to determine whether it might be a suitable tool for investigating S1Rs in rat models of disease and to also gain more information about the potential of this radiotracer for eventual clinical translation.
MATERIALS AND METHODS
General
If not otherwise stated, chemicals were purchased from commercial sources and used without further purification. Haloperidol and BD1047 for rat studies were purchased from Sigma Aldrich, and Haloperidol Injections USP (Bedford Laboratories) was used for monkeys. 18F-FTC-146 was synthesized as previously described (21), with a minimum specific radioactivity of 247 ± 143 GBq/μmol at the end of bombardment (n = 17) and radiochemical and chemical purities greater than 99%. Radiochemistry and semipreparative high-performance liquid chromatography (HPLC) were performed using a TRACERlab FX-FN module (GE Healthcare) with an ancillary Dionex 680 pump and KNAUER ultraviolet detector K-2001. Analytical HPLC was performed on an Agilent 1200 series quaternary pump equipped with an autosampler, a photodiode array detector, and a model 105S single-channel radiation (Carroll & Ramsey Associates) detector. Isolated radiochemical yields and doses for animal studies were measured using a CRC-15 PET dose calibrator (Capintec). PET/CT imaging of rats was performed using a microPET/CT hybrid Inveon scanner (Siemens). Imaging of squirrel monkeys was performed using a microPET R4 model scanner (Siemens) fitted with a computer-controlled bed, 10.8-cm transaxial and 8-cm axial field of view, and no septa. All PET images were reconstructed with a 2-dimensional ordered-subsets expectation maximization algorithm and coregistered with CT or MR images using the Inveon Research Work-place image analysis software (version 4.0; Siemens). For metabolite analysis, a Dionex UltiMate 3000 HPLC system including ultraviolet detector outfitted with an autosampler and Bioscan Flow-Count radioactivity detector was used.
Animals
All experimental procedures involving animals were approved by Stanford University Institutional Animal Care and Use Committee. Adult Sprague–Dawley male rats had access to food and water ad libitum and were kept under a 12-h light–dark cycle. Socially housed squirrel monkeys lived in large species-appropriate caging in climate-controlled rooms maintained at approximately 26°C with a 12-h light–dark cycle. Cages were cleaned daily, and monkeys were provisioned with ad libitum fresh drinking water, commercial monkey chow, and fruit and vegetable supplements. Species-appropriate environmental enrichment was provided to all monkeys at all times.
Ex Vivo Biodistribution and Stability in Rats
Rats were administered 18F-FTC-146 (11.0–18.5 MBq) via tail vein injection and then sacrificed by bilateral thoracotomy while under anesthesia at predetermined time points (5, 15, 30, and 60 min after injection of the tracer; n = 4 for each time point). To evaluate specificity of 18F-FTC-146, biodistribution studies were performed after rats were pretreated with known S1R ligands—that is, rats were injected with haloperidol (1 mg/kg, n = 4) or BD1047 (1 mg/kg, n = 4) via tail vein 10 min before radioligand administration and sacrificed 60 min later.
Blood samples (300–500 μL) were collected via cardiac puncture and placed in heparinized tubes (BD Microtainer; BD Biosciences). After thoracotomy of the anesthetized rats, organs (brain, heart, kidney, liver, lung, muscle, pancreas, skull, intestine, spleen, stomach, and femur bone) were quickly removed, weighed, and placed in tubes for γ counting. The brain of each rat was partially frozen on dry ice, and specific regions (cerebellum, frontal cortex, occipital cortex) were dissected, blotted, and weighed. The radioactivity in weighed organs was assessed via automated γ counter (Cobra II; Packard) and decay-corrected to the time of radiotracer injection using diluted aliquots of the initial administered dose as standards.
The stability of 18F-FTC-146 in rat plasma and brain homogenates at 15, 30, and 60 min after injection of the radiotracer (44.0–51.8 MBq) was determined using previously reported methods (21); however, in these studies methanol was used instead of acetonitrile for extractions and a Bio-Rad radioactivity detector was used to enable more sensitive detection of potential radiometabolites. Additionally, we verified 100% recovery of radioactivity injected onto the HPLC column.
Rat PET/CT Imaging and Ex Vivo Autoradiography
Rats were anesthetized using isoflurane gas (3.0%–3.5% for induction and 2.0%–3.5% for maintenance). Static PET scans (10 min) were acquired at 50 min after the intravenous administration of 18F-FTC-146 (32–46 MBq) (n = 3). Blocking studies involved intravenous pretreatment of rats with BD1047 (1 mg/kg, n = 3) 10 min before tracer administration. A CT image was acquired just before each PET scan to provide an anatomic reference frame for the respective PET data.
