

Abstract
Although differentiation of the isomeric Asn deamidation products (Asp and isoAsp) at the peptide level by electron capture dissociation (ECD) has been well established, isoAsp identification at the intact protein level remains a challenging task. Here, a comprehensive top-down deamidation study is presented using the protein beta2-microglobulin (β2M) as the model system. Of the three deamidation sites identified in the aged β2M, isoAsp formation was detected at only one site by the top-down ECD analysis. The absence of diagnostic ions likely resulted from an increased number of competing fragmentation channels and a decreased likelihood of product ion separation in ECD of proteins. To overcome this difficulty, an MS3 approach was applied where a protein ion was first fragmented by collisionally activated dissociation (CAD) and a resulting product ion was isolated and further analyzed by ECD. IsoAsp formation at all three deamidation sites was successfully identified by this CAD-ECD approach. Further, the abundance of the isoAsp diagnostic ion was found to increase linearly with the extent of deamidation. These results demonstrated the potential of ECD in the detection and quantitative analysis of isoAsp formation using the top-down approach.
Introduction
Asparagine (Asn) deamidation plays an important role in aging and many protein misfolding diseases; it may also influence the stability of protein drugs.1 Under neutral and basic conditions, Asn deamidation generates a mixture of isomeric acidic products, containing aspartic acid (Asp) and isoaspartic acid (isoAsp).2 Of the two, isoAsp formation has a more deleterious effect on the stability and function of the protein, because it inserts an extra methylene group into the polypeptide backbone, significantly altering the protein structure.3 Thus, it is important not only to detect Asn deamidation but also to differentiate its isomeric products. Detection of deamidation is straightforward by mass spectrometry (MS) methods on the basis of the resulting mass shift of +0.984 Da per deamidation site. Several analytical methods exist for the differentiation of Asp and isoAsp residues, but each has its own limitations.4 Furthermore, most sample preparation methods can generate artifactual deamidation, which often must be distinguished from the native deamidation.5 Recently, a tandem mass spectrometry (MS/MS) method was developed for the identification and quantification of isoAsp formation based on the presence and relative abundance of the isoAsp-specific diagnostic ion(s) (c+57 and z•-57) generated by either electron capture dissociation (ECD) or electron transfer dissociation (ETD).6 A high-throughput nanoLC-ESI-MS/MS (ECD) method was also developed for proteome-wide isoAsp mapping, which used the accurate mass of the isoAsp diagnostic fragment, as well as several other spectral and chromatographic parameters, to identify isoAsp-containing peptides.7 However, such bottom-up approaches require enzymatic digestion which could potentially introduce additional artifactual deamidation and other post-translational modifications.5a, b A top-down approach8 is thus preferred, wherein the intact protein is analyzed directly, providing a more accurate account of the biologically significant deamidation by eliminating additional sample preparation steps associated with enzymatic digestion. To date, however, isoAsp identification by ECD has only been demonstrated at the peptide level, with the largest reported being the Amyloid beta 1-42 fragment.9
In this study, in vitro Asn deamidation and isoAsp formation in the protein beta2-microglobulin (β2M) was investigated by top-down ECD analysis. Human β2M has important immunological functions. It is the non-covalently bound light chain of the major histocompatibility complex (MHC) class I, which is required for antigen presentation to induce immunological responses. When an MHC class I molecule undergoes degradation, the heavy chain embedded in the cell surface is absorbed by endocytosis, releasing the monomeric β2M into the circulation. Most of these β2M molecules are filtered through the kidney glomeruli into the kidney tubules, where they are degraded by the tubular cells by pinocytosis.10 In serum, β2M is normally present at a low concentration of about 1.5-3 mg/L.11 However, in renal failure patients, the concentration of β2M can rise 60-times higher and amyloid fibrils can be formed and deposited at the joints of patients inducing amyloidosis.10a Although the native monomeric β2M does not readily form fibrils under physiological conditions, fibril formation can be accelerated in acidic solutions, in the presence of trace amount of pre-existing fibrils as “seeds”, or in the presence of copper ions, Cu2+.10, 12 In addition, it was reported that β2M deamidation at Asn17 can lead to fibril formation at low pH.13
Human β2M is a small (~11.7 KDa), highly soluble, monomeric extracellular protein containing 99 amino acid residues.14 The native wild type β2M contains seven β-sheets and a single disulfide bond connecting Cys25 and Cys80.10a, 15 β2M contains five Asn residues, including two with the fast-deamidating -NG- sequence.1b, 16 β2M’s relatively small size and the presence of facile deamidation sites make it an ideal model system for testing the potential of ECD in top-down isoAsp analysis.
Experimental
Materials
Sequencing grade trypsin was purchased from Roche Applied Science (Indianapolis, IN). HPLC grade water and methanol were purchased from Honeywell/Burdick & Jackson (Muskegon, MI). Human β2M and all other chemicals were purchased from Sigma Aldrich (St. Louis, MO).
Reductive Alkylation and In Vitro Aging
Human β2M was reduced and alkylated, as described previously.5a Briefly, β2M was reduced in 6 M urea/50 mM ammonium bicarbonate buffer at pH 8, with a ten-fold molar excess of dithiothreitol over disulfide bonds. The resulting mixture was incubated at 37 °C for one hour. Iodoacetamide was then added to the mixture in a ten-fold molar excess over disulfide bonds, followed by incubation in the dark at room temperature for one hour. It is important to note that artifactual deamidation could occur during prolonged reductive alkylation carried out at pH 8, which may be avoided by performing reductive alkylation under slightly acidic condition, using an acid-compatible reducing agent such as TCEP (tris(2-carboxyethyl)phosphine).17 The reduced and alkylated β2M protein was dried in a speed vacuum and further purified by a home-made solid-phase microextraction tip packed with Poros 50 R1 (Applied Biosystems, Foster City, CA). The fractions eluted at 40% acetonitrile were tested by electrospray ionization Fourier-transform ion cyclotron resonance mass spectrometry (ESI FTICR MS). The reduced and alkylated β2M protein was then aged in vitro in 0.1 M ammonium bicarbonate buffer at pH 8.3, 37 °C for five days. A parallel time-course study was carried out by incubating the reduced and alkylated β2M in 50 mM ammonium bicarbonate buffer (pH 7.8), at 37 °C, with or without the presence of trypsin. Aliquots were taken after 2, 4, 8, and 24 hours of incubation, desalted, and subject to MS analysis as described below.
