

Abstract
The evolutionary relationship and functional correlation between human formyl peptide receptors (FPRs) and their mouse counterparts remain incompletely understood. We examined three members of the mouse formyl peptide receptor subfamily (mFprs) and found that they differ in agonist preference and cellular distributions. When stably expressed in transfected rat basophilic leukemia (RBL-2H3) cells, mFpr1 was readily activated by N-formylated peptides derived from Listeria monocytogenes (fMIVTLF), Staphylococcus aureus (fMIFL), and mitochondria (fMMYALF). In contrast, the Escherichia coli–derived fMLF was 1000-fold less potent. The aforementioned peptides were much less efficacious at mFpr2, which responded better to the synthetic hexapeptide WKYMVm, the synthetic agonists Quin-C1 (a substituted quinazolinone), and compound 43 (a nitrosylated pyrazolone derivative). Saturation binding assays showed that mFpr1 and mFpr2 were expressed at similar levels on the cell surface, although their affinity for N-formyl-Met-Leu-Phe-Ile-Ile-Lys-fluorescein isothiocyanate varied by more than 1000-fold [dissociation constant (Kd) values of 2.8 nM for mFpr1 and 4.8 μM for mFpr2]). Contrary to these receptors, mFpr-rs1 responded poorly to all the previously mentioned peptides that were tested. Fluorescent microscopy revealed an intracellular distribution pattern of mFpr-rs1. On the basis of these results, we conclude that mFpr1 is an ortholog of human FPR1 with certain pharmacologic properties of human FPR2/ALX, whereas mFpr2 has much lower affinity for formyl peptides. The intracellular distribution of mFpr-rs1 suggests an evolutionary correlation with human FPR3.
Introduction
The G-protein–coupled formyl peptide receptors (FPRs) contribute to the migration of phagocytes to sites of infection and inflammation. The human FPR gene family has three identified members, FPR1, FPR2, and FPR3, which encode three distinct receptors (Migeotte et al., 2006; Rabiet et al., 2007; Ye et al., 2009). Among the three receptors, FPR1 and FPR2/ALX display high similarity (69%) in primary sequences. They also share several agonists, including the synthetic hexapeptide WKYMVm (Trp-Lys-Tyr-Met-Val-D-Met-NH2) and non-peptide molecules such as compound 43 (Le et al., 1999; Forsman et al., 2011). N-formylated peptides derived from bacteria or mitochondria are most potent chemoattractants for FPR1, triggering such phagocytic activities as chemotaxis, calcium mobilization, degranulation, and release of superoxide anions (Le et al., 2002). The tripeptide fMLF (N-formyl-Met-Leu-Phe) derived from Escherichia coli is the shortest high-affinity agonist for FPR1. As for FPR2/ALX, other bacteria or mitochondria-derived formyl peptides are more potent agonists than fMLF (Rabiet et al., 2005). FPR3 responds poorly to formyl peptides except fMMYALF (N-formyl-Met-Met-Tyr-Ala-Leu-Phe), a hexapeptide derived from mitochondrial NADH dehydrogenase subunits 4, which displayed an EC50 of 1 × 10−6 M in a calcium mobilization assay (Rabiet et al., 2005). Of note, FPR2/ALX is a highly promiscuous receptor that can interact with agonists of various structures, including small proteins, peptides, and synthetic molecules, such as serum amyloid A, lipoxin A4, and a substituted quinazolinone Quin-C1 [4-butoxy-N-(2-[4-methoxy-phenyl]-4-oxo-1,4-dihydro-2H–uinazolin-3-yl)-benzamide] (He et al., 2003; Nanamori et al., 2004; Chiang et al., 2006). In contrast, FPR3 has only a few high-affinity endogenous ligands, including F2L, an acetylated amino-terminal peptide of the heme-binding protein (Migeotte et al., 2005). FPR3 has shown unique constitutive internalization (Rabiet et al., 2011), and its physiologic function remains largely unknown.
The mouse Fpr gene family is more complex and contains at least eight related genes, including Fpr1, Fpr2, Fpr-rs1, Fpr-rs3, Fpr-rs4, Fpr-rs5, Fpr-rs6, Fpr-rs7, and Fpr-rs8 (Gao et al., 1998; Ye et al., 2009; Tiffany et al., 2011). The first three genes code for receptors (mFpr1, mFpr2, and mFpr-rs1) that are found in leukocytes (Gao and Murphy, 1993; Gao et al., 1999; Hartt et al., 1999; Southgate et al., 2008). Fpr1−/− neutrophils showed susceptibility to Listeria infection, suggesting that it has an important function in host defense against Listeria (Gao et al., 1999). mFpr1 was highly responsive to fMIFL (N-formyl-Met-Ile-Phe-Leu), while it showed low-affinity binding of fMLF (Southgate et al., 2008). Moreover, its putative ligand binding domains resemble those of FPR2/ALX rather than FPR1. mFpr2 and mFpr-rs1 show sequence similarity to human FPR2/ALX and FPR3, respectively. It is presently unclear whether Fpr-rs1 or Fpr2 is the ortholog of FPR2/ALX because both receptors are reported to respond to LXA4 (Takano et al., 1997; Vaughn et al., 2002). Additionally, mFpr2 has been found to be less responsive to fMIFL but serves as a receptor for F2L, a highly potent and specific agonist for human FPR3 (Gao et al., 2007; Southgate et al., 2008). These observations suggest that human FPRs, especially FPR1 and FPR3, have better defined and more specialized ligand binding properties than the mouse receptors.
The present study was undertaken to clarify the functional and binding properties of selected mouse Fpr family members. Using rat basophilic leukemia (RBL-2H3) cells stably transfected to express mFpr1, mFpr2, and mFpr-rs1, respectively, we compared the cellular response to formyl peptides and selected synthetic agonists (Table 1). Saturation and competition binding assays were also performed with these agonists to further elucidate the pharmacologic features of the mouse receptors.
TABLE 1.
