

Abstract
We report the discovery and characterization of a series of benzisothiazolone inhibitors of PHOSPHO1, a newly identified soluble phosphatase implicated in skeletal mineralization and soft tissue ossification abnormalities. High-throughput screening (HTS) of a small molecule library led to the identification of benzoisothiazolones as potent and selective inhibitors of PHOSPHO1. Critical structural requirements for activity were determined, and the compounds were subsequently derivatized and measured for in vitro activity and ADME parameters including metabolic stability and permeability. On the basis of its overall profile the benzoisothiazolone analogue 2q was selected as MLPCN probe ML086
Keywords: Phosphatase, Vascular calcification, Probe compound, ML086, PHOSPHO1
PHOSPHO1 is a recently identified orphan phosphatase that belongs to the family of halo-acid dehalogenases. It is a soluble phosphatase with specificity for phosphoethanolamine (P-Etn) and phosphocholine (P-Cho) present in matrix vesicles (MVs).1 PHOSPHO1 is responsible for increasing the local concentration of inorganic phosphate (Pi) inside MVs to change the phosphate:pyrophosphate (Pi/PPi) ratio to favor precipitation of hydroxyapatite (HA) seed crystals.2 Aberrations of the Pi/PPi ratio have been associated with numerous pathologies. Low extracellular PPi (ePPi) production has been identified as a cause in the development of severe medial vascular calcification (MVC) known as generalized arterial calcification of infancy (GACI; OMIM # 208000)3 as well as ossification of the posterior longitudinal ligaments of the spine (OPLL; OMIM # 602475) and osteoarthritis (OA).4 In addition, low ePPi transport manifests as ankylosing vertebral hyperostosis (DISH; OMIM # 106400), chondrocalcinosis (OMIM # 605145) and ankylosing spondylitis (AS; OMIM # 106300).5 On the other hand, accumulation of ePPi results in rickets or osteomalacia, known as hypophosphatasia (HPP; OMIM # 171760).6 Because of the accumulation of ePPi, HPP patients may also display chondrocalcinosis or calcium pyrophosphate dihydrate deposition (CPPD) disease (OMIM # 118600). However, many of these conditions have been linked to deficiencies in other transporters and phosphatases, most notably tissue-nonspecific alkaline phosphatase (TNAP) that is also found in the same biological compartment.7 There is therefore a significant need to identify small molecule compounds that can probe the function of these enzymes and provide a starting point for the development of therapeutic agents.
We previously reported the synthesis and optimization of selective small molecule inhibitors of TNAP that have been employed to investigate the role of TNAP in vascular calcification.8 We also disclosed the synthesis and characterization of a series of compounds that inhibit phosphomannose isomerase (PMI), an enzyme implicated in therapeutically important protein glycosylation processes.8a, 9 These small molecule probes, discovered using high-throughput screening (HTS) and chemical optimization through the Molecular Libraries Probe Production Centers Network (MLPCN; http://mli.nih.gov/mli/mlpcn/), have found utility for investigating the role of intracellular and extracellular phosphatases. The structure of the PMI inhibitor probe ML089 (1) is shown in Figure 1. We hypothesized that, in a similar manner, it would be possible to discover small molecule inhibitors of PHOSPHO1 that would help to delineate the role of this enzyme in skeletal mineralization and soft tissue ossification abnormalities at a fundamental level. Furthermore, selective PHOSPHO1 inhibitors would be useful tools to elucidate the mechanism of action for the aforementioned diseases. With this in mind a high-throughput screening (HTS) campaign was performed through the MLPCN.
Figure 1.

