

Abstract
Copper-catalyzed azide–alkyne cycloaddition (CuAAC) has found numerous applications in a variety of fields. We report here only modest differences in the reactivity of various classes of terminal alkynes under typical bioconjugative and preparative organic conditions. Propargyl compounds represent an excellent combination of azide reactivity, ease of installation, and cost. Electronically activated propiolamides are slightly more reactive, at the expense of increased propensity for Michael addition. Certain alkynes, including tertiary propargyl carbamates, are not suitable for bioconjugation due to copper-induced fragmentation. A fluorogenic probe based on such reactivity is available in one step from rhodamine 110 and can be useful for optimization of CuAAC conditions.
More than ten years have now passed since first reports of copper-catalyzed azide–alkyne cycloaddition (CuAAC) by Meldal1 and Fokin/Sharpless.2 The ability to reliably form a stable linkage between these two functional groups in a multitude of settings was immediately appreciated as a powerful tool, placing CuAAC at the center of the click chemistry arsenal.3
Azides and alkynes are rare in nature, making the CuAAC process of great use as a bioorthogonal transformation4 for making linkages to biological molecules,5 usually involving low concentrations of reagents and mild conditions. The limited compatibility of copper with living organisms has inspired the development of metal-free azide–alkyne cycloaddition with highly strained cyclooctynes.6,7 The discovery of other reactions that fit these requirements, most notably tetrazine–olefin cyclooaddition,8–10 quickly followed, and this field remains one of rapid development.11
We have optimized the ligand-accelerated CuAAC bioconjugation reaction under practical conditions, ameliorating some of the adverse effects of the ability of copper ions and ascorbate to generate species harmful to biomolecules.12 Using this procedure, we wanted to identify the substrates that enable the most rapid triazole formation. We assumed from limited anecdotal information that CuAAC reaction rates are more sensitive to the nature of the alkyne component than the azide. It has long been known that alkynes can be activated toward uncatalyzed [3+2] cycloaddition by conjugating the alkyne unit with electron-withdrawing ester groups due to a lowering of the LUMO energy of the alkyne.13,14 Thus, it is reasonable to test the idea that propiolate derivatives, ethynyl ketones, and ethynyl aldehydes could be more reactive in a copper-catalyzed AAC as well. However, acetylenic ketones, aldehydes and alkyl propiolates and can be rather difficult to functionalize.15 Most importantly, these substrates, particularly in the terminal alkyne form required for CuAAC, are too reactive as Michael acceptors16 to be bioorthogonal. We regarded propiolamides as having a good combination of synthetic accessibility, electronic activation of the CuAAC process, and attenuated Michael reactivity. The 2nd-order rate constant of the conjugate addition of a model cysteine-containing peptide to N-phenylpropiolamide can be estimated as approximately 0.13 M−1s−1 (pH 7.0).17 The reaction rates of maleimide18 and oxanorbornadiene reagents19,20 with thiols are more than 500 times greater.
Standard unactivated alkynes, often derived from propargyl building blocks, perform very well with good CuAAC catalysts and are used in most applications of the reaction. Propiolamides, among the first groups used for CuAAC,1 have been usually considered only when even greater reactivity is required, such as when especially low concentrations of reagents must be used, or when that particular structure type is desired for other reasons. Thus, a search for optimal yields in radiolabel synthesis among aromatic, aliphatic, propargylamido, and propiolate alkyne possibilities found the last to be most effective;21 and several demanding bioconjugative applications involving propiolamides have been reported.22–25 A literature search for carbamoyltriazoles, the product of CuAAC between propiolamides and azides, revealed that most have been used in larger screening efforts and without special consideration of their potentially high reactivity. Often these triazoles were made by aminolysis of corresponding methyl triazole-4-carboxylate, as exemplified by a synthesis of the anticonvulsant drug rufinamide.26 To date, no quantitative comparison of ligand-accelerated triazole-forming reactivity (LACuAAC) has been reported for candidate alkyne types including propiolamides and propargylic compounds, using any of the more active catalyst formulations.