The various agonists used in this study
| Sequence/Name | Origin |
|---|---|
| Formyl-MLF | Escherichia coli |
| Formyl-MLFK | Derivative of fMLF |
| Formyl-MLFE | Derivative of fMLF |
| Formyl-MLFW | Derivative of fMLF |
| Formyl-MIFL | Staphylococcus aureus |
| Formyl-MLFII | Derivative of fMIFL |
| Formyl-MLFIIK | Derivative of fMIFL |
| Formyl-MIVTLF | Listeria monocytogenes |
| Formyl-MMYALF | Mitochondria ND6 protein |
| WKYMVm | Peptide library screen |
Quin-C1 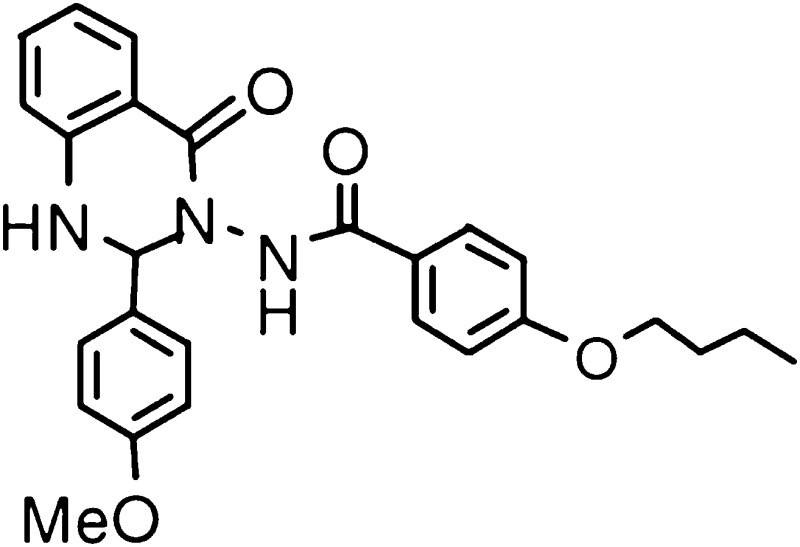
|
Small compound library screen |
Compound 43 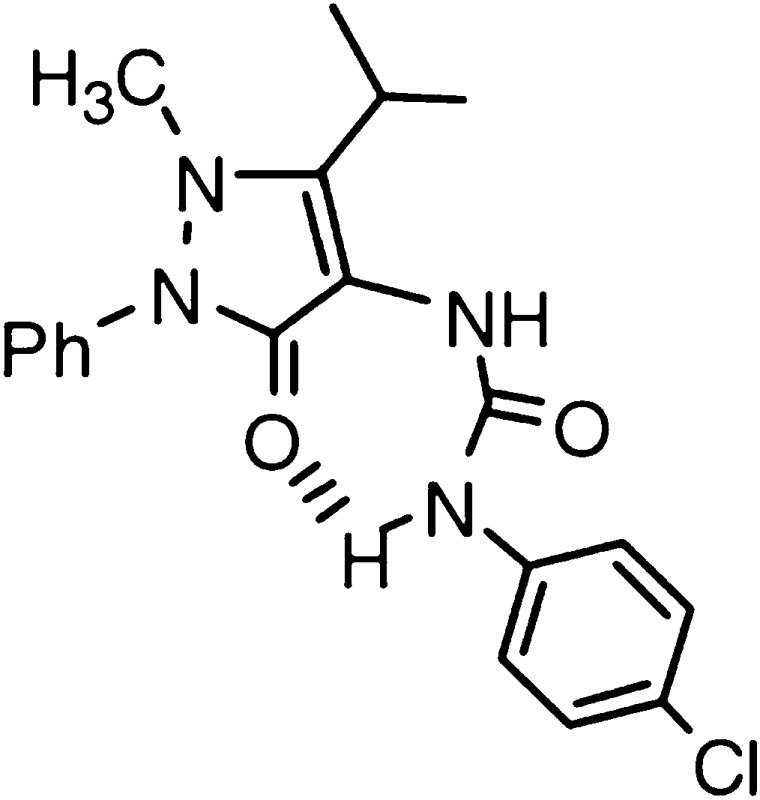
|
Small compound library screen |
fMLF, N-formyl-Met-Leu-Phe-Ala; ND6, NADH dehydrogenase 6.
Materials and Methods
Materials.
WKYMVm and other peptides except fMLF used in this study were synthesized at Shanghai Science Peptide Biologic Technology Co., Ltd. (Shanghai, China) and purified to ≥90% homogeneity. N-formyl-Met-Leu-Phe-Ile-Ile-Lys-fluorescein isothiocyanate (fMLFIIK-FITC) was synthesized, conjugated, and purified (≥96%) at Shanghai Science Peptide Biologic Technology Co., Ltd. fMLF (≥90% purity) was purchased from Sigma-Aldrich (St. Louis, MO). Quin-C1 was synthesized as previously described (Nanamori et al., 2004; Zhou et al., 2007). Compound 43 was synthesized in-house according to the method described (Burli et al., 2006). FLIPR calcium 5 reagent was obtained from Molecular Devices (Sunnyvale, CA). Other chemicals were purchased from Sigma-Aldrich.
Cell Culture.
The rat basophilic leukemia cell line RBL-2H3 (ATCC, Manassas, VA) was transfected with an expression vector SFFV.neo containing the mFpr1, mFpr2 or mFPR-rs1 cDNA, as previously described (He et al., 2000). Stable transfectants were selected with 250 μg/ml G418 (Invitrogen, Carlsbad, CA) after initial isolation with 500 μg/ml of G418. RBL stable cell lines were maintained at 37°C in 5% CO2 in Dulbecco’s modified Eagle’s medium supplemented with 20% heat-inactivated fetal bovine serum and 250 μg/ml G418.
Degranulation.
β-hexosaminidase release assay was performed as described (Nanamori et al., 2004). Briefly, RBL-2H3 cells (0.2 × 106/well) expressing mFpr1, mFpr2 or mFpr-rs1 were seeded in 24-well tissue culture plates for 24 hours. After washing twice with HBSS-HB (20 mM HEPES in Hank`s balanced salt solution [HBSS] with 0.5 mM CaCl2 and 1 mM MgCl2 supplemented with 0.1% bovine serum albumin, pH 7.4), cells were preincubated with 10 μM cytochalasin B for 15 minutes on ice and 15 minutes at 37°C successively. Agonists were then applied at desired concentrations. The reaction was terminated after a 10-minute stimulation by placing the plate on ice. β-Hexosaminidase release into the medium was determined by incubating 20 μl of supernatant or cell lysate with 10 μl of 1 mM p-nitrophenyl-N-acetyl-β-d-glucosamide in 0.1 M sodium citrate buffer (pH 4.5) at 37°C for 1 hour. At the end of the incubation, 200 μl of a 0.1 M Na2CO3/NaHCO3 buffer (pH = 10) were added. Absorbance was monitored at 405 nM in a FlexStation III Spectrometer (Molecular Devices). Values (means ± S.E.M.) were expressed as a percent of total β-hexosaminidase present in the cells.
Calcium Mobilization.
Cells were grown to 90% confluence in black wall/clear bottom 96-well assay plates. The calcium mobilization assay was performed using FLIPR calcium 5 reagent according to manufacturer’s protocol. After 1 hour incubation with the reagent (37°C, with 5% CO2), the agonists were added robotically, and samples were read in the FlexStation III with an excitation wavelength at 485 nm and an emission wavelength at 525 nm. Data were acquired by SoftMax Pro 6 (Molecular Devices) and analyzed with Origin 7.5 software (Northampton, MA). The dose response curves were plotted as means ± S.E.M. based on at least three experiments using 7–9 different concentrations of agonists.
Ligand Binding Assay.