Phosphomannose isomerase (PMI) probe ML089 (1) and the benzothiazolone scaffold of the PHOSPHO1 screening hits.
The screen employed a colorimetric assay based on the ability of PHOSPHO1 to liberate phosphate from P-Etn and its reaction with the Biomol Green reagent (Biomol International, Plymouth Meeting, PA, USA). The construct was designed to express PHOSPHO1 protein fused to a V5 epitope and 6 His-tag at the C-terminus. A diverse library of 288,481 compounds from the MLSMR collection was tested at a single concentration of 13.3 μM (PubChem AID 1565). This provided 3,164 compounds that showed greater than 60% activity in the single point assay, a hit rate of 1.1%. Hit confirmation was performed using the colorimetric assay to verify inhibitory activity against PHOSPHO1 in dose-response mode performed in duplicate using a 10-point 2-fold serial dilution of the hit compounds in DMSO. Inhibitors that were active in dose-response mode against PHOSPHO1 and soluble in the concentration range relevant to their potency were classified as confirmed hits. This led to the identification of several sub-micromolar inhibitors of PHOSPHO1 (see PubChem link to AID 1565 and 1666 for details).
It was noted that some of the confirmed hits fell into the benzoisothiazolone class of small molecules, such as 1, that were previously identified as PMI inhibitors. For example, the unsubstituted benzoisothiazolone 2a (Table 1) inhibited PHOSPHO1 with an IC50 value of 0.94μM while also inhibiting PMI (IC50 = 6.4 μM). We hypothesized that within this series it might be possible to optimize the potency of compounds against PHOSPHO1 while reducing or eliminating activity at PMI and PMM2. The benzothiazolone series was therefore prioritized for chemical optimization using both analogue by catalogue (ABC) and synthetic chemistry.
Table 1.
Summary of first round of SAR for compounds synthesized and tested for PHOSPHO1 inhibition as well as selectivity against related phosphatases.
| Cmpd. | Ar | R1 | PHOSPHO1a | PMIb IC50 [μM] | PMM2b |
|---|---|---|---|---|---|
| 2a |

|
H | 0.94 | 6.4 | > 20 |
| 2b |
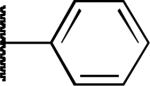
|
F | 0.79 | 1.3 | > 50 |
| 2c |

|
H | 1.3 | 3.9 | > 30 |
| 2d |
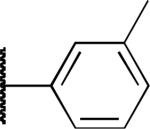
|
H | 2.7 | 6.0 | > 20 |
| 2e |
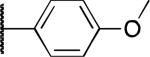
|
F | 6.7 | 1.0 | > 10 |
| 2f |

|
H | 4.9 | 3.4 | > 20 |
| 2g |
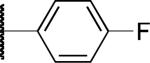
|
F | > 10 | 3.6 | > 50 |
| 2h |
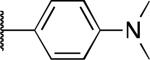
|
H | 11 | 8.4 | > 30 |
| 2i |

|
H | 5.2 | 4.8 | > 30 |
| 2j |
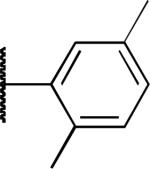
|
H | 4.0 | 6.6 | > 20 |
| 2k |

|
F | 3.3 | 1.1 | 7.3 |
| 21 |

|
F | 4.7 | 9.9 | > 20 |
| 2m |
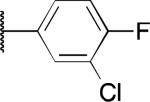
|
H | 1.8 | > 50 | > 100 |
| 2n |

|
H | 1.1 | 3.3 | 52 |
| 2o |

|
H | 0.82 | 4.9 | 12 |
| 2p |
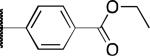
|
H | 1.2 | 23 | > 50 |
| 2q |

|
H | 0.14 | > 50 | > 50 |
| 2r |
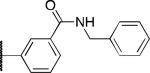
|
H | 2.3 | 2.3 | 18 |
See http://pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?aid=1666 for details of assay protocol.
See Dahl et al., 20118a for assay protocol.
Analogues of the benzoisothiazolone hits were synthesized by a method originally reported by Correa et al.10 that utilized a key cyclization step using phenyliodine bis(trifluoroacetate) (PIFA) to generate a N-acylnitrenium ion followed by intramolecular trapping by sulfur (Scheme 1a). The synthesis of the analogues was carried out in a semi-convergent manner. The required aniline precursors were either purchased or, in the case of the amides, prepared via EDCI mediated coupling of amines with the appropriate benzoic acid derivatives (Scheme 1b).11 This product was then coupled with methyl 2-mercaptobenzoate and subjected to the cyclization conditions (Scheme 1a). This synthetic methodology allowed for preparation of the benzoisothiazolone derivatives shown in Tables 1 and 2.12
Scheme 1.