Toward this end, we tested the performance of a panel of alkynes in a standard kinetic assay using the fluorogenic azidocoumarin of Wang and coworkers.27 We included several types of alkynes that are typically attached to molecules of interest, including secondary propiolamide A, tertiary propiolamide E, propargyl derivatives B–D and F–J, longer-chain alkyne L, and aromatic alkynes K and M (Scheme 1). In this survey we excluded poorly soluble and potentially volatile unfunctionalized alkynes.
Propiolamides were initially confirmed as good substrates for CuAAC using accelerating ligand THPTA (Figure 1).28 Tertiary propiolamide E reached >90% conversion within 1 hour even with 5 mM Cu catalyst, a lower concentration than we typically recommend.12,29,30 All alkyne substrates were quite reactive in the presence of 100 μM Cu+, reacting completely in less than 30 minutes. Under demanding conditions (10 μM Cu+) the differences were more distinct. Due to varying quantum yields, the comparison between kinetic traces of the reactions of coumarin azide with alkynes A–M is not straightforward. Therefore, we compare the relative rates by reporting the times required to reach 50% and 90% completion of maximum fluorescence in each case (Figure 1). Propiolamide A was the fastest, followed not by its analogue E, but by propargyl ethers B–D. N-propargylamide F, propargylamines G–I, and propargyl alcohol J all reacted reasonably well, while aromatic and aliphatic alkynes K–M were slower. The kinetic traces from which these data were extracted are shown in the Supporting Information.
Figure 1.

Performance of various alkyne substrates in the ligand-accelerated (LA) CuAAC process under bioconjugation conditions. (Top) Reaction scheme and structures of alkynes used. (Bottom) Time required to reach 50% and 90% of maximum fluorescence under various conditions.
Competition experiments performed under typical organic LACuAAC conditions (using ligand BimPy2 instead of THPTA)30 revealed that propiolamide E was more reactive than the aromatic and propargylic alkynes shown in Figure 2. In neither case (aqueous bioconjugation-like conditions, Figure 1, and organic synthesis-like conditions, Figure 2) were the observed differences in reactivity between alkynes great enough to warrant a firm recommendation to use one type of substrate over another. The relatively close reactivities of a variety of easily-installed terminal alkyne motifs serves to illustrate the robustness of these LACuAAC protocols.
Figure 2.
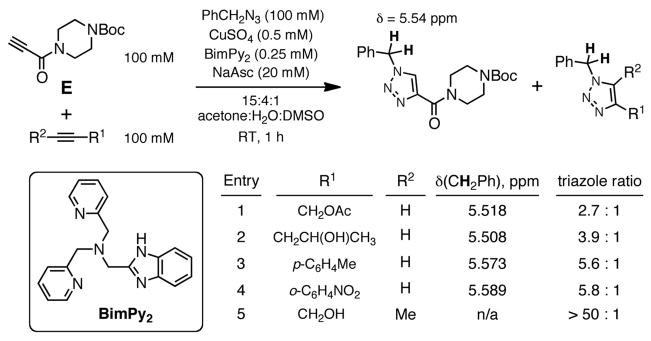
Competition experiment under typical organic conditions.