RBL-stable transfectants were harvested, washed, and resuspended at 1 × 106/ml in extracellular buffer (HBSS plus 0.5% bovine serum albumin and 20 mM HEPES, pH 7.4) on ice. Binding assays were performed in Falcon polypropylene tubes (Becton Dickinson, Franklin Lakes, NJ). For saturation binding assays, fMLFIIK-FITC was used at concentrations from 10−10 to 10−5 M. Total binding and nonspecific binding were measured in the absence and presence of the unlabeled ligands in excess (50 μM WKYMVm or 50 μM fMLFIIK, respectively). The cells were equilibrated for 1 hour on ice and then analyzed for mean fluorescent intensity on a FACScan flow cytometer (Becton Dickinson), with dead cells excluded by gating on forward and side scatter. The dissociation constant (Kd) was calculated for specific binding using one-site binding hyperbola nonlinear regression analysis as shown in the equation: Bound = Bmax ×[L] / ([L] + Kd), where Bmax is the maximal number of binding sites, Kd is the concentration of labeled ligand (fMLFIIK-FITC) required to reach half-maximal binding, and [L] is the concentration of the labeled ligand. Receptor density was estimated with FITC-conjugated bead standards of known fluorescein equivalents (Quantum FITC-5 Premix, Lot #9975, Bangs Laboratories, Fishers, IN). A correction factor of 1.52 was used according to a previous study that showed the difference in fluorescence intensity between free fluorescein and peptide-conjugated FITC and the quenching upon binding to receptor (Vilven et al., 1998). The receptor number per cell was derived from an analysis of specific binding of fMLFIIK-FITC at saturated concentrations. The relative affinities of the nonfluorescent ligands were measured in competitive binding assays, in which fMLFIIK-FITC (50 nM for mFpr1-RBL cells or 5 μM for mFpr2-RBL cells) was added on ice for 1 hour prior to the addition of increasing concentrations of the competitors. Samples were incubated for another 1 hour on ice, and competitive binding curves were obtained by flow cytometry. IC50 and Hill coefficient (nH) values were calculated by fitting data points to the standard four-parameter logistic function, using nonlinear regression analysis with Origin 7.5. Experimentally determined values are given as the means ± S.E.M.
Receptor Internalization.
Enhanced green fluorescent protein (EGFP)-tagged mFpr constructs were prepared by ligation of mFpr cDNAs to the N-terminus of the EGFP coding sequence. RBL-2H3 cells were transfected to express individual mFpr-EGFP constructs, and stable transfectants were selected. N-terminal and C-terminal FLAG-tagged mFpr constructs were prepared and expressed in RBL-2H3 cells. FLAG-tagged mFpr-rs1 receptors were labeled with anti-FLAG monoclonal antibody (Sigma-Aldrich) for 1 hour at room temperature. Cells were washed three times and incubated with green fluorescent Alexa 488-conjugated goat anti-rabbit antibody (Molecular Probes, Eugene, OR) for 30 minutes at room temperature. For internalization, HeLa cells were transiently transfected to express EGFP-tagged mFpr constructs with Lipofectamine 2000 (Invitrogen, Carlsbad, CA). The transfected cells were grown on a glass cover slip for 24 hours in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum. The cells were washed briefly with HBSS-HB and stimulated with the testing agonists at 37°C for 30 minutes. Internalization was terminated by addition of fixation buffer (4% paraformaldehyde in phosphate-buffered saline) followed by incubation at room temperature for 15 minutes. The cells were then washed twice with phosphate-buffered saline and nuclei were stained by 4,6-diamidino-2-phenylindole (DAPI, Beyotime, Shanghai) for 10 minutes at room temperature. Fluorescent images were taken on a Leica SP5 II inverted fluorescent confocal microscope.
Results
Agonists Induced Degranulation in RBL-2H3 Cells Expressing mFpr1 or mFpr2 but Not mFpr-rs1.
To establish an agonist response profile for the mouse Fpr family, stable transfectants expressing mFpr1 (NCBI: NP_038549.1), mFpr2 (NCBI: NP_032065.1) or mFpr-rs1 (NCBI: NP_032068.2) were stimulated with various FPR agonists, including two synthetic small molecules (Quin-C1, Compound 43), a d-methionine-containing peptide (WKYMVm), and N-formylated peptides containing bacterial or mitochondrial protein sequences (Table 1). The release of β-hexosaminidase was measured, and the results were compared with these agonists (Fig. 1, A–C). WKYMVm, Quin-C1, and compound 43 activated both mFpr1 and mFpr2 with similar potency. In contrast, mFpr-rs1 hardly responded to any of the agonists tested except WKYMVm, which elicited granule release above 100 nM. Interestingly, in degranulation assays, mFpr1 responded poorly to fMLF, and mFpr2 did not respond to fMLF up to 10 μM. The poor response to fMLF prompted us to determine whether formyl peptides with different sequences could be better agonists for these mouse receptors. As shown in Fig. 1D, all but fMLFK induced degranulation equal or better than fMLF at mFpr1, suggesting that peptide length and/or composition are factors that influence the response of this receptor. For instance, the tetrapeptides fMLFK, fMLFE, and fMLFW and the pentapeptide fMLFII respectively showed 1.5-, 3.6-, 5.1-, and 6.7-fold higher potency compared with fMLF when tested at 1 μM. The formyl peptides derived from S. aureus (fMIFL), L. monocytogenes (fMIVTLF), and human mitochondria (fMMYALF) also had higher potency for mFpr1 (Fig. 1D). In contrast, none of the seven peptides tested was a potent agonist for mFpr2, and only fMLFII and fMLFK could induce about 3.5–5% of β-hexosaminidase release through mFpr2 when used at 10 μM (Supplemental Fig. 1C). These observations suggest that detection of N-formylated peptides may not be a primary function of mFpr2 and mFpr-rs1.
Fig. 1.

Degranulation induced by agonists through mFpr1, mFpr2 or mFpr-rs1 receptors. Release of β-hexosaminidase by fMLF, WKYMVm, Quin-C1, and compound 43 at indicated concentrations were measured in RBL cells expressing mFpr1 (A), mFpr2 (B), or mFpr-rs1 (C), respectively. Various formyl peptides (1 μM) were compared for the induction of β-hexosaminidase secretion (D). Values are mean ± S.E.M. of single duplicate determinations and representative of at least three separate experiments.
The Three mFprs Responded Differently to Agonists in Calcium Mobilization Assays.
Agonists were further assayed for their ability to trigger calcium mobilization through activation of three chosen members of the mouse Fpr family. In Fig. 2, representative tracings showing relative concentrations of intracellular Ca2+ are displayed in the upper panels, and dose response curves are shown in the lower panels for the individual agonists tested. The untransfected RBL-2H3 cells did not respond to any one of the tested ligands, even when used at a high concentration of 10 μM. In comparison, the three receptor-expressing cell lines displayed varied responses to these agonists. The synthetic chemical compound 43 preferentially activated mFpr1 over mFpr2 with a 10-fold higher potency (EC50 187 nM versus. 1.2 μM; Fig. 2A; Table 2). Another synthetic compound, Quin-C1 (Fig. 2B), was less potent than compound 43, but its potency on mFpr1 and mFpr2 was similar. On the basis of the maximal calcium response as well as degranulation data, both Quin-C1 and compound 43 are highly efficacious at these receptors. In fact, their efficacies are only surpassed by WKYMVm, which is highly efficacious for these receptors with an EC50 value of 82 pM for mFpr1 and 449 pM for mFpr2 (Supplemental Fig. 2A).