Mass Spectrometry Analysis
All MS experiments were performed on a 12 T hybrid Qh-FTICR mass spectrometer (solariX™, Bruker Daltonics, Billerica, MA) equipped with a hollow cathode dispenser and a 25 W continuous wave CO2 laser (Synrad model J48-2, Mukilteo, WA). The intact β2M protein was introduced into the mass spectrometer by nano-electrospray out of a pulled fused silica capillary with a 1 μm orifice at 5 μM concentration in an electrospray solution containing 49.5:49.5:1 methanol:H2O:formic acid. The reduced and alkylated β2M protein, either unaged or aged, was washed twice with water and analyzed similarly at the same concentration. For top-down ECD analyses, molecular ions in a specific charge state were isolated by the front-end mass filtering quadrupole and fragmented in the ICR cell by irradiation of low-energy electrons for ~20 ms with the cathode bias set at -1 V and the heating current at 1.6 A. For MS3 experiments, molecular ions of all charge states were first dissociated by collisionally activated dissociation (CAD) in the funnel-skimmer region, and a fragment ion was isolated by the quadrupole and further fragmented by ECD in the FTICR cell. The collision energy was tuned to maximize the abundance of the specific fragment ion of interest. For activated ion (AI)-ECD experiments, ion activation was achieved by IR irradiation from the CO2 laser prior to the ECD event. Because of the ion magnetron motion, the delay between the ion trapping event and the IR laser trigger had to be adjusted to achieve optimum ion-electron overlap at the onset of the electron irradiation.18 All tandem mass spectra were internally calibrated with the precursor ion and a few fragment ions assigned with high confidence using the DataAnalysis™ software (Bruker Daltonics, Billerica, MA). Peak lists were generated by the SNAP™ (Sophisticated Numerical Annotation Procedure) algorithm19 and interpreted manually with a typical mass accuracy of less than 1 ppm. The high mass accuracy obtained for all assigned ions over the complete m/z range validates the choice of internal calibrants, as a misassignment of the calibrant here would cause major errors (>50 ppm) in the peak assignment tables. All ESI-MS spectra of intact proteins were externally calibrated to avoid possible bias in internal calibration when assigning the isotope index to selected internal reference peaks, and the resulting mass accuracy was better than 3 ppm.
Results and discussion
ESI-MS Analysis of the β2M Protein
The ESI mass spectra of the native and the reduced/alkylated β2M (Figures S1a and S1b, supporting information) show charge state distributions from 7+ to 12+ and from 10+ to 16+, respectively. The shift towards higher charge states for the reduced/alkylated β2M is consistent with its more unfolded structure as expected after the disulfide bond breakage. Each charge state contains two isotopic clusters corresponding to the molecular ion and its singly oxidized form, respectively. The ~16 Da mass increase resulted from the oxidization of the C-terminal methionine residue, as confirmed by the tandem mass spectra (not shown). The 116.24 Da mass difference between the two molecular ions corresponds to the reduction of the disulfide bond and the addition of two carbamidomethyl groups, confirming the success of the reductive alkylation.
Figure S1c shows the ESI mass spectrum of the reduced/alkylated and aged β2M (hereafter referred to as the aged β2M, for short). The inset shows the enlarged region of its 14+ charge state. The superior mass resolving power of the FTICR instrument allows accurate determination of the monoisotopic peak position (using the SNAP algorithm). The monoisotopic neutral mass of the aged β2M was determined to be 11840.80 Da, which represents a +3 Da mass shift after the in vitro aging, suggesting that deamidation may have occurred at three Asn sites.
Top-Down ECD Analysis of the Aged β2M Protein
The ECD spectrum of the isolated unoxidized form of the aged β2M in its 14+ charge state is shown in Figure 1 (lower panels). For clarity, the spectrum is divided into four segments, and only sequence ions of the c- and z-types are labeled in the spectrum. The cleavage map is shown in the top panel, representing 88 out of the potential 93 inter-residue N-Cα bond cleavages (or 94.6% cleavage coverage), excluding cleavages N-terminal to a proline residue. Based on the mass difference of fragment ions in the ECD spectra of the unaged and the aged proteins (Figure S2), the three deamidation sites were localized to the Asn17, Asn42, and Asn83 residues. Further, a peak corresponding to the isoAsp diagnostic ion, (c16+57)3+, was observed with a mass accuracy of 0.3 ppm (Figure 1, inset), confirming the formation of isoAsp at the Asn17 deamidation site in the aged protein, which demonstrated the utility of ECD in isoAsp analysis using the top-down approach.
Figure 1.
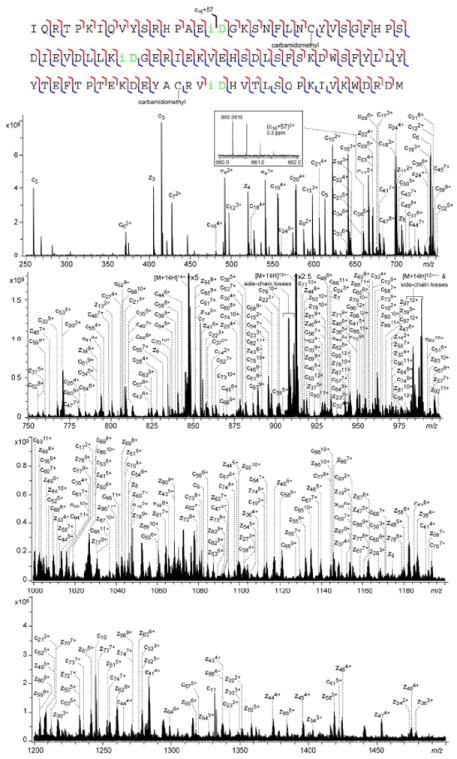
Top-down ECD spectrum of the reduced/alkylated and aged β2M in 14+ charge state, shown in four segments for clarity. The cleavage map is shown on top of the spectrum. The inset shows the N-terminal diagnostic ion for isoAsp17.