As evident from Figures 1 and 2, electronic activation of the alkyne does not bring about as great a rate increase in LACuAAC as it does in the uncatalyzed reaction (methyl propiolate reacts with phenyl azide 32 times faster than phenylacetylene).13 However, the electron-poor propiolamides are capable of altering the spectral properties of attached fluorophores, and thus may prove useful for development of fluorogenic substrates for LACuAAC. For example, we observed that N-(pyren-1-ylmethyl)propiolamide (PyrPRA) exhibited an 8-fold increase in fluorescence intensity at 394 nm upon reaction with a model azide γ-L-azidohomoalanine (AHA), suggesting that PyrPRA may be quenched by a photoinduced electron-transfer mechanism (Fig. 3A,B). Well-structured emission bands at 370–430 nm are typically associated with monomeric excited pyrene species (Fig 3B). Interestingly, upon reaction of PyrPRA with a polyvalent azide (Qβ virus-like particles bearing several hundred azide groups on the 28-nm-diameter surface31), the intensity of monomer emission at 394 nm grew slightly, but excimer fluorescence at 488 nm increased 3-fold (Fig. 3C). This suggests that excimers are formed from nearby pyrene units on the nanoparticle scaffold. Further development of these results requires a more polar derivative, as precipitation of PyrPRA was observed even when significant amounts of DMSO co-solvent were used. Other fluorogenic alkynes have been reported based on coumarin,27,32 naphthalimide,33 benzothiazole,34,35 benzoxadiazole,36 and cyclooctyne37 scaffolds.
Figure 3.

(A) CuAAC reaction of propiolamide PyrPRA with azidohomoalanine. (B) Emission spectra (excitation at 348 nm) before (PyrPRA) and after (PyrTRZ) CuAAC reaction with AHA. (C) Time course of polyvalent Qβ nanoparticle-azide labeling with PyrPRA (excitation at 348 nm, emission at 394 and 488 nm).
Over the years of active use of CuAAC, a few reports on the failure of certain substrates to convert cleanly and quantitatively to the desired triazoles have emerged. One of the best known cases is the formation of bis-triazoles by oxidative coupling of triazolylcopper intermediate, an occasional outcome under bioconjugative conditions that use low concentration of reagents.38 Other complications arise from azide-independent reactivity of copper acetylides. For example, 4-pentynoic acid and its derivatives such as propargyl glycine may give enol lactone side products using either CuBr or Cu(II)/ascorbate as a source of Cu(I).39 Predictably, this occurs for free acids but not for 4-pentynoic esters and amides. Using accelerating ligand THPTA, this pathway appears to be suppressed to a significant extent, as evidenced by the observed CuAAC reactivity of 4-pentynoic acid L.
Propargylic esters, carbamates, and carbonates readily lose their leaving groups upon formation of the corresponding Cu-acetylide to form copper-stabilized propargyl cations, which can be readily trapped by nucleophiles present in the reaction mixture. If the reaction is performed in water, propargylic alcohols are formed.40,41 This pathway is not a major one for primary propargyl substrates (Fig. 2, entry 1), but becomes dominant if the propargyl cation is stabilized further by methyl groups. We applied this reaction to the tertiary propargyl carbamate (Ynoc group) to create a copper-sensitive fluorogenic probe, as shown in Figure 4. Under typical aqueous CuAAC conditions, both carbamate groups of the readily available rhodamine alkynyl carbamate Ynoc2Rho were cleaved. The resulting release of Rhodamine 110 induced an 1800-fold increase of fluorescence within 30 minutes at 50 μM Cu+ only in the presence of THPTA (5:1 L:Cu+).
Figure 4.
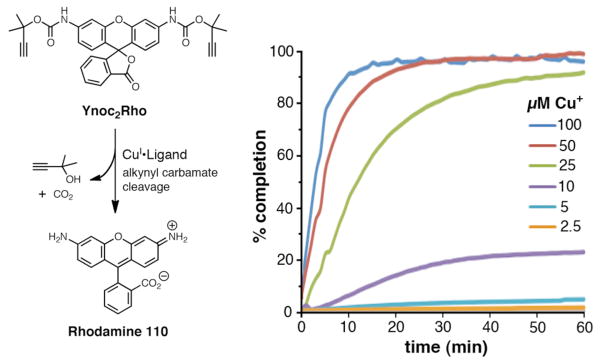
(Top) Cu(I)-induced cleavage of alkynyl carbamate releases fluorescent Rhodamine 110. (Bottom) Kinetic traces of Ynoc2Rho cleavage in the presence of indicated amount of catalyst (5:1 THPTA:Cu+) at 37°C. In the absence of THPTA, the reaction is more than 100 times slower (Supporting Information).