After PET imaging, rats were perfused with saline (20 mL) and sacrificed via bilateral thoracotomy while under anesthesia. Each rat brain was subsequently removed and embedded in optimal-cutting-temperature compound (Tissue-Tek) so that coronal brain sections could be obtained for ex vivo autoradiography. Autoradiography was conducted using previously described methods (21). The anatomy of brain sections was confirmed by nissl staining (cresyl violet acetate; Sigma Aldrich), whereas muscle sections were stained with hematoxylin and eosin (Fisher).
Monkey PET and MR Imaging
Brain PET images of 4 squirrel monkeys (Saimiri sciureus), 2 males (960–1,200 g) and 2 females (680–780 g), were acquired. Monkeys were prepared for imaging by sedation with an intramuscular injection of ketamine hydrochloride (20 mg/kg; Vedco Inc.), followed by subcutaneous injection of atropine (0.04 mg/kg; Butler Schein). Once sedated, 20 mL of Lactated Ringer’s solution (Hospira) was given subcutaneously between the scapulae, and ophthalmic ointment was placed in both eyes. A 24-gauge intravenous catheter (abbocath-T) was inserted into the saphenous vein of one or both legs and secured with skin glue and cloth tape. A 13-cm, 0.4-mL, microbore extension set with a T-connector and injection port (Abbott Laboratories) was added to the catheter, and 3M Vetwrap bandage tape was used to further secure the catheter and extension to the leg. Patency of the intravenous catheter was maintained with sterile heparinized saline. Monkeys were positioned on the PET scanner bed using a custom-made semicylindric acrylic cradle (a 10-cm-diameter × 46-cm-long cylindric tube from Tap Plastics was halved to make a semicylindric cradle). Passive maintenance of normal body temperature (~38.5°C) during anesthesia was supported by wrapping the monkeys in a warm water blanket (Gaymar Industries) and placing a reflective blanket (REI) around the water blanket. Velcro safety belts were used to secure the cradle, monkey, and water blanket in place on the PET scanner bed. During the imaging procedure, anesthesia was maintained with isoflurane gas (0.5%–1.5%; Butler Schein) that was delivered with a gas anesthesia machine (VetEquip) via a facemask (AC-09508-medium; Kent Scientific Corp.). Heart rate, oxygen saturation, respiration rate, and body temperature were monitored continuously throughout the procedure from preimaging preparation through full recovery from anesthesia. Food was withheld starting from 1900 h (lights out) the night before the morning of the procedure.
The acquisition of dynamic PET data (20 × 15, 20 × 30, 10 × 60, 10 × 120, and 15 × 300 s) was commenced just before intravenous administration of sterile-filtered (GV filter, 0.22 μm, 33 mm; Millipore) 18F-FTC-146 (92.5–111 MBq) in 500 μL of sterile heparinized saline, followed by 500 μL of sterile heparinized saline to flush the catheter, and yielded 75 frames over a 120-min period. Blocking studies involved pretreating monkeys with haloperidol (0.1 mg/kg) 10 min before radioligand administration.
Throughout baseline PET scans, blood samples (300 μL) were collected via the femoral vein at 2, 5, 15, 40, 50, 75, and 110 min and were analyzed for stability of 18F-FTC-146 in plasma using previously described methods (21).
Each monkey underwent brain MR imaging on a 3T Signa MR scanner (GE Healthcare) using a wrist coil (BC-10 wrist coil; Mayo Clinic GE Healthcare). Whole-brain T1-weighted images were acquired in the coronal plane with a 3-dimensional inversion recovery-prepared fast spoiled gradient pulse sequence on the Signa 3T scanner (GE Healthcare): repetition time, 8.8 ms; echo time, 3.0 ms; inversion time, 300 ms; flip angle, 15; number of excitations, 4; matrix, 160 × 160; field of view, 8 cm; voxel size, 0.5 × 0.5 × 0.5 mm; slice thickness, 0.5 mm; gap, 0 mm; bandwidth, 15.6 kHz; and total scan time; 18 min. For each scan, monkeys were initially sedated with a subcutaneous injection of ketamine hydrochloride (20 mg/kg), atropine sulfate (0.04 mg/kg), and xylazine (2 mg/kg) (Vedco). Anesthesia was maintained as needed with isoflurane gas (0.5%–1.5%) via face-mask (AC-09508-medium; Kent Scientific Corp.), and passive maintenance of normal body temperature was supported by wrapping the monkeys in a warm water blanket (Gaymar Industries) secured in place by blankets and a custom plastic cradle. Earplugs were applied for protection from noise generated by the scanner. Heart rate, oxygen saturation, and body temperature were continuously monitored throughout the scan until the monkey was fully recovered. A Registered Veterinary Technician was present during PET and MR imaging preparations and imaging sessions.