General synthetic sequences for the synthesis of small molecule PHOSPHO1 inhibitors. aPIFA phenyliodine bis(trifluoroacetate).
Table 2.
Data for analogues with a sulfonamide substituent at the 3-position of the phenyl ring.
| Cmpd. | Ar | PHOSPHO1a | PMIb IC50 [μM] | PMM2b |
|---|---|---|---|---|
| 2s |

|
0.50 | 2.8 | 13 |
| 2t |

|
0.56 | > 50 | > 100 |
| 2u |
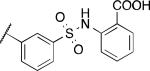
|
0.81 | 0.71 | 5.2 |
| 2v |
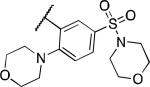
|
1.2 | 2.6 | 9.9 |
| 2w |
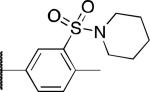
|
1.8 | 3.1 | 11 |
| 2x |
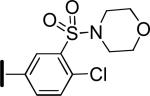
|
1.1 | 8.6 | 63 |
| 2y |
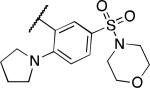
|
7.5 | 7.5 | 30 |
See http://pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?aid=1666 for details of assay protocol.
See Dahl et al., 20118a for assay protocol.
The potency and selectivity of the benzoisothiazolone analogues were assessed by in vitro enzymatic assays using purified human PHOSPHO1, PMI, or PMM213 to establish a preliminary SAR. As noted previously, the hit compound 2a inhibited PHOSPHO1 with good potency but also significantly inhibits PMI (Table 1). Fluoro substitution (R1 = F) of the benzoisothiazolone moiety, as in 2b, provides a marginal improvement in potency at PHOSPHO1 but also increased potency at PMI (IC50 = 1.3 μM). Monomethyl substitution of the phenyl ring, as in 2c and 2d, lessened potency at PHOSPHO1 while retaining the unwanted activity at PMI. Substitution at the 4-position of the phenyl ring with methoxy (2e), fluoro (2f, 2g), or NMe2 (2h) effectively worsened potency against PHOSPHO1 while in general increasing potency at PMI. Substitution at the 3-position with chloro, as in 2i, provided a compound with similar potency at both PHOSPHO1 and PMI. Interestingly, none of the initial set of compounds 2a-2i had significant activity at PMM2. The 2,5- or 2,3-dimethyl substitution patterns (2j and 2l, respectively) gave compounds that were similarly potent at both PHOSPHO1 and PMI. Introduction of R1= fluoro, as in 2k, unfortunately enhanced potency at both PMI and PMM2. In contrast to 2i, the 3-chloro-4-fluorophenyl derivative 2m was potent at PHOSPHO1 but inactive at both PMI and PMM2. Carboxylic acid substitution at the 3-position (2n) provided a potent PHOSPHO1 inhibitor with micromolar activity at PMI and no activity at PMM2. The methyl ester derivative 2o exhibited sub-micromolar potency at PHOSPHO1 but also micromolar activity at both PMI and PMM2, whereas the ethyl ester derivative 2p was essentially devoid of activity at PMI and PMM2. The breakthrough came, however, with the dimethyl amide derivative 2q, exhibiting an IC50 value of 140 nM at PHOSPHO1 and no activity at PMI or PMM2. Interestingly, the corresponding benzylamide derivative 2r, while potent at PHOSPHO1 also showed activity at PMI and PMM2.