The similarity of the response of Ynoc cleavage and CuAAC reactions to the nature of the catalyst is striking even though the former uses no azide and does not generate triazole. Nonetheless, just like the CuAAC reaction, the Ynoc2Rho probe did not react in the presence of 1 mM of other soft ions, including Hg2+ and Ag+,42 and responded very poorly to Cu+ without an accelerating ligand (Supporting Information). Both Ynoc2Rho cleavage and CuAAC were most efficient in the presence of THPTA (5:1 L:Cu+). In both cases, BimPy2, a strong copper chelator, worked well only at ligand:Cu+ ratios less than or equal to 1:1.29 Ynoc2Rho could therefore be a useful tool for ligand screening for CuAAC and related alkyne-dependent processes. In addition, Ynoc should be a useful protecting group for amines, since it is removed under very mild aqueous conditions with catalytic copper(I), in contrast to the strongly acidic conditions required for cleavage of the related tert-butoxycarbonyl (Boc) group.
We did not explore the reactivity of azides, and until recently there were no reports of significant effects of azide structure on the reaction rate,43 excluding such activated species as carbonyl- and sulfonylazides. However, Ting et al. have recently described the use of 2-picolyl azides for bioconjugation.44 The chelating effect allows the reaction to proceed at low micromolar concentration of reactants without accelerating ligands, but with added benefits from their use.
Here we compared the performance of common non-Michael-reactive alkyne building blocks, resulting in the conclusion that moderate changes in the electronic nature of the alkyne do not have a dramatic effect on CuAAC rate when good accelerating ligands are used. The results, recommendations, and potential pitfalls are summarized below.
Most alkyne substrates behave well under optimized CuAAC conditions, as seems to be the case from the wide use of the process. Substrates derived from simple alkyne building blocks, such as propargyl ethers, N-propargylamides, and propiolamides are recommended over more expensive and slower aryl- and alkylacetylenes.
Propiolamides are very good CuAAC substrates, particularly when Michael reactivity with biological nucleophiles is not a concern. The electron-poor nature of propiolamides may serve well for the development of turn-on fluorogenic substrates for CuAAC, as illustrated by the dequenching of pyrene-propiolamide (PyrPRA) substrate upon reaction with an azide.
Tertiary propargyl esters and carbamates should not be used in CuAAC process. Mild and catalytic copper-induced cleavage of Ynoc carbamate may be useful when traditional protecting groups for an amino group cannot be used due to senstitivity of the substrtate to the cleavage protocols.
Future ligand screens may be facilitated by the introduction of Ynoc2Rho, a fluorogenic probe containing cleavable alkynyl carbamate groups that responds to Cu(I) when an appropriate accelerating ligand is present.
Supplementary Material
Acknowledgments
This work was supported by the NIH (F31CA165653 to A. A. K.), NSF (CH-1011796), and an Eli Lilly Fellowship to A. A. K.