Human Serum and Liver Microsome Studies
Previously described methods (21) were used to evaluate stability of 18F-FTC-146 in human serum samples and stability of nonradioactive FTC-146 in human liver microsomes.
Statistical Analyses
Statistical examinations were performed using IBM SPSS statistics software (version 20). Uptake of 18F-FTC-146 in different rat organs in baseline and blocking studies were compared by 1-way, between-group, ANOVA. Post hoc comparisons for statistically significant results were performed by the Tukey test. The Student t test was used to compare differences in uptake between rat organs at a given time point.
RESULTS
Rat Studies
The biodistribution of 18F-FTC-146 (11.0–18.5 MBq) in rats was investigated via ex vivo γ counting of harvested tissues at predetermined time points after radiotracer administration (Fig. 1). The mean percentage injected dose per gram (%ID/g) in blood and tissues at each time point is depicted in Figure 1. The highest uptake at 5 min after injection of 18F-FTC-146 was observed in the lung (4.91 ± 0.63 %ID/g), kidney (2.14 ± 0.29 %ID/g), pancreas (1.58 ± 0.21 %ID/g), spleen (1.34 ± 0.35 %ID/g), and brain (different regions ranged between 1.10 ± 0.11 and 1.71 ± 0.35 %ID/g). From 5 to 60 min after injection, the accumulation of 18F-FTC-146 in the lungs decreased by 75% (P < 0.01); whereas, radiotracer uptake in the pancreas, liver, and spleen did not significantly wash out throughout the course of the study (P = 0.31, 0.18, and 0.27, respectively). Uptake in the brain peaked at 15 min, remained relatively high until 30 min, and then began to gradually wash out between 30 and 60 min. Negligible uptake was observed in blood and muscle throughout the duration of the study, and accumulation of 18F-FTC-146 in bone was significantly lower than that observed in the brain—that is, radioactivity in the rat brain was 4-fold higher than that in the femur/skull at 30 min and 2.6-fold higher at 60 min, P < 0.05 (Fig. 1; Supplemental Table 1 [supplemental materials are available at http://jnm.snmjournals.org]).
FIGURE 1.

Biodistribution of 18F-FTC-146 (11.0–18.5 MBq) in rats at 5, 15, 30, and 60 min after injection of radioligand (n = 4 for each time point). Error bars represent SEM.
Blocking biodistribution studies (Fig. 2), whereby rats were pretreated with either haloperidol (1 mg/kg, n = 4) or BD1047 (1 mg/kg, n = 4) at 10 min before radiotracer administration, demonstrated a significant decrease in radioactivity in all regions of rat brain (Fig. 2) at 60 min after injection of 18F-FTC-146 (~85% decrease, P < 0.001). Additionally, both blocking compounds resulted in attenuated uptake of 18F-FTC-146 in the pancreas, spleen, and liver (>80%, >65%, and >70% reduction, respectively).
FIGURE 2.
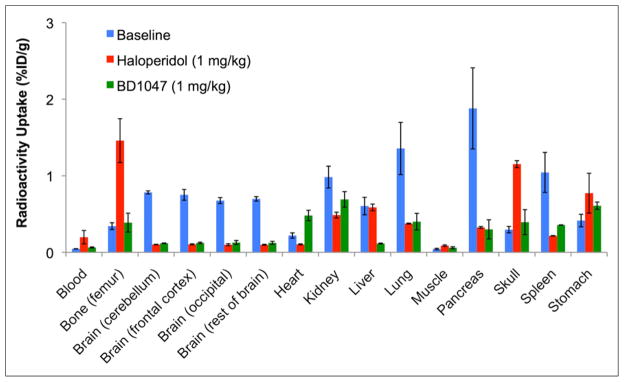
Biodistribution of 18F-FTC-146 (11.0–18.5 MBq) at 60 min after injection of radioligand in baseline rats (no pretreatment) (n = 4) and rats pretreated with either haloperidol (1 mg/kg; n = 4) or BD1047 (1 mg/kg; n = 4). Error bars represent SEM.
Baseline PET/CT images (Fig. 3A) demonstrate the ability of 18F-FTC-146 to cross the rat blood–brain barrier (BBB) and accumulate in brain tissue, although it is difficult to identify which exact brain regions from the images alone. Pretreatment with S1R-specific ligand BD1047 (1 mg/kg) resulted in a marked reduction in 18F-FTC-146 accumulation in the rat brain (Fig. 3B), compared with baseline images (Fig. 3A) (i.e., 86% reduction at 50–60 min after injection). PET/CT images also reveal some accumulation of radioactivity in the rat skull. Specifically, we found that the ratio of brain-to-skull uptake at 60 min was 3.1 ± 0.2 (Supplemental Table 1).
FIGURE 3.