However, no isoAsp diagnostic ions were observed at either the Asn42 or the Asn83 deamidation site. This difficulty in isoAsp identification in intact proteins likely arises from the significantly increased number of available fragmentation channels that compete with diagnostic ion formation as the size of the protein increases. Moreover, the fragment ion pair formed after the N-Cα bond cleavage could still be held together by extensive noncovalent interactions present in large proteins, preventing fragment ion separation and detection, further hindering the observation of diagnostic ions. These problems can be alleviated by performing the ECD analysis on a smaller piece of the protein. Here, an MS3 (CAD/ECD) method was developed where the protein first underwent CAD and a fragment ion containing deamidation site(s) of interest was further selected and analyzed by ECD. Such gas-phase “digestion” can avoid the problem of artifactual deamidation encountered in traditional bottom-up approach that utilizes solution-phase enzymatic digestion to break down the proteins.
Identification of isoAsp17 by NSD-ECD MS3 Analysis and Potential Interference from Sequence Scrambling
Figure S3 shows the CAD spectrum of the aged β2M generated by fragmentation in the funnel/skimmer region at a collision energy of 30 eV. No precursor ion selection was applied as the fragmentation occurred in a region before the mass filtering quadrupole. As a result, this nozzle-skimmer dissociation (NSD) spectrum contains many more peaks than that typically obtained by low-energy CAD of isolated precursor ions in a single charge state. Three fragment ions were chosen for further ECD analysis: the Asn17-containing b224+, the Asn17- and Asn42-containing b639+, and the Asn83-containing y285+.
ECD of the b224+ ion generated from the aged β2M provided a near complete series of inter-residue N-Cα bond cleavages in the N-terminal region of the aged β2M protein (Figure 2, bottom panel), as summarized in the cleavage map (Figure 2, top panel). The isoAsp diagnostic (c16+57)2+ and (z6-57)+ ions were also observed (Figure 2, insets), with a mass accuracy of 0.3 and 0.1 ppm, respectively, once again confirming the formation of isoAsp17. Thus, ECD can be successfully applied towards isoAsp identification not only in intact proteins, but also in fragments of a protein generated by gas-phase dissociation methods, as demonstrated here using the NSD-ECD MS3 approach.
Figure 2.
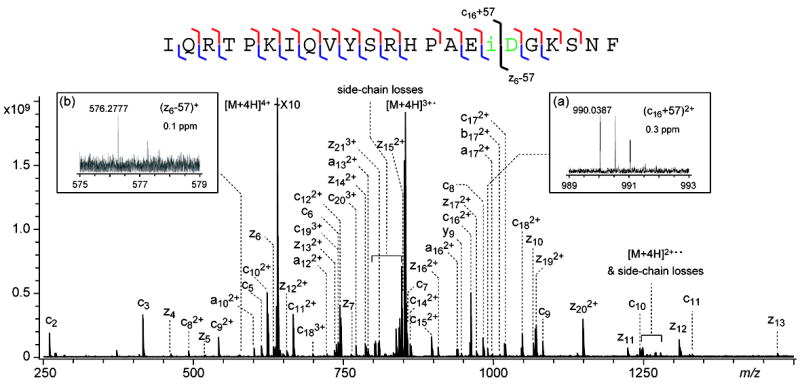
NSD-ECD spectrum of the b224+ ion generated from the aged β2M, with the cleavage map shown on top of the spectrum. The inset shows the N-terminal diagnostic ion for isoAsp17.
A potential problem for such MS3 approach could arise if the selected b ion undergoes sequence scrambling before ECD. It is well known that some b ions can form a macrocyclic structure via head-to-tail cyclization. These macrocycles can reopen at different positions, generating a series of sequence-permutated linear b-ions, which could give rise to non-direct sequence ions when further fragmented by CAD.20 Sequence-scrambled fragment ions have also been observed in ECD of small bn ions (n = 5 to 10).21 Despite its frequent occurrence in small and intermediate-sized bn ions (n ≤ 10), sequence scrambling was found to be statistically insignificant in a recent shotgun proteomics study by Zubarev and coworkers.22 Based on the analysis of 26,775 CAD spectra of doubly protonated tryptic peptides acquired with high mass resolution, they concluded that sequence scrambling via b-ion cyclization “can be safely ignored as a possible source of error in sequence assignment.” Siu and coworkers also investigated sequence scrambling in CAD of a series of tryptic peptides from standard proteins. Although non-direct sequence ions were observed in 35% of the peptides studied, inclusion of these scrambled masses in database searches had little effect on protein matching scores, which led the authors to conclude that “sequence scrambling is unlikely to impact negatively on the accuracy of automated peptide and protein identifications in proteomics”.23 It is important to note that, although the presence of one or a few sequence scrambled fragment ions may not significantly impact the protein ID in proteomics, isoAsp analysis does not have the same statistical benefit, as it relies on the presence of a single (or a pair of complementary) diagnostic ion(s), often in low abundance, for isoAsp identification. Whereas chance of a diagnostic ion’s having the same m/z value as that of a direct sequence ion is very low, chance of a spurious match could significantly increase when the b-ion sequence is permuted before ECD.