Footnotes
Improved synthetic procedure for the THPTA ligand, synthesis of alkyne substrates, conditions for “bioconjugative” and “organic” CuAAC (Tables S1 and S2), kinetic traces for the data in Fig. 1 (Fig. S2), fluorescent response of Ynoc2Rho to other metals (Fig. S3). The material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Tornøe CW, Christensen C, Meldal M. Peptidotriazoles on Solid Phase: [1,2,3]-Triazoles by Regiospecific Copper(I)-Catalyzed 1,3-Dipolar Cycloadditions of Terminal Alkynes to Azides. J Org Chem. 2002;67:3057–3062. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 2.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective Ligation of Azides and Terminal Alkynes. Angew Chem, Int Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 3.Kolb HC, Finn MG, Sharpless KB. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew Chem Int Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 4.Sletten EM, Bertozzi CR. Bioorthogonal Chemistry: Fishing for Selectivity in a Sea of Functionality. Angew Chem Int Ed. 2009;48:6974–6998. doi: 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang Q, Chan TR, Hilgraf R, Fokin VV, Sharpless KB, Finn MG. Bioconjugation by Copper(I)-Catalyzed Azide-Alkyne [3+2] Cycloaddition. J Am Chem Soc. 2003;125:3192–3193. doi: 10.1021/ja021381e. [DOI] [PubMed] [Google Scholar]
- 6.Agard NJ, Prescher JA, Bertozzi CR. A Strain-Promoted [3+2] Azide-Alkyne Cycloaddition for Covalent Modification of Biomolecules in Living Systems. J Am Chem Soc. 2004;126:15046–15047. doi: 10.1021/ja044996f. [DOI] [PubMed] [Google Scholar]
- 7.Laughlin ST, Baskin JM, Amacher SL, Bertozzi CR. In Vivo Imaging of Membrane-Associated Glycans in Developing Zebrafish. Science. 2008;320:664–667. doi: 10.1126/science.1155106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blackman ML, Royzen M, Fox JM. Tetrazine Ligation: Fast Bioconjugation Based on Inverse-Electron-Demand Diels–Alder Reactivity. J Am Chem Soc. 2008;130:13518–13519. doi: 10.1021/ja8053805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Devaraj NK, Weissleder R, Hildebrand SA. Tetrazine-Based Cycloadditions: Application to Pretargeted Live Cell Labeling. Bioconjugate Chem. 2008;19:2297–2299. doi: 10.1021/bc8004446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Devaraj NK, Weissleder R. Biomedical Applications of Tetrazine Cycloadditions. Accounts Chem Res. 2011;44:816–827. doi: 10.1021/ar200037t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Debets MF, Van Berkel SS, Dommerholt J, Dirks AJ, Rutjes FPJT, Van Delft FL. Bioconjugation with Strained Alkenes and Alkynes. Accounts Chem Res. 2011;44:805–815. doi: 10.1021/ar200059z. [DOI] [PubMed] [Google Scholar]
- 12.Hong V, Presolski SI, Ma C, Finn MG. Analysis and Optimization of Copper-Catalyzed Azide-Alkyne Cycloaddition for Bioconjugation. Angew Chem, Int Ed. 2009;48:9879–9883. doi: 10.1002/anie.200905087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huisgen R. Kinetics and Mechanism of 1,3-Dipolar Cycloadditions. Angew Chem, Int Ed Engl. 1962;2:633–645. [Google Scholar]
- 14.Huisgen R. 1,3-Dipolar Cycloaddition - Introduction, Survey, Mechanism. In: Padwa A, editor. 1,3-Dipolar Cycloaddition Chemistry. Vol. 1. Wiley; New York: 1984. pp. 1–176. [Google Scholar]
- 15.Nagel M, Hansen HJ. Synthesis of Polyalkylphenyl Prop-2-ynoates and Their Flash Vacuum Pyrolysis to Polyalkylcyclohepta[b]furan-2(2H)-ones. Helv Chim Acta. 2000;83:1022–1048. [Google Scholar]