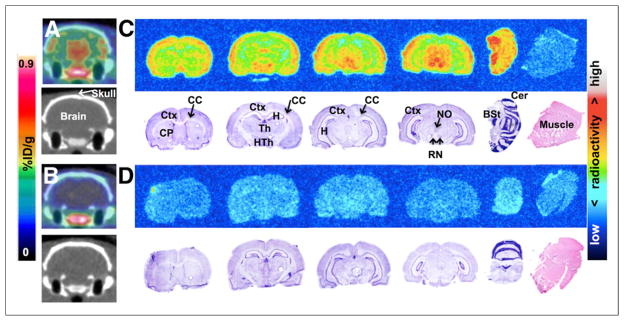
Rat brain PET/CT and ex vivo autoradiography. (A) Baseline coronal PET/CT images 50–60 min after intravenous administration of 18F-FTC-146 (30–35 MBq). (B) PET/CT images from blocking study, involving pretreatment with S1R-specific ligand BD1047 (1 mg/kg) 10 min before radioligand (30–35 MBq) injection. (C) Autoradiography (top) and nissl/H&E staining (bottom) of coronal brain and muscle sections from baseline study. (D) Autoradiography (top) and nissl/H&E staining (bottom) of coronal brain and muscle sections from blocking study. BSt = brain stem; CC = corpus callosum; Cer = cerebellum; CP = caudate-putamen; Ctx = cortex; H&E = hematoxylin and eosin; H = hippocampus; HTh = hypothalamus; NO = nucleus oculomotor; RN = red nucleus; Th = Thalamus.
The distribution of 18F-FTC-146 in rat brain was further evaluated by ex vivo autoradiography and was shown to mainly accumulate in the brain stem, cerebellum, cortex, hypothalamus, nucleus oculomotor, red nucleus, and thalamus, with some accumulation in the caudate putamen and hippocampus (Fig. 3C). There was little, if any, uptake of 18F-FTC-146 in the corpus callosum and muscle. Autoradiography results from the BD1047 blocking study (Fig. 3D) demonstrate a reduction of 18F-FTC-146 uptake in the brain to the level observed in muscle.
Ex vivo stability studies of 18F-FTC-146 in rat plasma and brain homogenates revealed a rapid decline of percentage intact radiotracer in plasma over time (27.1% ± 5.3%, 25.3% ± 1.3%, and 19.5% ± 4.4% at 15, 30, and 60 min, respectively); however, only intact 18F-FTC-146 was observed in the brain at all time points evaluated (Fig. 4). The average extracted radioactivity was 75% (n = 3), 76% (n = 3), and 70% (n = 3) for 15-, 30-, and 60-min plasma samples, respectively, and 71% (n = 3), 65% (n = 3), and 69% (n = 3) for 15-, 30-, and 60-min brain samples.
FIGURE 4.

Percentage of intact radiotracer in rat plasma and brain at 15, 30, and 60 min after injection of 18F-FTC-146 (44.0–51.8 MBq). Error bars represent SEM.
Monkey Studies
Preliminary brain PET imaging of squirrel monkeys was performed to evaluate the regional distribution and brain permeability of 18F-FTC-146 in a nonhuman primate. Both baseline and blocking studies were performed. The former consisted of a 120-min dynamic PET scan after administration of 18F-FTC-146 (93–111 MBq), whereas the latter involved pretreating the monkey with haloperidol (0.1 mg/kg) 10 min before radioligand injection.
Baseline brain PET/MR images (Fig. 5) show that 18F-FTC-146 was able to penetrate the monkey BBB and accumulate in the cingulate, frontal cortex, parietal cortex, temporal cortex, occipital cortex, hippocampus, striatum, cerebellum, and vermis. PET images also revealed some accumulation of radioactivity in the monkey skull (Fig. 6). The brain-to-skull ratio at 60 min after injection was found to be 1.2 ± 0.1 (Supplemental Table 1). Time–activity curves in Figure 6 illustrate that 18F-FTC-146 uptake reached an average maximum of approximately 0.4 %ID/g in all analyzed brain regions at 15 min after injection and then gradually washed out to a level of approximately 0.25 %ID/g over the remainder of the scan time. At 60 min after injection, the highest levels of radioligand uptake were seen in the cingulate (0.30 %ID/g), vermis (0.30 %ID/g), frontal cortex (0.28 %ID/g), cerebellum (0.27 %ID/g), and occipital cortex (0.27 %ID/g), whereas the lowest uptake was observed in the striatum (0.20 %ID/g) (Fig. 6). Pretreatment of monkeys with haloperidol attenuated the binding of 18F-FTC-146 by approximately 68%–77% at 60–75 min after injection in all brain regions (apart from the occipital cortex, which was decreased by 53%).
FIGURE 5.