Fig. 2.

Calcium mobilization in RBL-mFpr cells stimulated with various agonists. Cells were loaded with FLIPR Calcium 5 reagent and analyzed for changes of intracellular calcium in response to agonist stimulation. (A) Quin-C1, (B) compound 43, (C) fMMYALF, (D) fMIVTLF, (E) fMLFK, (F) fMLFE, (G) fMLFW, and (H) fMLFII. The upper panels show typical calcium traces in response to the indicated agonist concentration. The lower panels are dose-dependent curves, which were based on peak Ca2+ increase at indicated agonist concentrations and shown as mean ± S.E.M. representing >3 separate experiments.
TABLE 2.
Calcium mobilization in RBL cells expressing mFpr1 or mFpr2 induced by various agonists in a concentration-dependent manner
The EC50 values and the maximum effects (relative Ca2+ flux) shown are based on >3 independent experiments.
| Agonists | mFpr1 | mFpr2 | ||
|---|---|---|---|---|
| EC50 (M) | MaxE | EC50 (M) | MaxE | |
| fMLF | 2.3 ± (2.7) × 10−5 | 2.6 ± 0.2 | ND | ND |
| fMLFE | 6.7 ± (1.1) × 10−9 | 3.6 ± 0.4 | ND | ND |
| fMLFK | 5.1 ± (1.0) × 10−7 | 3.6 ± 0.3 | 2.3 ± (.67) × 10−6 | 3.0 ± 0.3 |
| fMLFW | 1.1 ± (.16) × 10−9 | 4.6 ± 0.3 | 3.3 ± (.45) × 10−6 | 3.1 ± 0.3 |
| fMIFL | 3.5 ± (.46) × 10−10 | 4.8 ± 0.4 | 5.3 ± (2.6) × 10−4 | 1.7 ± 0.2 |
| fMLFII | 3.1 ± (.32) × 10−11 | 4.8 ± 0.3 | 9.1 ± (3.7) × 10−7 | 3.3 ± 0.2 |
| fMLFIIK | 4.6 ± (1.1) × 10−10 | 3.4 ± 0.1 | 9.0 ± (7.4) × 10−7 | 3.7 ± 0.1 |
| fMIVTLF | 2.1 ± (.45) × 10−11 | 4.7 ± 0.4 | 2.8 ± (.55) × 10−6 | 2.6 ± 0.1 |
| fMMYALF | 9.8 ± (4.2) × 10−11 | 4.1 ± 0.2 | 4.7 ± (3.1) × 10−6 | 2.4 ± 0.4 |
| WKYMVm | 8.2 ± (1.9) × 10−11 | 4.6 ± 0.3 | 4.5 ± (1.2) × 10−10 | 4.0 ± 0.2 |
| Quin C1 | 2.4 ± (.33) × 10−6 | 4.4 ± 0.2 | 6.3 ± (.92) × 10−7 | 3.6 ± 0.3 |
| Compound 43 | 1.9 ± (.25) × 10−9 | 4.4 ± 0.4 | 1.2 ± (.17) × 10−7 | 3.0 ± 0.3 |
ND, not determined.
Among the native agonists tested, fMMYALF (mitocondria) and fMIVTLF (L. monocytogenes) had the highest potency for mFpr1 (EC50 = 21 and 98 pM), and they triggered intracellular Ca2+ flux even more efficaciously than WKYMVm (Fig. 2, C and D). As previously reported (Southgate et al., 2008), the S. aureus-derived fMIFL stimulated a marked Ca2+ increase in mFpr1-RBL cells, with an EC50 in the high picomolar range (350 pM; Supplemental Fig. 2C). fMLFII and fMLFIIK, both derived from fMLFI (S. aureus) with C-terminal extensions, showed similar potency on mFpr1-RBL cells in calcium mobilization assays (EC50 310 and 460 pM, respectively; Fig. 2H; Supplemental Fig. 2D). In agreement with previous findings (Southgate et al., 2008), mFpr1 preferred these bacterial and mitochondrial formyl peptides over E. coli-derived fMLF (EC50 = 23 μM, Supplemental Fig. 2B), fMLFK (EC50 = 510 nM), fMLFE (EC50 = 6.7 nM) and fMLFW (EC50 = 1.1 nM) (Fig. 2, E-G; Table 2).
In contrast to mFpr1, mFpr2 responded poorly to all the formyl peptides tested. The hexapeptides fMIVTLF and fMMYALF had EC50 values of 2.8 μM and 4.7 μM in calcium flux assays, respectively. The other two peptides, fMLFII (Fig. 2H) and fMLFIIK (Supplemental Fig. 2D), exhibited somewhat higher potency, with EC50 of 910 nM and 900 nM, respectively. The tetrapeptides fMLFW and fMLFK were less potent at mFpr2, with EC50 values in micromolar range (3.3 and 2.3 μM; Fig. 2, E and G). Whereas fMIFL was active on mFpr2 only above 10 μM (EC50 = 53 μM), the E. coli-derived peptide fMLF (Supplemental Fig. 2B) and its longer derivative fMLFE (Fig. 2F) were essentially inactive at mFpr2.
Throughout these experiments, the mFpr-rs1-RBL cells were insensitive to all agonists except WKYMVm, which induced calcium flux at a very high concentration (Supplemental Fig. 2A). Because of the variability, we next examined cellular distribution of the receptors in transfected RBL cells.
The Cellular Distribution Profile of mFpr-rs1 Differs from That of mFpr1 and mFpr2.
To determine whether the mouse receptors are expressed on the cell surface, the green fluorescent protein EGFP was fused to the C-terminus of the mouse Fpr proteins. The resulting chimeric receptors were either expressed in RBL cells for selection of stable transfectants, or transiently expressed in HeLa cells. Like human FPR1 and FPR2/ALX, mFpr1-EGFP and mFpr2-EGFP chimeric receptors were found mostly on the cell surface in the transfected RBL-2H3 cells (Fig. 3, A and B) and HeLa cells (Fig. 3, E and F). Moreover, mFpr1-EGFP and mFpr2-EGFP internalized upon agonist stimulation in HeLa cells (Fig. 3, G–N). The level of internalization correlates approximately with the potency of the agonist as measured in parallel degranulation and calcium mobilization assays.
Fig. 3.

Cell surface expression and internalization of mouse Fpr receptors. (A) mFpr1-EGFP, (B) mFpr2-EGFP, or (C and D) mFpr-rs1-EGFP expressed in RBL cells. The scale bar for (A–C) is shown (A), and the images were captured using a 63× oil immersion objective, whereas (D) was captured under a 40× dry objective, and the scale bar is shown in the figure. Internalization of mFpr1-EGFP or mFpr2-EGFP in HeLa cells, before (E and F) and after 30 minutes of stimulation with fMLF (G and H), fMLFK (I and J), WKYMVm (K and L), Quin-C1 (M and N), or compound 43 (O and P), at indicated concentrations. The scale bar for (E–P) is shown in (E), and the images were captured using a 63× oil immersion objective.