On the other hand, MS3 is rarely needed for sequence analysis in the terminal regions, because they are usually adequately covered by MS2 experiments. Likewise, it is also relatively easy to identify isoAsp residues in the terminal regions by MS2, as demonstrated by the successful detection of isoAsp17 in the aged β2M protein shown above. Therefore, one should have concern only for the possibility of sequence scrambling among large b-ions that contain isoAsp residues in the middle of the protein sequence, where the MS3 approach is likely needed. Conceivably, sequence scrambling should become less a problem as the size of the b-ions increases, because of the unfavorable entropical factor for head-to-tail cyclization of larger b-ions. However, Polfer and coworkers have recently reported that the relative intensity of non-direct sequence ions in low-energy CAD spectra of tryptic peptides increases with the peptide length.24 This seemingly surprising result can be partially attributed to the fact that the tandem mass spectra analyzed were obtained by performing low-energy CAD in a linear ion trap, where first-generation b-ions of various lengths could potentially cyclize and produce a large variety of sequence scrambled secondary fragment ions. In fact, most of the unambiguously identified non-direct sequence ions in the Polfer study were at lower mass, and likely formed from b-ions of smaller sizes. By contrast, in the NSD-ECD approach used here, a single b-ion was isolated for ECD analysis, which does not cause further sequence scrambling. Thus, one needs only to consider cyclization of the original b-ion, which is expected to become insignificant when its size is sufficiently large. In the present study, nearly all peaks in the ECD spectrum of the b224+ ion of aged β2M can be identified with high confidence as primary sequence ions, and the signal recorded for the assigned isoAsp diagnostic peak has far higher intensity than the few unassigned peaks present. Our result, combined with the literature reports on the lack of direct evidence for macrocycle formation in large b-ions (with 20+ residues) reported in literature, suggests that sequence scrambling is unlikely to be an issue for isoAsp analysis using the NSD-ECD approach adopted here.
Identification of isoAsp83 by NSD-ECD MS3 Analysis and Potential Interference from Co-isolation
Figure 3 shows the ECD spectrum of the y285+ ion from the aged β2M, which contains the Asn83 deamidation site. A complete series of inter-residue N-Cα bond cleavages, excluding the one N-terminal to a proline residue, were observed. In comparison, four of the five missed cleavages in the top-down ECD analysis of the intact protein occurred in this same span of the C-terminal region of the protein (Figure 1). The fifth cleavage, missed by top-down, was recovered in the ECD spectrum of the b224+ ion (Figure 2). Thus, the MS3 approach can be a valuable tool for improving sequence coverage in protein sequence analysis. Similar improvement in sequence coverage by MS3 has been reported using an NSD-CAD approach on an LTQ-Orbitrap instrument.25 More importantly, and pertinent to the deamidation study, complementary isoAsp83 diagnostic (z17-57)2+, (z17-57)3+ and (c11+57)2+ ions were identified with high mass accuracy (<0.2 ppm) in the ECD spectrum of the y285+ ion (Figure 3, insets), but were absent in the top-down ECD spectrum, illustrating the utility and the necessity of the NSD-ECD MS3 approach in isoAsp analysis of intact proteins.
Figure 3.
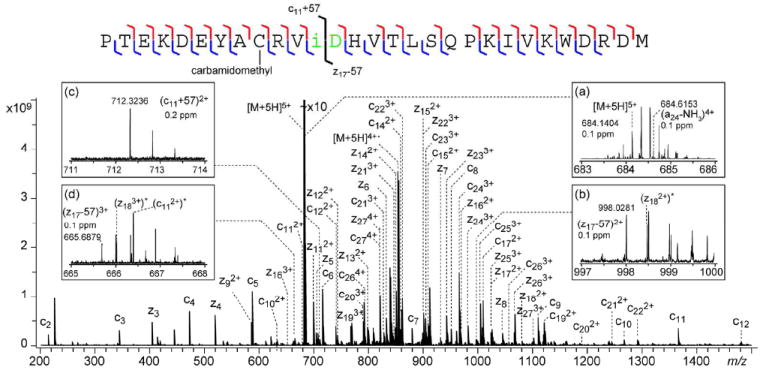
NSD-ECD spectrum of the y285+ ion generated from the aged β2M, with the cleavage map shown on top of the spectrum. Inset (a) shows the enlarged region at the precursor ion m/z, insets (b-d) show the N- and C-terminal diagnostic ions for isoAsp83. * denotes peaks resulting from ECD of the co-isolated (a24-NH3)4+ ion.
The majority of the peaks in Figure 3 can be assigned as primary sequence ions from the y285+ ion, and only those of the c- and z-types are labeled in the spectrum for clarity. However, a significant number of peaks cannot be interpreted as common ECD fragment ions (a-, b-, c-, y-, z-, or w-ions) from the y285+ ion. Because y ions are not known to undergo sequence scrambling, these unassigned ions are likely derived from different precursor ion(s). As shown in Figure S3, the y285+ ion was present in very low abundance in the NSD spectrum of the aged β2M. Thus, extended external accumulation of the isolated y285+ ion was needed to boost its population to a level appropriate for ECD analysis. As a result, other low-abundance fragment ions of similar m/z, even those absent in the NSD spectrum because of the space charge limit of the collision cell, could be co-isolated and accumulated externally to reach an abundance level high enough to generate ECD fragments. The top-right inset of Figure 3 shows the zoomed-in region of the y285+ ECD spectrum near its precursor ion m/z, where the presence of a second isotope cluster is evident in addition to the isotope cluster of the y285+ ion. The monoisotopic mass of this 4+ ion matches that of the a24 – NH3 ion among all common CAD fragments that could be generated from the aged β2M. This assignment is further confirmed by the observation of many low-abundance ECD fragments that can be attributed to ECD of the (a24 – NH3)4+ precursor. The top-left and bottom-right insets of Figure 3 show the enlarged region of the C-terminal isoAsp83 diagnostic ions, where interference peaks, labeled with an asterisk, from ECD of the (a24 –NH3)4+ ion are evident. The complete list of assigned peaks can be found in Table S1. In the example shown here, the A+1 isotope of the isoAsp diagnostic ions is 71 mDa heavier than the monoisotopic peak of the interfering (z183+)* peak, which can be easily distinguished by the FTICR mass analyzer. Although it is not common that an interfering fragment ion from a co-isolated precursor ion has an m/z value indistinguishable (by an FTICR analyzer) from that of the isoAsp diagnostic ion, it is important to examine such a possibility when co-isolation is evident. In the rare case when the interfering fragment cannot be differentiated from the diagnostic ion, it is possible to perform ECD analysis on a different NSD fragment ion (either in a different charge state or with a different length) to avoid such interference.