Monkey brain PET/MR images from baseline dynamic imaging. Summed axial images are shown for 30–45, 60–75, and 105–120 min after administration of 18F-FTC-146 (92.5–111 MBq). Brain regions of interest are shown in MR images (top panel) and are labeled as (1) cingulate cortex, (2) frontal cortex, (3) striatum, (4) parietal cortex, (5) occipital cortex, (6) temporal cortex, (7) hippocampus, (8) cerebellum, and (9) vermis. ROIs = regions of interest.
FIGURE 6.

Monkey brain PET/MR images 60–75 min after injection of radiotracer, and time–activity curves demonstrating uptake of radiotracer in different brain regions for baseline (18F-FTC-146) and blocking (18F-FTC-146 + haloperidol 0.1 mg/kg) studies. p.i. = after injection.
HPLC analyses of monkey plasma samples indicated the presence of 1 polar metabolite with 60% intact 18F-FTC-146 at 5 min and 35% intact 18F-FTC-146 at 15 min, reaching a lower plateau of 15% intact parent tracer between 50 and 110 min (Supplemental Fig. 1). The average extracted radioactivity was 65% (n = 2), 55% (n = 2), 52% (n = 2), and 50% (n = 2) for 5-, 15-, 50-, and 110-min plasma samples, respectively.
Human Serum Stability and Liver Microsome Studies
Radiochromatograms from stability analyses of 18F-FTC-146 in human serum (Supplemental Fig. 2) displayed 1 single radioactive peak at the same retention time as 18F-FTC-146 at all time points evaluated (5 min through to 120 min).
The metabolic stability of FTC-146 was further evaluated by determining its half-life (t1/2) in human liver microsomes. Interestingly, FTC-146 was found to have a longer t1/2 in human liver microsomes (18.2 min) than our previously reported studies in mouse and rat liver microsomes (Table 1).
TABLE 1.
t1/2 of 18F-FTC-146 in Mouse, Rat, and Human Liver Microsomes
| Liver microsomes | t1/2 (min) |
|---|---|
| Mouse | 4.2 ± 0.6 |
| Rat | 12.6 ± 1.9 |
| Human | 18.2 ± 3.6 |
Errors are expressed as SEM.
DISCUSSION
S1Rs are key regulators of multiple neurochemical pathways and have been linked to certain neurologic disorders (9). The study of S1Rs in both normal and disease states in living subjects through the use of PET radioligands is of great value and will likely increase our understanding of S1R-associated conditions such as dementia and pain. The goal of this study was to characterize 18F-FTC-146, a previously reported S1R-targeting PET radioligand, in rats and monkeys with the aim of assessing its potential as an imaging tool for studying S1Rs in these species and also determining whether this tracer might be a suitable candidate for eventual clinical translation.
Rat ex vivo γ counting results revealed that 18F-FTC-146 was able to rapidly cross the rat BBB and accumulate in the brain (Fig. 1), which is known to contain a high density of S1Rs (23). Additionally, 18F-FTC-146 was shown to accumulate in peripheral organs containing S1Rs, including the liver, pancreas, and spleen (4). Uptake in bone, muscle, and blood was significantly lower than 18F-FTC-146 uptake in rat brain regions at all time points investigated (P < 0.005, <0.001, and <0.001, respectively), indicating that 18F-FTC-146 should enable relatively high signal-to-background brain PET imaging in rats. Blocking biodistribution studies (Fig. 2) resulted in significant attenuation of radioactivity in the brain (P < 0.001), suggesting specific binding of 18F-FTC-146 to S1Rs in this organ. There was also a significant reduction in radiotracer binding in the kidney (P < 0.05) and spleen (P < 0.05) after pretreatment with haloperidol, whereas pretreatment with BD1047 resulted in significant reduction in the liver (P < 0.05), pancreas (P < 0.05), and heart (P < 0.005). Disparities in blocking results for haloperidol and BD1047 may be due to the different affinities these compounds have for S1Rs or their differential binding profiles. Haloperidol not only binds to S1Rs but also dopamine receptor type 1/type 2, histamine H1, 5-hydroxytryptamine type 2 (5-HT2) receptors, α2 adrenergic receptors, S2R, and vesicular acetylcholine transporter. BD1047, on the other hand, has high affinity for S1Rs (Ki = 0.93 nM), approximately 50-fold lower affinity for S2Rs (Ki = 47 nM), and only low micromolar affinity for 5-HT2 receptors.
Increased radioactive signal in the bone/skull of rats pretreated with haloperidol, compared with baseline rats (Fig. 2), may be due to increased availability of 18F-FTC-146 in blood, resulting in increased metabolism or defluorination and hence increased bone uptake. It is unclear why BD1047 pretreated rats did not display this same trend.