In contrast, mFpr-rs1-EGFP showed a more uniform cellular distribution profile. This pattern remained unchanged when expressed in RBL-2H3 (Fig. 3, C and D), HeLa cells (Supplemental Fig. 3) and HEK293 cells (unpublished data). To exclude the possibility that the minimal cell surface expression might result from unwanted frame shift or early transcription termination, full-length sequencing was performed, and a normal reading frame was confirmed. Additional mFpr-rs1 expression plasmids were constructed by tagging the receptor with a much smaller FLAG sequence (N-DYKDDDDK-C) to either the N- or C-terminus of mFpr-rs1. HeLa cells were transfected to express mFpr-rs1-N-FLAG or mFpr-rs1-C-FLAG. Thirty-six hours after transfection, cells were incubated with an anti-FLAG antibody and labeled with Alexa Fluro 488-conjugated secondary antibody. The mFpr-rs1-N-FLAG and mFpr-rs1-C-FLAG receptors were mostly visualized inside the cells (Supplemental Fig. 3), suggesting that the expression pattern of mFpr-rs1 receptor did not change inadvertently because of the addition of an EGFP.
The Mouse Formyl Peptide Receptors Have Distinct Patterns of Agonist Binding.
To determine the binding affinity of the three mouse receptors, a FITC-conjugated fMLFIIK was synthesized, and saturation binding assays were performed using flow cytometry (see Materials and Methods). The untransfected RBL cells produced little background binding to the fluorescent ligand (unpublished data). Unlike radiolabeled ligands, nonspecific binding was relatively low with the FITC-conjugated fMLFIIK (Fig. 4A). A dissociation constant (Kd) of 2.8 ± 0.4 nM and a binding capacity (Bmax) of 90,468 ± 7805 binding sites per cell were derived for the mFpr1-RBL cells. Scatchard plot (inlet) and Hill plot (not shown) confirmed a one-site binding model. In contrast, total binding to either mFpr2 or mFpr-rs1 was low, thus no significant specific binding was measurable within the same nanomolar concentration range of fMLFIIK-FITC (0–100 nM), as with the mFpr1-RBL cells. However, an increase in ligand binding to mFpr2 or mFpr-rs1 was detected in the micromolar concentration range, which approached saturation at 10 μM or above despite a high background caused by nonspecific binding (Fig. 4, B and C). The estimated Kd values based on these experiments were 4.8 ± 0.2 and 6.2 ± 0.3 μM, respectively, suggesting low-affinity binding for fMLFIIK to mFpr2 and mFpr-rs1. The number of total binding sites was estimated with 7 μM fMLFIIK-FITC, using FITC-conjugated standard beads as reference. The results suggested that approximately 100,000 mFpr2 receptors were expressed in each transfected RBL cells, which is slightly more than the expression of mFpr1 (unpublished data). In contrast, only 5000–9000 mFpr-rs1 were found on the surface of transfected RBL cells, which is about 10% of the mFpr1 and mFpr2 in the transfected RBL cells. This observation is consistent with the results from fluorescent microscopy (Fig. 3, C and D) and support the notion that a significant portion of mFpr1-rs1 is present inside the cells.
Fig. 4.

Binding assays with RBL-2H3 cells stably transfected by mFpr1, mFpr2, or mFpr-rs1. Saturation, nonspecific binding, and specific binding of fMLFIIK-FITC to (A) mFpr1-, (B) mFpr2-, or (C) mFpr-rs1- expressing RBL cells. The inlet shows Scatchard analysis of the data in (A). Data were analyzed with Origin 7.5 software (Northampton, MA), and the results are shown as means ± S.E.M. with >3 experiments.
To compare the pharmacologic properties of agonists from a variety of sources, competitive binding assays were performed using RBL cells stably expressing mFpr1 with a fixed amount of fMLFIIK-FITC (50 nM) and variable concentrations of unlabeled agonists (Fig. 5A). Table 3 summarizes the IC50 values of these competitive ligands. Among the agonists tested, fMLFII showed the highest potency to displace fMLFIIK-FITC from mFpr1-RBL cells (IC50 = 5.4 nM). An extended version of this peptide, fMLFIIK, displayed a 40-fold less potency (IC50 = 216 nM). Consistent with the results from functional assays, WKYMVm and fMIVTLF were also strong competitors of fMLFIIK (IC50 values of 11.7 nM and 24.4 nM), whereas fMLFW, fMMYALF, and fMIFL displaced fMLFIIK-FITC with lower potency (IC50 = 153 nM, 272 nM, and 415 nM, respectively). The synthetic chemicals compound 43 and Quin-C1 were weaker competitors (IC50 = 7.8 μM and 8.5 μM, respectively) despite their agonistic activities in functional assays. This may result from the fact that peptides and the chemicals occupy binding sites that are only partially overlapping. Finally, fMLF and fMLFK showed inability to cause any displacement below 10 μM (IC50 = 46.6 and 405 μM; Fig. 5; Table 3).
Fig. 5.

Competition binding assays performed on (A) mFpr1-RBL and (B) mFpr2-RBL cells. Binding of fMLFIIK-FITC (50 nM for mFpr1-RBL and 5 μM for mFpr2-RBL cells) was competitively displaced by increasing concentrations of agonists, including synthetic ligands WKYMVm, Quin-C1, and Compound 43 (left); E. coli-derived fMLF and derivatives fMLFK, fMLFE, fMLFW, and fMLFII (middle); and other bacterial formyl peptides fMIFL, fMIVTLF, and mitochondrial fMMYALF (right). Data were analyzed as described in the legend for Fig. 4. The results are shown as means ± S.E.M. with >3 experiments.
TABLE 3.
Various agonists displacing fMLFIIK-FITC binding to mFpr1-RBL or mFpr2-RBL cells with different affinity
Shown are the means of IC50 values and Hill coefficient (nH) of unlabeled ligands in competition binding assays conducted with at least 3 independent experiments as described.
| Agonist | mFpr1-RBL | mFpr2-RBL | ||
|---|---|---|---|---|
| IC50 (M) | nH | IC50 (M) | nH | |
| WKYMVm | 1.2 ± (0.06) × 10−8 | 1.2 ± 0.1 | 7.1 ± (1.6) × 10−7 | 0.6 ± 0.1 |
| fMLF | 4.7 ± (0.98) × 10−5 | 1.0 ± 0.1 | ND | ND |
| fMLFK | 4.1 ± (17) × 10−4 | 1.1 ± 0.5 | > 5.6 × 10−5 | ND |
| fMLFE | 8.8 ± (1.1) × 10−6 | 0.6 ± 0.1 | ND | ND |
| fMLFW | 1.5 ± (0.05) × 10−7 | 0.9 ± 0.03 | ND | ND |
| fMLFII | 5.4 ± (0.43) × 10−9 | 0.8 ± 0.03 | 3.8 ± (0.72) × 10−6 | ND |
| fMIFL | 4.2 ± (0.57) × 10−7 | 0.7 ± 0.1 | ND | ND |
| fMIVTLF | 2.4 ± (0.24) × 10−8 | 0.8 ± 0.06 | 6.0 ± (4.0) × 10−6 | 0.5 ± 0.2 |
| fMMYALF | 2.7 ± (0.22) × 10−7 | 0.7 ± 0.04 | ND | ND |
| fMLFIIK | 2.2 ± (0.14) × 10−7 | 0.8 ± 0.03 | 6.4 ± (2.7) × 10−6 | 0.9 ± 0.1 |
| MLFIIK | ND | ND | ND | ND |
| Quin C1 | 8.5 ± (0.52) × 10−6 | 0.6 ± 0.06 | > 1.4 × 10−4 | ND |
| Compound 43 | 7.8 ± (0.28) × 10−6 | 0.7 ± 0.04 | > 6.1 × 10−4 | ND |
ND, not determined.