Based on the promising data for the first set of compounds, and in particular 2q, we next tested a series of analogues containing a sulfonamide moiety at the 3-position of the phenyl ring. The results of these efforts are shown in Table 2. Several analogues in this series exhibited good potency as PHOSPHO1 inhibitors, with the dimethyl (2s) and diethyl (2t) analogues being especially potent. Interestingly, while compound 2s was active at PMI and PMM2 at micromolar levels, the diethyl sulfonamide 2t was devoid of activity at these phosphatases. The anthranilic acid sulfonamide 2u exhibited submicromolar potency at both PHOSPHO1 and PMI. Sulfonamide derivatives 2v-2y were less potent at PHOSPHO1 and all had some level of activity at PMI and PMM2.
Select PHOSPHO1 inhibitors (2n, 2o, 2q, 2s) were comprehensively profiled in in vitro absorption, distribution, metabolism and excretion (ADME) assays (Table 3).14 The data in Table 3 provide insight into the drug-likeness and potential for systemic activity of compounds, thus enabling advanced testing and future target validation efforts. The selected compounds were shown to have properties indicative of the potential for oral availability including acceptable metabolic stability, good permeability across artificial lipid membranes, and good solubility. No significant cell toxicity could be detected for any of the analogues. With these data indicative of drug-like behavior, good potency and selectivity, this series may be suitable for in vivo proof-of-concept studies. Of note is the esterification of the carboxylic group on 2o, which essentially forms a pro-drug prone to facile metabolic cleavage and subsequent formation of 2n. Since 2o exhibits improved permeability parameters compared with 2n, the data suggest that the development of a series of pro-drug analogues may be a viable approach to develop compounds with in vivo activity.
Table 3.
Characterization of a selection of synthesized analogues for drug-like properties in a range of in vitro ADME assays. For details of assay protocols see Khan et al., 2011 14
| Cmpd. | Structure | Aqueous Solubility in pION's buffer [μg/ml] | PAMPA Permeability [×10−6 cm/s] Acceptor pH 7.4 | Plasma Protein Binding [% Bound] | Plasma Stability [% Remaining at 3hrs] Plasma: 1× PBS, pH 7.4, 1:1 1×PBS, pH 7.4 | Hepatic Microsome Stability [% Remaining at 1hr] (NADPH minus) | Toxicity towards Fa2N-4 Immortalized Human Hepatocytes LC50 [μM] | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PH 5.0 | PH 6.2 | PH 7.4 | PH 5.0 | PH 6.2 | PH 7.4 | Human 1 μM / 10 μM | Mouse 1 μM / 10 μM | Human | Mouse | Human | Mouse | |||
| 2n |