PET/CT rat brain images (Fig. 3) from both baseline and blocking studies corresponded well with ex vivo autoradiography results, showing specific accumulation of 18F-FTC-146 in S1R-rich brain regions (e.g., brain stem, cortex, hypothalamus, mid brain/mesencephalon, cerebellum, red nucleus, and nucleus oculomotor) (16,24,25). The fact that pretreatment with BD1047 reduced the level of 18F-FTC-146 uptake in these brain structures to the level of uptake in muscle (known to contain low levels of S1Rs) (Fig. 3) provides evidence that the observed signal in baseline brain studies is due to specific binding to S1Rs.
Ex vivo analysis of 18F-FTC-146 stability in the brain revealed that only intact radiotracer was present at 15, 30, and 60 min, whereas rapid metabolism of residual 18F-FTC-146 was observed in rat plasma (Fig. 4). From this data, we can conclude that no radiolabeled metabolites were able to cross the BBB and that the radioactive signal observed in PET images and autoradiography was likely due to intact 18F-FTC-146 in the brain. It is important to note that brain-to-blood ratios at 15, 30, and 60 min were 23.7, 23.6, and 15.8, respectively, illustrating the high relative amount of tracer in brain (our target region), compared with blood. These results, along with acceptable brain-to-skull ratios (Supplemental Table 1), favorable kinetics, and good in vivo specificity for S1Rs, suggest that 18F-FTC-146 is suitable for imaging S1Rs in rat brain (at least up until 60 min). Currently, we are evaluating the utility of 18F-FTC-146 for visualizing S1R alterations in a rat model of neuropathic pain, in which our initial results appear promising. Full details of these studies will be reported in a forthcoming manuscript.
Preliminary PET/MR imaging studies of squirrel monkey brain showed that 18F-FTC-146 uptake was widely distributed throughout the brain, with marked accumulation in the cortical structures, cingulate, cerebellum, mid brain, and vermis (Fig. 5)—all regions known to contain high levels of S1Rs in monkeys (26). This binding profile appears similar to the uptake of 18F-FTC-146 we observed in the rat brain (Fig. 3) and also similar to the uptake of previously reported S1R radioligands 11C-SA4503 (27), 18F-FE-SA4503 (28), and 18F-FE-SA5845 (28), in awake Rhesus monkeys.
Pretreatment with haloperidol (0.1 mg/kg) reduced the binding of 18F-FTC-146 in most brain regions at 60–75 min after injection by 68%–77% relative to baseline (Fig. 6), suggesting specific binding to S1Rs in monkey brain. Our results were comparable to other reported haloperidol blocking studies using 11C-SA4503 (27), 18F-FE-SA4503 (28), and 18F-FE-SA5845 (28) in awake Rhesus monkeys, whereby the decrease in radioligand brain uptake after 1 mg of haloperidol per kilogram was 40%–50%, 40%–55%, and 60%–70%, respectively.
HPLC analyses of monkey plasma samples (Supplemental Fig. 1) showed a metabolic profile similar to what we observed in rat and mouse plasma (21)—that is, a diminishing amount of intact 18F-FTC-146 over time, with formation of 1 polar radiometabolite. This metabolic profile appears similar to that reported for 11C-SA4503 (27), 18F-FE-SA4503 (28), and 18F-FE-SA5845 (28) in monkey plasma (i.e., 31% unchanged 11C-SA4503 at 90 min after injection, whereas for 18F-FE-SA4503 and 18F-FE-SA5845 there was 23% and 14% parent compound present in plasma at 120 min after injection of tracer, respectively).
In the current monkey PET studies, accumulation of radioactivity was observed not only in the S1R-rich regions of the brain but also in skull (Fig. 6; Supplemental Table 1). As mentioned in the introduction, bone uptake due to radiotracer metabolism is unfavorable due to creating difficulties with quantitation of brain PET signal, which can affect the eventual utility of the radiotracer.
Although the metabolism of 18F-FTC-146 is a limitation of this tracer in rodent and nonhuman primate applications, this does not necessarily reflect what would occur in human studies, because there are examples of radiotracers in the literature that have defluorinated in nonhuman primates but have demonstrated stability in human studies (22). Furthermore, the fact that FTC-146 is more stable in human liver microsomes, compared with mouse and rat liver microsomes (Table 1), is a good indication that it might be more stable in humans. As we continue to gather information about this radiotracer concerning its metabolic stability and clinical translatability, we are simultaneously exploring different derivatives of 18F-FTC-146 to stabilize the C-F bond or adjust hydrophilicity.