Since mFpr2 binds fMLFIIK-FITC with lower affinity than mFpr1, a concentration of 5 μM was used in competition binding assays (Fig. 5B). Table 3 showed that the IC50 values for mFpr2-RBL cells were at least 1∼2 orders of magnitude larger than those for mFpr1-RBL cells. WKYMVm was the strongest competitor (IC50 = 708 nM), although it induced incomplete displacement (<80%) of fMLFIIK-FITC, suggesting that its binding site partially overlaps with the site for formyl peptides on mFpr2. Quin-C1 was 4.5-fold more potent than compound 43, with the respective IC50 values of 137 μM and 610 μM. The peptides fMLFII, fMIVTLF, and fMLFIIK displaced fMLFIIK-FITC from mFpr2 with micromolar concentrations (IC50 = 3.8, 4.7, and 6.4 μM, respectively). In comparison, fMLF, fMLFE, fMLFW, fMIFL, and fMMYALF were incapable of displacing fMLFIIK-FITC; however, fMLFK at very high concentrations (>50 μM) showed limited potency in a competition binding assay (Table 3; Fig. 5B). In addition, as expected, the nonformylated peptide MLFIIK caused no displacement on either mFpr1-RBL or mFpr2-RBL cells (Fig. 6), suggesting that the N-formyl group is essential for the peptide fMLFIIK to bind mFpr1 and mFpr2. We were unable to perform meaningful competitive binding assays on mFpr-rs1 due to the limited cell surface expression and low affinity for the agonist.
Fig. 6.

Comparison of formyl and nonformyl peptides in competitive binding assay. The ability of nonformylated peptide MLFIIK and N-formylated fMLFIIK to compete with fMLFIIK-FITC for binding to (A) mFpr1- and (B) mFpr2-RBL cells was determined. All data were analyzed as described in the legend of Fig. 4.
Discussion
So far, most functional studies of the FPRs have been conducted in human neutrophils, and some early studies were conducted using rabbit neutrophils. In comparison, much less is known about the FPRs from other species. In mice, the number of genes coding for putative Fpr family receptors far exceeds that in humans. Although there are eight members in the mouse Fpr gene family, not all of them have been identified to encode formyl peptide receptors. For instance, Fpr-rs5 (ΨFpr-rs3) is a pseudogene, and Fpr-rs8 (ΨFpr-rs2) has been recently characterized as a constitutively expressed gene that may affect longevity (Tiffany et al., 2011). mFpr-rs3, mFpr-rs4, 6, and mFpr-rs7 were reported to be expressed by vomeronasal sensory neurons and function as chemoreceptors (Liberles et al., 2009; Riviere et al., 2009). The remaining 3 genes (Fpr1, Fpr2, and Fpr-rs1), which are expressed in mouse phagocytic leukocytes, are most similar to human FPRs at protein sequence level. Generally, they have been considered to be the orthologs of human FPR members. However, the promiscuous binding property of these three receptors and their complex evolutionary relationship make it difficult to accurately define orthologous correlation between members of the human and mouse formyl peptide receptor gene families. With rapid advancement in mouse genetics that has already helped to unveil the functional roles played by selected Fpr genes (Gao et al., 1999; Chen et al., 2010; Dufton et al., 2010), it will be possible to use mouse Fpr as models for study of human diseases. Therefore, it is important to gain a better understanding of the pharmacologic properties of the mouse Fpr family members.
The three mFpr family members studied in this work exhibit high sequence homology to the human FPRs, and their tissue distribution profile also resembles that of the human FPRs. It has been suggested that, despite being closest (76%) in primary sequence to hFPR1, mFPR1 shares several features found in human FPR2/ALX. For example, both mFpr1 and human FPR2/ALX are low-affinity receptors for the E.coli-derived fMLF, yet they respond well to formyl peptides derived from S. aureus (fMIFL), L. monocytogenes (fMIVTLF), and mitochondria (fMMYALF) (Rabiet et al., 2005; Southgate et al., 2008). Of note, fMLF is a major chemotactic peptide in E.coli culture supernatant, but it is not the only one that stimulates neutrophil response. In this work, we found that mFpr1 displays higher affinity for fMLF derivatives in the order of fMLFW > fMLFE > fMLFK > fMLF. This observation indicates that, besides the N-formyl group, the addition of amino acids to the C-terminus of formyl peptides may promote binding to mFpr1. There is also evidence that longer peptides, such as fMLFII and fMLFIIK, often behave better in functional and binding assays.
The second mouse receptor, mFpr2, is believed to be a low-affinity receptor for formyl peptides (Hartt et al., 1999). It has a more restricted specificity for the peptides tested in this work, but responds better to nonpeptides, such as Quin-C1 and compound 43. Besides, mFpr2 responds well to WKYMVm, a peptide that contains a d-methionine. These findings suggest that the native ligands for mFpr2 may not be formylated peptides. Studies have shown that some of the mouse Fprs, including mFpr2 and mFpr-rs1, are receptors for lipoxin A4 (Takano et al., 1997; Vaughn et al., 2002). Recent knockout studies have shown that mFpr2 plays an important role in airway inflammation and immune response (Chen et al., 2010). Hence, a more detailed characterization of this receptor is of potential interest as it may provide a useful animal model for the study of human lung diseases. Regardless of its low affinity for fMLF, mFpr2 retains some capability in binding formyl peptides. Longer formyl peptides, such as fMLFK, fMLFII, and fMLFIIK, are better agonists for this receptor. The relatively low efficacy and affinity of fMMYALF, fMIVTLF, and fMIFL at mFpr2 suggests that the sequence as well as side chains of C-terminal residues in these peptides can be more crucial than their length for binding to mFpr2.
Sequence comparison has shown similarities and differences between the mouse and human FPRs and among the three mouse Fprs tested in this study. Notably, some important residues known to be critical for the interaction with formylated peptides at their C-terminus (Mills et al., 2000), such as Arg205, are not present in all receptors. In human FPR3, a histidine takes place at position 205, but whether this substitution is sufficient to alter ligand binding specificity remains unclear because an arginine is found at the same position in all three mFprs tested. Likewise, residues at positions 83–85 and 284 (based on the human FPR1 sequence) are known to be involved in binding of formyl peptides (Quehenberger et al., 1997; Mills et al., 1998; Lala et al., 1999) and are substituted to various degrees in the human FPRs and mouse Fprs tested so far. These observations pose challenges to sequence-based modeling and to structure–function studies relying on site-directed mutagenesis. The complexity and diversity of ligands for the FPR family provides an excellent model to study G-protein–coupled receptor structures and functions.