|
> 27 | > 27 | > 27 | 157 | 18 | <4.4 | n.d. / 59.4 | 18.8 /30.1 | 50.2 55.5 |
100 71.2 |
100 | 100 | > 50 |
| 2o |
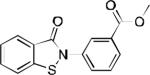
|
12.6 | 18.9 | 20.8 | 1294 | 1231 | 1300 | 41.2 / 57.3 | 48.0 / 37.6 | 15.4 49.3 |
59.6 55.2 |
72.4 31.5 |
45.9 44.6 |
> 50 |
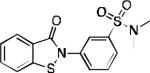
| 2q |
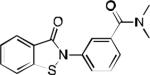
|
> 30 | > 30 | > 30 | 94 | 92 | 97 | 41.3 /44.2 | 48.0 / 46.7 | 18.9 60.0 |
76.2 56.9 |
45.7 | 56.6 | > 50 |
| 2s |
|
23.4 | 24.6 | 23.8 | 781 | 817 | 777 | n.d. / 56.5 | 35.6 / 34.1 | 11.4 32.3 |
65.3 40.0 |
72.4 | 100 | > 50 |
Compound 2q was additionally tested for selectivity against the enzymes TNAP and ectonucleotide pyrophosphatase/phosphodiesterase-1 (NPP-1) (Table 4). Gratifyingly, 2q was found to be devoid of any significant inhibitory activity against these counter targets. With respect to testing in other screens through the MLPCN program, 2q (CID16749996) was found to have weak activity in a screen for inhibitors of the mevalonate pathway in streptococcus pneumonia (AID1028). As shown in in Table 4, 2q was at least 170-fold selective for PHOSPHO1 versus counter targets.
Table 4.
Activity of 2q (MLPCN Probe ML-086) on the target (PHOSPHO1) compared with counter-target enzymes and a pathway screen.
| PHOSPHO1 | TNAP | NPP-1 | PMI | PMM2 | AID1028a | |
|---|---|---|---|---|---|---|
| IC50 [μM] | 0.14 | > 100 | > 30 | 62 | 76 | 24 |
| Selectivity | - | > 719 | > 215 | 442 | 549 | 176 |
In conclusion, we have identified and developed a series of PHOSPHO1 inhibitors with sub-micromolar potencies and promising drug-like properties. Medicinal chemistry efforts were applied to the optimization of a benzoisothialozone core scaffold initially identified through HTS. A carefully guided SAR study yielded a series of highly active inhibitors with proven ability to inhibit PHOSPHO1 in ex vivo models.15 On the basis of its overall profile compound 2q was selected as MLPCN probe ML086. This work provides an example of a successful strategy using medicinal chemistry to develop a useful biological probe. Furthermore, the drug-like properties of the resulting compounds provide an opportunity to lay the foundation for the development of therapeutic agents suitable for the treatment of diseases caused by MVC.
Acknowledgments
This work was supported by NIH grants HG005033 (NIGMS/NIMH) and an American Recovery and Reinvestment Act (ARRA) Challenge grant RC1HL10899 from the National Heart, Lung and Blood Institute (NHLB).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.a Houston B, Seawright E, Jefferies D, Hoogland E, Lester D, Whitehead C, Farquharson C. Identification and cloning of a novel phosphatase expressed at high levels in differentiating growth plate chondrocytes. Biochim Biophys Acta. 1999;1448(3):500–6. doi: 10.1016/s0167-4889(98)00153-0. [DOI] [PubMed] [Google Scholar]; b Houston B, Stewart AJ, Farquharson C. PHOSPHO1-A novel phosphatase specifically expressed at sites of mineralisation in bone and cartilage. Bone. 2004;34(4):629–37. doi: 10.1016/j.bone.2003.12.023. [DOI] [PubMed] [Google Scholar]; c Roberts SJ, Stewart AJ, Sadler PJ, Farquharson C. Human PHOSPHO1 exhibits high specific phosphoethanolamine and phosphocholine phosphatase activities. Biochem J. 2004;382(Pt 1):59–65. doi: 10.1042/BJ20040511. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Stewart AJ, Roberts SJ, Seawright E, Davey MG, Fleming RH, Farquharson C. The presence of PHOSPHO1 in matrix vesicles and its developmental expression prior to skeletal mineralization. Bone. 2006;39(5):1000–7. doi: 10.1016/j.bone.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 2.Roberts S, Narisawa S, Harmey D, Millan JL, Farquharson C. Functional involvement of PHOSPHO1 in matrix vesicle-mediated skeletal mineralization. J Bone Miner Res. 2007;22(4):617–27. doi: 10.1359/jbmr.070108. [DOI] [PubMed] [Google Scholar]