Compared with other available S1R PET radiotracers (e.g., 11C-SA4503 and 18F-fluspidine), 18F-FTC-146 appears to have several key advantages, including its picomolar affinity (2.5 pm) for S1Rs; high brain uptake in mouse, rat, and monkey; favorable kinetic profile; and the fact that it has the highest selectivity for S1Rs over S2Rs of any reported S1R radiotracer.
CONCLUSION
Results from the present experiments in rats and nonhuman primates show that this radioligand accumulates in S1R-rich regions of the brain and that the PET signal appears to reflect specific binding to S1Rs in vivo. These results together with the fact that 18F-FTC-146 has the highest in vitro affinity and selectivity of any radioligand for S1R over S2R to date indicate the enormous potential of 18F-FTC-146 as an in vivo S1R imaging agent thus warranting further investigation. Our efforts to evaluate the utility of 18F-FTC-146 for investigating S1R-related disease and prospects for eventual clinical translation are ongoing.
Supplementary Material
Acknowledgments
We appreciate the technical help of Wendy Baumgardner RVT (Stanford University) for her assistance with nonhuman primate studies and Dr. Frezghi Habte (Stanford University) and Dr. Tim Doyle (Small Animal Imaging Facility, Stanford University) for their help with image analysis and imaging setup/instrument troubleshooting, respectively.
Footnotes
DISCLOSURE
This project was supported by NCI ICMIC P50 grant CA114747, NIDA grant DA023205, and NIGMS grant P20 GM104932. No other potential conflict of interest relevant to this article was reported.
References
- 1.Maurice T, Su TP. The pharmacology of sigma-1 receptors. Pharmacol Ther. 2009;124:195–206. doi: 10.1016/j.pharmthera.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guitart X, Codony X, Monroy X. Sigma receptors: biology and therapeutic potential. Psychopharmacology (Berl) 2004;174:301–319. doi: 10.1007/s00213-004-1920-9. [DOI] [PubMed] [Google Scholar]
- 3.Walker JM, Bowen WD, Walker FO, Matsumoto RR, De Costa B, Rice KC. Sigma receptors: biology and function. Pharmacol Rev. 1990;42:355–402. [PubMed] [Google Scholar]
- 4.Hayashi T, Su TP. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell. 2007;131:596–610. doi: 10.1016/j.cell.2007.08.036. [DOI] [PubMed] [Google Scholar]
- 5.Hellewell SB, Bruce A, Feinstein G, Orringer J, Williams W, Bowen WD. Rat liver and kidney contain high densities of sigma 1 and sigma 2 receptors: characterization by ligand binding and photoaffinity labeling. Eur J Pharmacol. 1994;268:9–18. doi: 10.1016/0922-4106(94)90115-5. [DOI] [PubMed] [Google Scholar]
- 6.Quirion R, Bowen WD, Itzhak Y, et al. A proposal for the classification of sigma binding sites. Trends Pharmacol Sci. 1992;13:85–86. doi: 10.1016/0165-6147(92)90030-a. [DOI] [PubMed] [Google Scholar]
- 7.Su TP, Hayashi T, Maurice T, Buch S, Ruoho AE. The sigma-1 receptor chaperone as an inter-organelle signaling modulator. Trends Pharmacol Sci. 2010;31:557–566. doi: 10.1016/j.tips.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsai SY, Hayashi T, Harvey BK, et al. Sigma-1 receptors regulate hippocampal dendritic spine formation via a free radical-sensitive mechanism involving Rac1xGTP pathway. Proc Natl Acad Sci USA. 2009;106:22468–22473. doi: 10.1073/pnas.0909089106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ishikawa M, Hashimoto K. The role of sigma-1 receptors in the pathophysiology of neuropsychiatric diseases. J Receptor Ligand Channel Res. 2009;2010:25–36. [Google Scholar]
- 10.Uchida N, Ujike H, Tanaka Y, et al. A variant of the sigma receptor type-1 gene is a protective factor for Alzheimer disease. Am J Geriatr Psychiatry. 2005;13:1062–1066. doi: 10.1176/appi.ajgp.13.12.1062. [DOI] [PubMed] [Google Scholar]
- 11.Jansen KL, Faull RL, Storey P, Leslie RA. Loss of sigma binding sites in the CA1 area of the anterior hippocampus in Alzheimer’s disease correlates with CA1 pyramidal cell loss. Brain Res. 1993;623:299–302. doi: 10.1016/0006-8993(93)91441-t. [DOI] [PubMed] [Google Scholar]