Identification of isoAsp42 by NSD-AI-ECD MS3 Analysis
As in the case of the Asn83 deamidation site, isoAsp formation at Asn42 cannot be confirmed by top-down ECD analysis of the intact aged β2M. Even with the MS3 approach, detection of isoAsp42 is still very challenging. Unlike isoAsp17 and isoAsp83 which are located near the protein termini, isoAsp42 is located in the middle of the protein sequence, and most isoAsp42-containing NSD fragments were fairly large. None of the isoAsp42-containing NSD fragments analyzed produced isoAsp diagnostic ion(s) under ECD, which may be attributed to either slow product separation or restricted conformational access. Both restrictions may be lifted when the precursor ion is activated prior to ECD analysis. Figure 4 shows the AI-ECD spectrum of the b639+ ion, obtained by irradiating the precursor ion with 10.6 μm IR light from the CO2 laser (95% power) for 400 ms right before the ECD event. For clarity, the spectrum is divided into two segments, and only c- and z-type ions are labeled. Insets a and b show the enlarged region of the expected N-terminal isoAsp42 diagnostic ion in 6+ charge state in the (a) ECD and (b) AI-ECD spectrum of the b639+ ion. Whereas the ECD spectrum shows only noise in this region (inset a), the presence of a 6+ fragment ion is evident in the AI-ECD spectrum (inset b). The theoretical isotopic distribution of the (c41+57)6+ ion, represented by circles in inset b, matches well in m/z values with the isotope cluster observed experimentally, although the intensity match is only fair, as the ion abundance is too low to reach the statistical distribution. Because the monoisotopic peak is barely observable above the noise level, the experimental monoisotopic m/z value is derived from the A+1 and A+2 peaks; its measured mass is within 0.5 ppm of the theoretical value. IsoAsp formation at the Asn42 deamidation site is further confirmed by the presence of a C-terminal diagnostic ion, (z22-57)2+, in both the AI-ECD (0.8 ppm, inset c) and ECD (0.3 ppm, inset d) spectra, although the S/N ratio is also fairly low.
Figure 4.
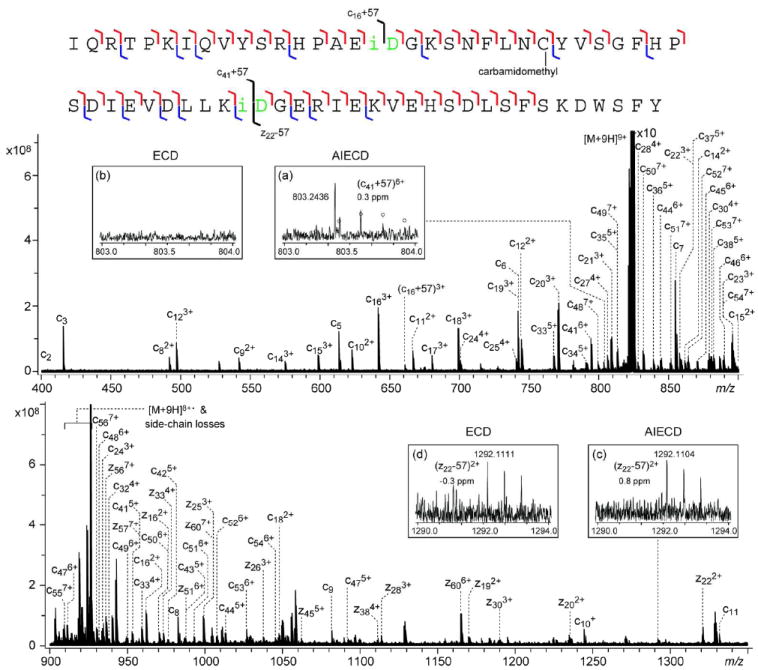
NSD-AI-ECD spectrum of the b639+ ion generated from the aged β2M, shown in two segments for clarity. The cleavage map is shown on top of the spectrum. Insets (a) and (b) show the comparison between the NSD-AI-ECD and NSD-ECD spectra at the N-terminal isoAsp42 diagnostic ion region; insets (c) and (d) show the comparison at the C-terminal isoAsp42 diagnostic ion region.
Poor S/N ratio in NSD-AI-ECD spectra can be partially attributed to the difficulty in maintaining the optimum delay between ion and electron injection for best ion-electron overlap and maximum ECD efficiency. Because the frequency of ion magnetron motion is dependent on the ion population,18 fluctuation in ion abundance due to changes in efficiency of ionization, fragmentation or isolation can lead to significant decrease in ECD efficiency if the delay is not adjusted accordingly. This is a particularly severe problem when a prolonged period of IR irradiation is needed for efficient ion activation, during which time ions may complete multiple cycles of magnetron motion, and a small change in magnetron frequency could lead to a significant change in ion-electron overlap at the time of electron injection. This problem is further aggravated by the necessity of averaging multiple scans to achieve better S/N ratio, during which span ion abundance may change appreciably. In principle, problems associated with ion abundance variation inside the ICR cell could be mitigated by using the automatic gain control (AGC), although AGC was not implemented in this study. Nonetheless, the presence of complementary diagnostic ions assigned with high mass accuracy provides compelling evidence for isoAsp42 formation.