- 3.McKusick VA. Mendelian inheritance in man and its online version, OMIM. Am J Hum Genet. 2007;80(4):588–604. doi: 10.1086/514346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a Terkeltaub RA. Inorganic pyrophosphate generation and disposition in pathophysiology. Am J Physiol Cell Physiol. 2001;281(1):C1–C11. doi: 10.1152/ajpcell.2001.281.1.C1. [DOI] [PubMed] [Google Scholar]; b Rutsch F, Ruf N, Vaingankar S, Toliat MR, Suk A, Hohne W, Schauer G, Lehmann M, Roscioli T, Schnabel D, Epplen JT, Knisely A, Superti-Furga A, McGill J, Filippone M, Sinaiko AR, Vallance H, Hinrichs B, Smith W, Ferre M, Terkeltaub R, Nurnberg P. Mutations in ENPP1 are associated with ‘idiopathic’ infantile arterial calcification. Nat Genet. 2003;34(4):379–81. doi: 10.1038/ng1221. [DOI] [PubMed] [Google Scholar]; c Rutsch F, Vaingankar S, Johnson K, Goldfine I, Maddux B, Schauerte P, Kalhoff H, Sano K, Boisvert WA, Superti-Furga A, Terkeltaub R. PC-1 nucleoside triphosphate pyrophosphohydrolase deficiency in idiopathic infantile arterial calcification. Am J Pathol. 2001;158(2):543–54. doi: 10.1016/S0002-9440(10)63996-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsui HW, Inman RD, Paterson AD, Reveille JD, Tsui FW. ANKH variants associated with ankylosing spondylitis: gender differences. Arthritis Res Ther. 2005;7(3):R513–25. doi: 10.1186/ar1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a Whyte MP. Physiological role of alkaline phosphatase explored in hypophosphatasia. Ann N Y Acad Sci. 2010;1192:190–200. doi: 10.1111/j.1749-6632.2010.05387.x. [DOI] [PubMed] [Google Scholar]; b Whyte MP, Greenberg CR, Salman NJ, Bober MB, McAlister WH, Wenkert D, Van Sickle BJ, Simmons JH, Edgar TS, Bauer ML, Hamdan MA, Bishop N, Lutz RE, McGinn M, Craig S, Moore JN, Taylor JW, Cleveland RH, Cranley WR, Lim R, Thacher TD, Mayhew JE, Downs M, Millan JL, Skrinar AM, Crine P, Landy H. Enzyme-replacement therapy in life-threatening hypophosphatasia. N Engl J Med. 2012;366(10):904–13. doi: 10.1056/NEJMoa1106173. [DOI] [PubMed] [Google Scholar]
- 7.a Bobryshev YV, Orekhov AN, Sobenin I, Chistiakov DA. Role of Bone-Type Tissue-Nonspecific Alkaline Phosphatase and PHOSPO1 in Vascular Calcification. Current pharmaceutical design. 2014 doi: 10.2174/1381612820666140212193011. [DOI] [PubMed] [Google Scholar]; b Kiffer-Moreira T, Narisawa S. The use of tissue-nonspecific alkaline phosphatase (TNAP) and PHOSPHO1 inhibitors to affect mineralization by cultured cells. Methods in molecular biology (Clifton, NJ) 2013;1053:125–134. doi: 10.1007/978-1-62703-562-0_8. [DOI] [PubMed] [Google Scholar]
- 8.a Dahl R, Sergienko EA, Su Y, Mostofi YS, Yang L, Simao AM, Narisawa S, Brown B, Mangravita-Novo A, Vicchiarelli M, Smith LH, O'Neill WC, Millan JL, Cosford ND. Discovery and validation of a series of aryl sulfonamides as selective inhibitors of tissue-nonspecific alkaline phosphatase (TNAP). J Med Chem. 2009;52(21):6919–25. doi: 10.1021/jm900383s. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Sidique S, Ardecky R, Su Y, Narisawa S, Brown B, Millan JL, Sergienko E, Cosford NDP. Design and synthesis of pyrazole derivatives as potent and selective inhibitors of tissue-nonspecific alkaline phosphatase (TNAP). Bioorg Med Chem Lett. 2009;19(1):222–225. doi: 10.1016/j.bmcl.2008.10.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sharma V, Ichikawa M, He P, Scott DA, Bravo Y, Dahl R, Ng BG, Cosford ND, Freeze HH. Phosphomannose isomerase inhibitors improve N-glycosylation in selected phosphomannomutase-deficient fibroblasts. J Biol Chem. 2011;286(45):39431–8. doi: 10.1074/jbc.M111.285502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Correa A, Tellitu I, Dominguez E, SanMartin R. Novel alternative for the N-S bond formation and its application to the synthesis of benzisothiazol-3-ones. Org Lett. 2006;8(21):4811–3. doi: 10.1021/ol061867q. [DOI] [PubMed] [Google Scholar]