In this study, we found that Quin-C1, a substituted quinazolinone, activated both mFpr2 and mFpr1 with almost equal potency in calcium flux assay. Compound 43, a nitrosylated pyrazolone derivative, was 100-fold more potent at mFpr1 in functional and binding assays. These findings are in agreement with a previous study by Dahlgren and colleagues that predicted shared binding between mFpr1 and mFpr2 for compound 43 (Forsman et al., 2011). The synthetic small molecules are clearly different than formyl peptides in terms of binding site selection on the receptors. As we have shown in this study, Quin-C1 and compound 43 are less effective in competing with fMLFIIK-FITC for binding to the two receptors, despite their relatively high potency in functional assays. We have previously shown that Quin-C1 could not effectively displace radiolabeled WKYMVm in human FPR2/ALX binding assay, suggesting that their binding sites are only partially overlapping (Nanamori et al., 2004). It will be interesting to further investigate whether these synthetic molecules influence FPR family receptors through allosteric modulation.
The third mouse receptor studied, mFpr-rs1, shares similar structural features with mFpr2 and mFpr1 but differs from them with its unique intracellular distribution profile. Unlike mFpr1 and mFpr2, which are found mostly on cell surface, mFpr-rs1 was hardly detectable on plasma membrane in transfected RBL-2H3, HeLa, and HEK293 cells. Instead, it was mostly visualized inside the transfected cells. The observed intracellular distribution in this work was not due to artifact. In fact, the human FPR3 has the propensity of intracellular localization, as observed in blood monocytes (Migeotte et al., 2005). A possible mechanism for the intracellular localization of human FPR3 was reported recently (Rabiet et al., 2011). Therefore, mFpr-rs1 resembles human FPR3 in cellular distribution. The weak cell response to agonists found in this study might be attributable to its low expression on cell surface. Interestingly, peptides, such as F2L, which is derived from the heme-binding protein (Migeotte et al., 2005), probably act through binding to a cell surface receptor. Whether there are additional and possibly intracellular agonists for mFpr-rs1 and human FPR3 remains an interesting question. Such agonists might be lipophilic, able to permeate the cell membrane to gain access to the intracellular pool of receptors. It is also possible that an agonist might recruit mFpr-rs1 from cytoplasm to the plasma membrane upon binding to the available receptors, resulting in an enrichment of cell surface receptors. However, none of the agonists tested in this work seem to possess this property.
In conclusion, we examined the pharmacologic properties of three mFpr family members in transfected cells. Our results show that mFrp1 is an ortholog of the human FPR1, and both are high affinity receptors for N-formyl peptides. Major differences between these two receptors are that mFpr1 displays relatively low affinity for fMLF, and that it responds well to the synthetic molecule Quin-C1, whereas the human FPR1 does not (Nanamori et al., 2004). mFpr1 shares the latter property with human FPR2/ALX. Our results also show that, in general, formyl peptides are weak agonists for mFpr2, suggesting that its native ligand may not be a formylated peptide. Finally, we report for the first time that mFpr-rs1 has limited cell surface expression, a property shared with the human FPR3. Therefore, mFpr-rs1 may be an ortholog of human FPR3. In this regard, the term mFpr3 is appropriate, although the natural ligand for the intracellular receptors remains to be determined. A ligand for human FPR3, F2L, has been shown to activate mFpr2, suggesting an overlapping feature of mFpr2 and human FPR3 (Gao et al., 2007). Intracellular G-protein–coupled receptors are rare, and their functions and signaling mechanisms are poorly understood. It is hopeful that a better understanding of the mouse Fpr family will facilitate the use of genetics tools for study of human FPRs and their pathophysiologic functions.
Supplementary Material
Acknowledgments
The authors thank Drs. Erica Troksa and Masakatsu Nanamori for preparation of the peptides.
Abbreviations
- EGFP
enhanced green fluorescent protein
- FITC
fluorescein isothiocyanate
- fMIFL
N-formyl-Met-Ile-Phe-Leu
- fMLF
N-formyl-Met-Leu-Phe
- fMLFIIK
N-formyl-Met-Leu-Phe-Ile-Ile-Lys
- fMMYALF
N-formyl-Met-Met-Tyr-Ala-Leu-Phe
- FPR
formyl peptide receptor
- HBSS
Hanks’s balanced saline solution
- mFpr1
mouse formyl peptide receptor 1
- mFpr2
mouse formyl peptide receptor 2
- mFpr-rs1
mouse formyl peptide receptor-related sequence 1
- RBL
rat basophilic leukemia
- WKYMVm
Trp-Lys-Tyr-Met-Val-D-Met-NH2
Authorship Contributions
Participated in research design: He and Ye.
Conducted experiments: He and Liao.
Contributed new reagents or analytic tools: Z.G. Wang, Z.L. Wang, Zhou, and M.W.Wang.
Performed data analysis: He, Liao, and Ye.
Wrote or contributed to the writing of the manuscript: He and Ye.
Footnotes
This work was supported in part by National Basic Research Program of China (973 Program) [Grant 2012CB518001] and Chinese Postdoctoral Science Fund [Grant12Z102060002] (H.Q.H.).