Quantification of the isoAsp content by top-down analysis
The extent of deamidation can be quantified by measuring the relative abundance of the deamidation product and the undeamidated species. In top-down analysis, the mass resolving power is usually not sufficient to separate the monoisotopic precursor peak of the Asp/isoAsp-containing species from the 13C isotope peak of the Asn-containing species. In this case, quantification can be achieved by deconvoluting the isotopic distribution, as demonstrated in an in vitro deamidation study of ribonuclease B.26 The accuracy depends on how closely the observed isotopic pattern resembles the theoretical one, which is affected by the number of ions and signal averaging. Accurate quantification is difficult for low-degree Asn deamidation, and in fragment ions containing several potential deamidation sites, which requires multivariable fitting.
Quantification of the isoAsp content by MS is more challenging. An elegant approach was recently proposed by Zhou and coworkers, who utilized selective protein isoaspartate methyltransferase (PIMT)-mediated 18O-labeling of isoaspartic acid for isoAsp quantification.27 Although this assay is well suited for identification and quantification of isoAsp formation resulting from Asp isomerization, it is not suitable for isoAsp detection at facile Asn deamidation sites, as 18O-labeled Asp/isoAsp can also be produced by rapid Asn deamidation during the hydrolysis step. Alternatively, isoAsp quantification can be achieved based on the relative abundance of the diagnostic ion(s) produced in ECD at the peptide level.6c However, both methods mentioned above adopt the bottom-up approach, and artifactual in vitro deamidation can occur during enzymatic digestion.5a Figure S4 shows the isotopic pattern of the Asn17-containing peptide HPAENGK, generated by tryptic digestion for 2, 4, 8, and 24 hours, respectively. For this small tryptic peptide, the monoisotopic peak of the deamidated product can be baseline separated from the 13C isotope peak of the undeamidated species (Figure S4b, inset). Even with a short 2-hr digestion, nearly 30% of Asn17 residues were already deamidated. IsoAsp diagnostic c4+57 ion (m/z 509.2232) was also observed in the ECD spectrum of this 2-hr digestion mixture, illustrating the adverse effect of hydrolysis during proteolysis on the detection and quantification of isoAsp formation.
In contrast, in the in vitro aging experiment without the presence of trypsin, no isoAsp17 diagnostic ion, (c16+57)3+, was observed in the ECD spectrum of the unaged β2M (Figure S5a). Further, the relative abundance of the diagnostic ion increased as the incubation time increased (Figures S5b-e). The degree of deamidation can be estimated from the relative abundance of the tryptic peptides 13HPAED(/iD)GK19 and 13HPAENGK19 produced after the same incubation period in the parallel bottom-up aging experiment (Figure S4a). Figure 5a shows the linear relationship (R2 = 0.97) between the relative peak height of the diagnostic (c16+57)3+ ion (to c112+ ion) as measured in the top-down ECD analysis, and the degree of deamidation as measured in the bottom-up experiment, generated with technical triplicates. The slight, negative deviation from the line for points obtained at shorter incubation times can be attributed to the different deamidation rates of Asn17 in intact β2M and in the tryptic peptide. Alternatively, the degree of deamidation can be obtained directly from the top-down analysis by measuring the isotopic pattern shift of the c17 (or larger) ion after incubation. Figure 5b shows the same relative isoAsp diagnostic ion abundance versus % deamidation plot, with the degree of deamidation obtained from the top-down analysis instead. The linearity is far better (R2 = 0.99), as the % deamidation obtained this way is a more accurate measure of the deamidation extent in in vitro aging of the intact protein.
Figure 5.
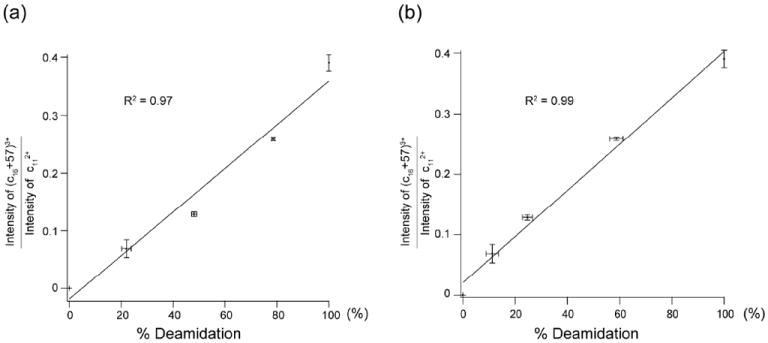
Plots of the relative abundance of the isoAsp17 diagnostic ion in top-down ECD spectra versus % deamidation as measured by (a) bottom-up analysis, and (b) top-down analysis.
It is important to note that all quantitative analyses require the generation of the calibration curve. For peptides, pure isoAsp peptide standards can be obtained either synthetically or by HPLC separation of the deamidation products. However, chemical synthesis of the isoAsp-containing protein standard is both costly and technically difficult, whereas biosynthesis is impossible because isoAsp is not coded in any genome. Further, HPLC separation of the Asp- and isoAsp-isomers at the protein level is challenging as their affinity difference is likely very small. One possible solution is to generate the calibration line in a two-step manner. First, the intact protein standard is incubated for an extensive time period to ensure 100% deamidation of the Asn residues of interest. The resultant deamidated protein is then subject to parallel ECD analyses using both the top-down and bottom-up approaches. The isoAsp content in the tryptic peptide mixture can be quantified using the strategy as described previously,6c which will then be used to establish the relationship between the relative abundance of the isoAsp diagnostic ion in top-down analysis and the relative isoAsp content.