- 11.a Chemler SR, Trauner D, Danishefsky SJ. The B-Alkyl Suzuki-Miyaura Cross-Coupling Reaction: Development, Mechanistic Study, and Applications in Natural Product Synthesis A list of abbreviations can be found at the end of the article. Angew Chem Int Ed Engl. 2001;40(24):4544–4568. doi: 10.1002/1521-3773(20011217)40:24<4544::aid-anie4544>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]; b Kotha S, Lahiri K. Application of the Suzuki-Miyaura cross-coupling reaction for the modification of phenylalanine peptides. Biopolymers. 2003;69(4):517–28. doi: 10.1002/bip.10420. [DOI] [PubMed] [Google Scholar]
- 12.General synthesis of PHOSPHO1 inhibitors: To a stirred solution of benzoic acid derivative 5 (300mg, 2.18 mmol) in CH2Cl2 was added an amine (266 mg, 3.27 mmol) followed by the coupling agents; HOBT (442 mg, 3.27 mmol) and EDCI (630 mg, 3.27 mmol) and then Hunig's base (1.14 mL, 6.54 mmol). The reaction was stirred at room temperature over night, then quenched with saturated NaHCO3 solution and CH2Cl2, then dried over Na2SO4. The solvents were removed by rotary evaporation, no further purification was needed. To a stirred solution of the aniline 6 (260 mg, 1.58 mmol) in CH2Cl2 at 0 °C under nitrogen was added AlMe3 (2 mL, 1 M in THF) dropwise and the reaction was slowly warmed to room temperature. The mixture was stirred continuously for an additional 30 min. Methyl thiosalicylate (130 µL, 0.790 mmol) was added and the reaction was heated to 60 °C and then heated under reflux overnight. The reaction was quenched with HCl (5% aq.) and CH2Cl2 was added (50 mL). The organic layer was separated and washed with saturated NaHCO3 solution, then brine, and dried over Na2SO4. The solvents were removed by rotary evaporation. The products were purified by flash chromatography or reverse phase HPLC and lyophilized to provide thiols 4 which were determined to be > 95% pure by HPLC-UV, HPLC-MS, and 1H NMR. A solution of PIFA in CH2Cl2 was added to 0 °C solution of the benzamide (250 mg, 0.833 mmol) and TFA (0.185 mL, 16 M) in CH2Cl2. The reaction mixture was stirred at room temperature overnight. The solvents were removed in vacuo and the product isolated by flash chromatography or reverse phase HPLC and lyophilized to provide the final compounds (2) which were determined to be > 95% pure by HPLC-UV, HPLC-MS, and 1H NMR.
- 13.a Chung TD, Sergienko E, Millan JL. Assay format as a critical success factor for identification of novel inhibitor chemotypes of tissue-nonspecific alkaline phosphatase from high-throughput screening. Molecules. 2010;15(5):3010–37. doi: 10.3390/molecules15053010. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Dahl R, Bravo Y, Sharma V, Ichikawa M, Dhanya RP, Hedrick M, Brown B, Rascon J, Vicchiarelli M, Mangravita-Novo A, Yang L, Stonich D, Su Y, Smith LH, Sergienko E, Freeze HH, Cosford ND. Potent, selective, and orally available benzoisothiazolone phosphomannose isomerase inhibitors as probes for congenital disorder of glycosylation ia. J Med Chem. 2011;54(10):3661–8. doi: 10.1021/jm101401a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khan PM, Correa RG, Divlianska DB, Peddibhotla S, Sessions EH, Magnuson G, Brown B, Suyama E, Yuan H, Mangravita-Novo A, Vicchiarelli M, Su Y, Vasile S, Smith LH, Diaz PW, Reed JC, Roth GP. Identification of Inhibitors of NOD1-Induced Nuclear Factor-kappaB Activation. Acs Med Chem Lett. 2011;2(10):780–785. doi: 10.1021/ml200158b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kiffer-Moreira T, Yadav MC, Zhu D, Narisawa S, Sheen C, Stec B, Cosford ND, Dahl R, Farquharson C, Hoylaerts MF, Macrae VE, Millan JL. Pharmacological inhibition of PHOSPHO1 suppresses vascular smooth muscle cell calcification. J Bone Miner Res. 2013;28(1):81–91. doi: 10.1002/jbmr.1733. [DOI] [PMC free article] [PubMed] [Google Scholar]