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Bürli RW, Xu H, Zou X, et al. (2006) Potent hFPRL1 (ALXR) agonists as potential anti-inflammatory agents. Bioorg Med Chem Lett 16:3713–3718 [DOI] [PubMed] [Google Scholar]
- Chen K, Le Y, Liu Y, Gong W, Ying G, Huang J, Yoshimura T, Tessarollo L, Wang JM. (2010) A critical role for the g protein-coupled receptor mFPR2 in airway inflammation and immune responses. J Immunol 184:3331–3335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang N, Serhan CN, Dahlén SE, Drazen JM, Hay DW, Rovati GE, Shimizu T, Yokomizo T, Brink C. (2006) The lipoxin receptor ALX: potent ligand-specific and stereoselective actions in vivo. Pharmacol Rev 58:463–487 [DOI] [PubMed] [Google Scholar]
- Dufton N, Hannon R, Brancaleone V, Dalli J, Patel HB, Gray M, D’Acquisto F, Buckingham JC, Perretti M, Flower RJ. (2010) Anti-inflammatory role of the murine formyl-peptide receptor 2: ligand-specific effects on leukocyte responses and experimental inflammation. J Immunol 184:2611–2619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsman H, Onnheim K, Andreasson E, Dahlgren C. (2011) What formyl peptide receptors, if any, are triggered by compound 43 and lipoxin A4? Scand J Immunol 74:227–234 [DOI] [PubMed] [Google Scholar]
- Gao JL, Chen H, Filie JD, Kozak CA, Murphy PM. (1998) Differential expansion of the N-formylpeptide receptor gene cluster in human and mouse. Genomics 51:270–276 [DOI] [PubMed] [Google Scholar]
- Gao JL, Guillabert A, Hu J, et al. (2007) F2L, a peptide derived from heme-binding protein, chemoattracts mouse neutrophils by specifically activating Fpr2, the low-affinity N-formylpeptide receptor. J Immunol 178:1450–1456 [DOI] [PubMed] [Google Scholar]
- Gao JL, Lee EJ, Murphy PM. (1999) Impaired antibacterial host defense in mice lacking the N-formylpeptide receptor. J Exp Med 189:657–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao JL, Murphy PM. (1993) Species and subtype variants of the N-formyl peptide chemotactic receptor reveal multiple important functional domains. J Biol Chem 268:25395–25401 [PubMed] [Google Scholar]
- Hartt JK, Barish G, Murphy PM, Gao JL. (1999) N-formylpeptides induce two distinct concentration optima for mouse neutrophil chemotaxis by differential interaction with two N-formylpeptide receptor (FPR) subtypes. Molecular characterization of FPR2, a second mouse neutrophil FPR. J Exp Med 190:741–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He R, Sang H, Ye RD. (2003) Serum amyloid A induces IL-8 secretion through a G protein-coupled receptor, FPRL1/LXA4R. Blood 101:1572–1581 [DOI] [PubMed] [Google Scholar]
- He R, Tan L, Browning DD, Wang JM, Ye RD. (2000) The synthetic peptide Trp-Lys-Tyr-Met-Val-D-Met is a potent chemotactic agonist for mouse formyl peptide receptor. J Immunol 165:4598–4605 [DOI] [PubMed] [Google Scholar]
- Lala A, Gwinn M, De Nardin E. (1999) Human formyl peptide receptor function role of conserved and nonconserved charged residues. Eur J Biochem 264:495–499 [DOI] [PubMed] [Google Scholar]
- Le Y, Gong W, Li B, Dunlop NM, Shen W, Su SB, Ye RD, Wang JM. (1999) Utilization of two seven-transmembrane, G protein-coupled receptors, formyl peptide receptor-like 1 and formyl peptide receptor, by the synthetic hexapeptide WKYMVm for human phagocyte activation. J Immunol 163:6777–6784 [PubMed] [Google Scholar]
- Le Y, Murphy PM, Wang JM. (2002) Formyl-peptide receptors revisited. Trends Immunol 23:541–548 [DOI] [PubMed] [Google Scholar]
- Liberles SD, Horowitz LF, Kuang D, Contos JJ, Wilson KL, Siltberg-Liberles J, Liberles DA, Buck LB. (2009) Formyl peptide receptors are candidate chemosensory receptors in the vomeronasal organ. Proc Natl Acad Sci USA 106:9842–9847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migeotte I, Communi D, Parmentier M. (2006) Formyl peptide receptors: a promiscuous subfamily of G protein-coupled receptors controlling immune responses. Cytokine Growth Factor Rev 17:501–519 [DOI] [PubMed] [Google Scholar]
- Migeotte I, Riboldi E, Franssen JD, et al. (2005) Identification and characterization of an endogenous chemotactic ligand specific for FPRL2. J Exp Med 201:83–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills JS, Miettinen HM, Barnidge D, Vlases MJ, Wimer-Mackin S, Dratz EA, Sunner J, Jesaitis AJ. (1998) Identification of a ligand binding site in the human neutrophil formyl peptide receptor using a site-specific fluorescent photoaffinity label and mass spectrometry. J Biol Chem 273:10428–10435 [DOI] [PubMed] [Google Scholar]
- Mills JS, Miettinen HM, Cummings D, Jesaitis AJ. (2000) Characterization of the binding site on the formyl peptide receptor using three receptor mutants and analogs of Met-Leu-Phe and Met-Met-Trp-Leu-Leu. J Biol Chem 275:39012–39017 [DOI] [PubMed] [Google Scholar]
- Nanamori M, Cheng X, Mei J, Sang H, Xuan Y, Zhou C, Wang MW, Ye RD. (2004) A novel nonpeptide ligand for formyl peptide receptor-like 1. Mol Pharmacol 66:1213–1222 [DOI] [PubMed] [Google Scholar]
- Quehenberger O, Pan ZK, Prossnitz ER, Cavanagh SL, Cochrane CG, Ye RD. (1997) Identification of an N-formyl peptide receptor ligand binding domain by a gain-of-function approach. Biochem Biophys Res Commun 238:377–381 [DOI] [PubMed] [Google Scholar]
- Rabiet MJ, Huet E, Boulay F. (2005) Human mitochondria-derived N-formylated peptides are novel agonists equally active on FPR and FPRL1, while Listeria monocytogenes-derived peptides preferentially activate FPR. Eur J Immunol 35:2486–2495 [DOI] [PubMed] [Google Scholar]
- Rabiet MJ, Huet E, Boulay F. (2007) The N-formyl peptide receptors and the anaphylatoxin C5a receptors: an overview. Biochimie 89:1089–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabiet MJ, Macari L, Dahlgren C, Boulay F. (2011) N-formyl peptide receptor 3 (FPR3) departs from the homologous FPR2/ALX receptor with regard to the major processes governing chemoattractant receptor regulation, expression at the cell surface, and phosphorylation. J Biol Chem 286:26718–26731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivière S, Challet L, Fluegge D, Spehr M, Rodriguez I. (2009) Formyl peptide receptor-like proteins are a novel family of vomeronasal chemosensors. Nature 459:574–577 [DOI] [PubMed] [Google Scholar]
- Southgate EL, He RL, Gao JL, Murphy PM, Nanamori M, Ye RD. (2008) Identification of formyl peptides from Listeria monocytogenes and Staphylococcus aureus as potent chemoattractants for mouse neutrophils. J Immunol 181:1429–1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano T, Fiore S, Maddox JF, Brady HR, Petasis NA, Serhan CN. (1997) Aspirin-triggered 15-epi-lipoxin A4 (LXA4) and LXA4 stable analogues are potent inhibitors of acute inflammation: evidence for anti-inflammatory receptors. J Exp Med 185:1693–1704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiffany HL, Gao JL, Roffe E, Sechler JM, Murphy PM. (2011) Characterization of Fpr-rs8, an atypical member of the mouse formyl peptide receptor gene family. J Innate Immun 3:519–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughn MW, Proske RJ, Haviland DL. (2002) Identification, cloning, and functional characterization of a murine lipoxin A4 receptor homologue gene. J Immunol 169:3363–3369 [DOI] [PubMed] [Google Scholar]
- Vilven JC, Domalewski M, Prossnitz ER, Ye RD, Muthukumaraswamy N, Harris RB, Freer RJ, Sklar LA. (1998) Strategies for positioning fluorescent probes and crosslinkers on formyl peptide ligands. J Recept Signal Transduct Res 18:187–221 [DOI] [PubMed] [Google Scholar]
- Ye RD, Boulay F, Wang JM, Dahlgren C, Gerard C, Parmentier M, Serhan CN, Murphy PM. (2009) International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) family. Pharmacol Rev 61:119–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C, Zhang S, Nanamori M, et al. (2007) Pharmacological characterization of a novel nonpeptide antagonist for formyl peptide receptor-like 1. Mol Pharmacol 72:976–983 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.