Conclusion
Using the protein β2M as the model system, our study demonstrated the potential of ECD in the detection and quantitative analysis of isoAsp formation at the intact protein level, which eliminates the need for enzymatic digestion prior to MS analysis and the potential for artifactual deamidation that could occur during proteolysis. Top-down ECD analysis identified three Asn deamidation sites, but only detected isoAsp formation at one site. An NSD-ECD MS3 approach was developed which resulted in improved sequence coverage and positive isoAsp identification at all three deamidation sites. Sequence scrambling was not observed among the b-ions chosen for ECD analysis, and is not expected to cause false isoAsp identification using the NSD-ECD approach. False positives due to ion co-isolation are also unlikely when using a mass analyzer with a sufficiently high resolving power. When the potential isoAsp formation site is in the middle of the protein sequence, ion activation may be needed to facilitate isoAsp diagnostic ion formation and detection. The top-down ECD and NSD-ECD MS3 approaches developed here appear to be well suited for deamidation studies involving proteins of relatively small size, but may become increasingly more challenging as the size of the protein increases. Alternative methods, such as the middle-down approach, may be better suited for studying deamidation of large proteins.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge the financial support from NIH P41 RR10888/GM104603, R01 GM078293, S10 RR025082, the Warwick Centre for Analytical Science (EPSRC funded EP/F034210/1), and the University of Warwick Department of Chemistry.
References
- 1.(a) Nilsson MR, Driscoll M, Raleigh DP. Protein Sci. 2002;11:342–349. doi: 10.1110/ps.48702. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Robinson NE, Robinson AB. Proc Natl Acad Sci U S A. 2001;98:944–949. doi: 10.1073/pnas.98.3.944. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Robinson AB. Mech Ageing Dev. 1979;9:225–236. doi: 10.1016/0047-6374(79)90101-5. [DOI] [PubMed] [Google Scholar]; (d) Takata T, Oxford JT, Demeler B, Lampi KJ. Protein Sci. 2008;17:1565–1575. doi: 10.1110/ps.035410.108. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Liu DTY. Trends Biotechnol. 1992;10:364–369. doi: 10.1016/0167-7799(92)90269-2. [DOI] [PubMed] [Google Scholar]; (f) Wakankar AA, Borchardt RT. J Pharm Sci. 2006;95:2321–2336. doi: 10.1002/jps.20740. [DOI] [PubMed] [Google Scholar]; (g) Lindner H, Helliger W. Exp Gerontol. 2001;36:1551–1563. doi: 10.1016/s0531-5565(01)00140-1. [DOI] [PubMed] [Google Scholar]
- 2.Wright HT. Crit Rev Biochem Mol Biol. 1991;26:1–52. doi: 10.3109/10409239109081719. [DOI] [PubMed] [Google Scholar]
- 3.Aswad DW, Paranandi MV, Schurter BT. J Pharmaceut Biomed Anal. 2000;21:1129–1136. doi: 10.1016/s0731-7085(99)00230-7. [DOI] [PubMed] [Google Scholar]
- 4.(a) Zhang W, Czupryn MJ. J Pharmaceut Biomed Anal. 2003;30:1479–1490. doi: 10.1016/s0731-7085(02)00479-x. [DOI] [PubMed] [Google Scholar]; (b) Chazin WJ, Kordel J, Thulin E, Hofmann T, Drakenberg T, Forsen S. Biochem. 1989;28:8646–8653. doi: 10.1021/bi00447a055. [DOI] [PubMed] [Google Scholar]; (c) Carlson AD, Riggin RM. Anal Biochem. 2000;278:150–155. doi: 10.1006/abio.1999.4421. [DOI] [PubMed] [Google Scholar]; (d) Castet S, Enjalbal C, Fulcrand P, Guichou JF, Martinez J, Aubagnac JL. Rapid Commun Mass Spectrom. 1996;10:1934–1938. [Google Scholar]; (e) Gonzalez LJ, Shimizu T, Satomi Y, Betancourt L, Besada V, Padron G, Orlando R, Shirasawa T, Shimonishi Y, Takao T. Rapid Commun Mass Spectrom. 2000;14:2092–2102. doi: 10.1002/1097-0231(20001130)14:22<2092::AID-RCM137>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 5.(a) Li XJ, Cournoyer JJ, Lin C, O’Connor PB. J Am Soc Mass Spectrom. 2008;19:855–864. doi: 10.1016/j.jasms.2008.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gaza-Bulseco G, Li B, Bulseco A, Liu H. Anal Chem. 2008;80:9491–9498. doi: 10.1021/ac801617u. [DOI] [PubMed] [Google Scholar]; (c) Xiao G, Bondarenko PV, Jacob J, Chu GC, Chelius D. Anal Chem. 2007;79:2714–2721. doi: 10.1021/ac0617870. [DOI] [PubMed] [Google Scholar]