- 16.Böhme A, Thaens D, Schramm F, Paschke A, Schüürmann G. Thiol Reactivity and Its Impact on the Ciliate Toxicity of alpha,beta-Unsaturated Aldehydes, Ketones, and Esters. Chem Res Toxicol. 2010;23:1905–1912. doi: 10.1021/tx100226n. [DOI] [PubMed] [Google Scholar]
- 17.Shiu HY, Chan TC, Ho CM, Liu Y, Wong MK, Che CM. Electron-Deficient Alkynes as Cleavable Reagents for the Modification of Cysteine-Containing Peptides in Aqueous Medium. Chem Eur J. 2009;15:3839–3850. doi: 10.1002/chem.200800669. [DOI] [PubMed] [Google Scholar]
- 18.Tournier EJM, Wallach J, Blond P. Sulfosuccinimidyl 4-(N-maleimidomethyl)-1-cyclohexane carboxylate as a bifunctional immobilization agent. Optimization of the coupling conditions. Analytica Chim Acta. 1998;361:33–44. [Google Scholar]
- 19.Kislukhin AA, Higginson CJ, Hong VP, Finn MG. Degradable Conjugates from Oxanobornadiene Reagents. J Am Chem Soc. 2012;134:6491–6497. doi: 10.1021/ja301491h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hong V, Kislukhin A, Finn MG. Thiol-Selective Fluorogenic Probes for Labeling and Release. J Am Chem Soc. 2009;131:9986–9994. doi: 10.1021/ja809345d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Glaser M, Arstad E. “Click Labeling” with 2-[18F]Fluoroethylazide for Positron Emission Tomography. Bioconjugate Chem. 2007;18:989–993. doi: 10.1021/bc060301j. [DOI] [PubMed] [Google Scholar]
- 22.Yang Y, Kluger R. Efficient CuAAC click formation of functional hemoglobin bistetramers. Chem Commun. 2010;46:7557–7559. doi: 10.1039/c0cc02023k. [DOI] [PubMed] [Google Scholar]
- 23.Billing JF, Nilsson UJ. C2-Symmetric Macrocyclic Carbohydrate/Amino Acid Hybrids through Copper(I)-Catalyzed Formation of 1,2,3-Triazoles. J Org Chem. 2005;70:4847–4850. doi: 10.1021/jo050585l. [DOI] [PubMed] [Google Scholar]
- 24.Wang J, Li H, Zou G, Wang LX. Novel template-assembled oligosaccharide clusters as epitope mimics for HIV-neutralizing antibody 2G12. Design, synthesis, and antibody binding study. Org Biomol Chem. 2007;5:1529–1540. doi: 10.1039/b702961f. [DOI] [PubMed] [Google Scholar]
- 25.Srinivasachari S, Fichter KM, Reineke TM. Polycationic β-Cyclodextrin “Click Clusters”: Monodisperse and Versatile Scaffolds for Nucleic Acid Delivery. J Am Chem Soc. 2008;130:4618–4627. doi: 10.1021/ja074597v. [DOI] [PubMed] [Google Scholar]
- 26.Mudd WH, Stevens EP. An efficient synthesis of rufinamide, an antiepileptic drug. Tetrahedron Lett. 2010;51:3229–3231. [Google Scholar]
- 27.Sivakumar K, Xie F, Cash BM, Long S, Barnhill HN, Wang Q. A Fluorogenic 1,3-Dipolar Cycloaddition Reaction of 3-Azidocoumarins and Acetylenes. Org Lett. 2004;6:4603–4606. doi: 10.1021/ol047955x. [DOI] [PubMed] [Google Scholar]
- 28.We also report in Supporting Information an improved synthesis of THPTA suitable for large scale production. The procedure uses (Ph3P)2CuOAc catalyst in organic solvent [ Shao C, Cheng G, Su D, Xu J, Wang X, Hu Y. Copper(I) Acetate: A Structurally Simple but Highly Efficient Dinuclear Catalyst for Copper-Catalyzed Azide-Alkyne Cycloaddition. Adv Synth Catal. 2010;352:1587–1592.Gonda Z, Novak Z. Highly Active Copper Catalysts for Azide-Alkyne Cycloaddition. Dalton Trans. 2010;39:726–729. doi: 10.1039/b920790m.], and provides material without the bis(triazole) contaminant that can inhibit the CuAAC reaction.