- 12.de la Puente B, Nadal X, Portillo-Salido E, et al. Sigma-1 receptors regulate activity-induced spinal sensitization and neuropathic pain after peripheral nerve injury. Pain. 2009;145:294–303. doi: 10.1016/j.pain.2009.05.013. [DOI] [PubMed] [Google Scholar]
- 13.Narayanan S, Mesangeau C, Poupaert JH, McCurdy CR. Sigma receptors and cocaine abuse. Curr Top Med Chem. 2011;11:1128–1150. doi: 10.2174/156802611795371323. [DOI] [PubMed] [Google Scholar]
- 14.Romero L, Zamanillo D, Nadal X, et al. Pharmacological properties of S1RA, a new sigma-1 receptor antagonist that inhibits neuropathic pain and activity-induced spinal sensitization. Br J Pharmacol. 2012;166:2289–2306. doi: 10.1111/j.1476-5381.2012.01942.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roh DH, Kim HW, Yoon SY, et al. Intrathecal injection of the sigma-1 receptor antagonist BD1047 blocks both mechanical allodynia and increases in spinal NR1 expression during the induction phase of rodent neuropathic pain. Anesthesiology. 2008;109:879–889. doi: 10.1097/ALN.0b013e3181895a83. [DOI] [PubMed] [Google Scholar]
- 16.Kawamura K, Ishiwata K, Tajima H, et al. In vivo evaluation of [11C]SA4503 as a PET ligand for mapping CNS sigma-1 receptors. Nucl Med Biol. 2000;27:255–261. doi: 10.1016/s0969-8051(00)00081-0. [DOI] [PubMed] [Google Scholar]
- 17.Kawamura K, Tsukada H, Shiba K, et al. Synthesis and evaluation of fluorine-18-labeled SA4503 as a selective sigma 1 receptor ligand for positron emission tomography. Nucl Med Biol. 2007;34:571–577. doi: 10.1016/j.nucmedbio.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 18.Waterhouse RN, Collier TL. In vivo evaluation of [18F]1-(3-fluoropropyl)-4-(4-cyanophenoxymethyl)piperidine: a selective sigma-1 receptor radioligand for PET. Nucl Med Biol. 1997;24:127–134. doi: 10.1016/s0969-8051(96)00184-9. [DOI] [PubMed] [Google Scholar]
- 19.Waterhouse RN, Zhao J, Stabin MG, et al. Preclinical acute toxicity studies and dosimetry estimates of the novel sigma-1 receptor radiotracer, [18F]SFE. Mol Imaging Biol. 2006;8:284–291. doi: 10.1007/s11307-006-0056-1. [DOI] [PubMed] [Google Scholar]
- 20.Fischer S, Wiese C, Maestrup EG, et al. Molecular imaging of sigma receptors: synthesis and evaluation of the potent sigma-1 selective radioligand [18F]fluspidine. Eur J Nucl Med Mol Imaging. 2011;38:540–551. doi: 10.1007/s00259-010-1658-z. [DOI] [PubMed] [Google Scholar]
- 21.James ML, Shen B, Zavaleta CL, et al. New positron emission tomography (PET) radioligand for imaging sigma-1 receptors in living subjects. J Med Chem. 2012;55:8272–8282. doi: 10.1021/jm300371c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pike VW. PET radiotracers: crossing the blood-brain barrier and surviving metabolism. Trends Pharmacol Sci. 2009;30:431–440. doi: 10.1016/j.tips.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cobos EJ, Entrena JM, Nieto FR, Cendan CM, Del Pozo E. Pharmacology and therapeutic potential of sigma-1 receptor ligands. Curr Neuropharmacol. 2008;6:344–366. doi: 10.2174/157015908787386113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bouchard P, Quirion R. [3H]1,3-di(2-tolyl)guanidine and [3H](+)pentazocine binding sites in the rat brain: autoradiographic visualization of the putative sigma 1 and sigma 2 receptor subtypes. Neuroscience. 1997;76:467–477. doi: 10.1016/s0306-4522(96)00221-7. [DOI] [PubMed] [Google Scholar]
- 25.Alonso G, Phan V, Guillemain I, et al. Immunocytochemical localization of the sigma-1 receptor in the adult rat central nervous system. Neuroscience. 2000;97:155–170. doi: 10.1016/s0306-4522(00)00014-2. [DOI] [PubMed] [Google Scholar]
- 26.Mash DC, Zabetian CP. Sigma receptors are associated with cortical limbic areas in the primate brain. Synapse. 1992;12:195–205. doi: 10.1002/syn.890120304. [DOI] [PubMed] [Google Scholar]
- 27.Ishiwata K, Tsukada H, Kawamura K, et al. Mapping of CNS sigma-1 receptors in the conscious monkey: preliminary PET study with [11C]SA4503. Synapse. 2001;40:235–237. doi: 10.1002/syn.1046. [DOI] [PubMed] [Google Scholar]
- 28.Elsinga PH, Tsukada H, Harada N, et al. Evaluation of [18F]fluorinated sigma receptor ligands in the conscious monkey brain. Synapse. 2004;52:29–37. doi: 10.1002/syn.20001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.