- 6.(a) Cournoyer JJ, Pittman JL, Ivleva VB, Fallows E, Waskell L, Costello CE, O’Connor PB. Protein Sci. 2005;14:452–463. doi: 10.1110/ps.041062905. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cournoyer JJ, Lin C, O’Connor PB. Anal Chem. 2006;78:1264–1271. doi: 10.1021/ac051691q. [DOI] [PubMed] [Google Scholar]; (c) Cournoyer JJ, Lin C, Bowman MJ, O’Connor PB. J Am Soc Mass Spectrom. 2007;18:48–56. doi: 10.1016/j.jasms.2006.08.008. [DOI] [PubMed] [Google Scholar]; (d) O’Connor PB, Cournoyer JJ, Pitteri SJ, Chrisman PA, McLuckey SA. J Am Soc Mass Spectrom. 2006;17:15–19. doi: 10.1016/j.jasms.2005.08.019. [DOI] [PubMed] [Google Scholar]; (e) Chan WYK, Chan TWD, O’Connor PB. J Am Soc Mass Spectrom. 2010;21:1012–1015. doi: 10.1016/j.jasms.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Yang H, Fung EYM, Zubarev AR, Zubarev RA. J Proteome Res. 2009;8:4615–4621. doi: 10.1021/pr900428m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yang H, Zubarev RA. Electrophoresis. 2010;31:1764–1772. doi: 10.1002/elps.201000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Chait BT. Science. 2006;314:65–66. doi: 10.1126/science.1133987. [DOI] [PubMed] [Google Scholar]; (b) Yates JR, Ruse CI, Nakorchevsky A. Annu Rev Biomed Eng. 2009;11:49–79. doi: 10.1146/annurev-bioeng-061008-124934. [DOI] [PubMed] [Google Scholar]
- 9.Sargaeva NP, Lin C, O’Connor PB. Anal Chem. 2009;81:9778–9786. doi: 10.1021/ac901677t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Radford SE, Gosal WS, Platt GW. Biochim Biophys Acta Proteins Proteomics. 2005;1753:51–63. doi: 10.1016/j.bbapap.2005.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Heegaard NHH. Amyloid-J Protein Fold Disord. 2009;16:151–173. [Google Scholar]
- 11.Drueke TB, Massy ZA. Semin Dial. 2009;22:378–380. doi: 10.1111/j.1525-139X.2009.00584.x. [DOI] [PubMed] [Google Scholar]
- 12.(a) Eakin CM, Miranker AD. Biochim Biophys Acta Proteins Proteomics. 2005;1753:92–99. doi: 10.1016/j.bbapap.2005.09.002. [DOI] [PubMed] [Google Scholar]; (b) Srikanth R, Mendoza VL, Bridgewater JD, Zhang GS, Vachet RW. Biochem. 2009;48:9871–9881. doi: 10.1021/bi901172y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Antwi K, Mahar M, Srikanth R, Olbris MR, Tyson JF, Vachet RW. Protein Sci. 2008;17:748–759. doi: 10.1110/ps.073249008. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Naiki H, Yamamoto S, Hasegawa K, Yamaguchi I, Goto Y, Gejyo F. Amyloid-J Protein Fold Disord. 2005;12:15–25. doi: 10.1080/13506120500032352. [DOI] [PubMed] [Google Scholar]
- 13.Kad NM, Thomson NH, Smith DP, Smith DA, Radford SE. J Mol Biol. 2001;313:559–571. doi: 10.1006/jmbi.2001.5071. [DOI] [PubMed] [Google Scholar]
- 14.Iwata K, Matsuura T, Sakurai K, Nakagawa A, Goto Y. J Biochem. 2007;142:413–419. doi: 10.1093/jb/mvm148. [DOI] [PubMed] [Google Scholar]
- 15.Borysik AJH, Radford SE, Ashcroft AE. J Biol Chem. 2004;279:27069–27077. doi: 10.1074/jbc.M401472200. [DOI] [PubMed] [Google Scholar]
- 16.(a) Robinson NE, Robinson AB. Proc Natl Acad Sci U S A. 2001;98:4367–4372. doi: 10.1073/pnas.071066498. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Robinson NE, Robinson AB, Merrifield RB. J Pept Res. 2001;57:483–493. doi: 10.1034/j.1399-3011.2001.00863.x. [DOI] [PubMed] [Google Scholar]; (c) Chelius D, Rehder DS, Bondarenko PV. Anal Chem. 2005;77:6004–6011. doi: 10.1021/ac050672d. [DOI] [PubMed] [Google Scholar]
- 17.Rüegg UT, Rudinger J. In: Methods Enzymol. Hirs CHW, Serge NT, editors. Vol. 47. Academic Press; 1977. pp. 111–116. [DOI] [PubMed] [Google Scholar]
- 18.Tsybin YO, Hendrickson CL, Beu SC, Marshall AG. Int J Mass spectrom. 2006;255-256:144–149. [Google Scholar]
- 19.Koster C, Holle A. ASMS annual conference; Dallas, TX. 1999. [Google Scholar]
- 20.(a) Bleiholder C, Osburn S, Williams TD, Suhai S, Van Stipdonk M, Harrison AG, Paizs B. J Am Chem Soc. 2008;130:17774–17789. doi: 10.1021/ja805074d. [DOI] [PubMed] [Google Scholar]; (b) Harrison AG, Young AB, Bleiholder C, Suhai S, Paizs B. J Am Chem Soc. 2006;128:10364–10365. doi: 10.1021/ja062440h. [DOI] [PubMed] [Google Scholar]
- 21.Li X, Huang Y, O’Connor P, Lin C. J Am Soc Mass Spectrom. 2011;22:245–254. doi: 10.1007/s13361-010-0036-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goloborodko A, Gorshkov M, Good D, Zubarev R. J Am Soc Mass Spectrom. 2011;22:1121–1124. doi: 10.1007/s13361-011-0130-z. [DOI] [PubMed] [Google Scholar]
- 23.Saminathan IS, Wang XS, Guo Y, Krakovska O, Voisin S, Hopkinson AC, Siu KWM. J Am Soc Mass Spectrom. 2010;21:2085–2094. doi: 10.1016/j.jasms.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 24.Yu L, Tan Y, Tsai Y, Goodlett DR, Polfer NC. J Proteome Res. 2011;10:2409–2416. doi: 10.1021/pr101235w. [DOI] [PubMed] [Google Scholar]
- 25.Théberge R, Infusini G, Tong W, McComb ME, Costello CE. Int J Mass spectrom. 2011;300:130–142. doi: 10.1016/j.ijms.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zabrouskov V, Han X, Welker E, Zhai H, Lin C, van Wijk KJ, Scheraga HA, McLafferty FW. Biochemistry. 2005;45:987–992. doi: 10.1021/bi0517584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu M, Cheetham J, Cauchon N, Ostovic J, Ni W, Ren D, Zhou ZS. Analytical Chemistry. 2011;84:1056–1062. doi: 10.1021/ac202652z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.