- 29.Rodionov VO, Presolski S, Díaz DD, Fokin VV, Finn MG. Ligand-Accelerated Cu-Catalyzed Azide-Alkyne Cycloaddition: a Mechanistic Report. J Am Chem Soc. 2007;129:12705–12712. doi: 10.1021/ja072679d. [DOI] [PubMed] [Google Scholar]
- 30.Presolski SI, Hong V, Cho SH, Finn MG. Tailored Ligand Acceleration of the Cu-Catalyzed Azide-Alkyne Cycloaddition Reaction: Practical and Mechanistic Implications. J Am Chem Soc. 2010;132:14570–14576. doi: 10.1021/ja105743g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sen Gupta S, Kuzelka J, Singh P, Lewis WG, Manchester M, Finn MG. Accelerated Bioorthogonal Conjugation: A Practical Method for the Ligation of Diverse Functional Molecules to a Polyvalent Virus Scaffold. Bioconjugate Chem. 2005;16:1572–1579. doi: 10.1021/bc050147l. [DOI] [PubMed] [Google Scholar]
- 32.Zhou Z, Fahrni CJ. A fluorogenic Probe for the Copper(I)-Catalyzed Azide-Alkyne [3+2] Cycloaddition: Modulation of the Fluorescence Emission via 3(n,π*)−1(π,π*) Inversion. J Am Chem Soc. 2004;126:8862–8863. doi: 10.1021/ja049684r. [DOI] [PubMed] [Google Scholar]
- 33.Sawa M, Hsu T, Itoh T, Sugiyama M, Hanson SR, Vogt PK, Wong CH. Glycoproteomic probes for fluorescent imaging of fucosylated glycans in vivo. Proc Natl Acad Sci USA. 2006;103:12371–12376. doi: 10.1073/pnas.0605418103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qi JJ, Han MS, Chang YC, Tung CH. Developing Visible Fluorogenic ‘Click-On’ Dyes for Cellular Imaging. Bioconjugate Chem. 2011;22:1758–1762. doi: 10.1021/bc200282t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qi JJ, Tung CH. Development of benzothiazole ‘click-on’ fluorogenic dyes. Bioorg Med Chem Lett. 2011;21:320–323. doi: 10.1016/j.bmcl.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Key JA, Cairo CW. Identification of fluorogenic and quenched benzoxadiazole reactive chromophores. Dyes and Pigments. 2011;88:95–102. [Google Scholar]
- 37.Friscourt F, Fahrni CJ, Boons GJ. A Fluorogenic Probe for the Catalyst-Free Detection of Azide-Tagged Molecules. J Am Chem Soc. 2012;134:18809–18815. doi: 10.1021/ja309000s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kwon M, Jang Y, Yoon S, Yang D, Jeon HB. Unusual Cu(I)-catalyzed 1,3-dipolar cycloaddition of acetylenic amides: formation of bistriazoles. Tetrahedron Lett. 2012;53:1606–1609. [Google Scholar]
- 39.Mindt TL, Schibli R. Cu(I)-Catalyzed Intramolecular Cyclization of Alkynoic Acids in Aqueous Media: A “Click Side Reaction”. J Org Chem. 2007;72:10247–10250. doi: 10.1021/jo702030e. [DOI] [PubMed] [Google Scholar]
- 40.Bertrand P, Gesson JP. Click Chemistry with O-Dimethylpropargylcarbamate for Preparation of pH-Sensitive Functional Groups. J Org Chem. 2007;72:3596–3599. doi: 10.1021/jo070131j. [DOI] [PubMed] [Google Scholar]
- 41.Ljungdahl N, Kann N. Transition-Metal-Catalyzed Propargylic Substitution. Angew Chem Int Ed. 2009;48:642–644. doi: 10.1002/anie.200804114. [DOI] [PubMed] [Google Scholar]
- 42.McNulty J, Keskar K. Discovery of a Robust and Efficient Homogeneous Silver(I) Catalyst for the Cycloaddition of Azides onto Terminal Alkynes. Eur J Org Chem. 2012:5462–5470. [Google Scholar]
- 43.See reference 28 for a study of reactivity vs. electronic effects in aromatic azides.
- 44.Uttamapinant C, Tangpeerachaikul A, Grecian S, Clarke S, Singh U, Slade P, Gee KR, Ting AY. Fast, Cell-Compatible Click Chemistry with Copper-Chelating Azides for Biomolecular Labeling. Angew Chem Int Ed. 2012;51:5852–5856. doi: 10.1002/anie.201108181. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.