

Abstract
Stat3 activation has been implicated as an important driver of brain metastasis in breast cancer, but the critical targets of Stat3 in this process are yet to be fully defined. In this study, we identified the lipid raft organizing protein Caveolin-1 (Cav-1) as a critical genetic target of Stat3 in this process. In human breast cancers, we found that activated Stat3 correlated with attenuation of Cav-1 in brain metastases relative to primary tumors. Cav-1 promoter activity and gene expression was increased by overexpressing an activated form of Stat3, but decreased by attenuation of Stat3 activity or expression. We identified putative Stat3-binding elements in the Cav-1 promoter and demonstrated a direct repression of Cav-1 transcription by Stat3. Reciprocally, we demonstrated that strategies to increase or decrease Cav-1 expression were sufficient to attenuate or promote breast cancer cell invasion. Further, increased expression of Cav-1 phenocopied the effects of Stat3 activation in blocking primary tumor growth and abrogating formation of brain metastases. Collectively, our findings provide clinical and mechanistic evidence that Cav-1 is a critical target for suppression by Stat3 in driving invasion and metastasis of breast cancer cells.
Keywords: Stat3, caveolin-1, SOCS-1, brain metastases, breast cancer, invasion
Introduction
Breast cancer is the most common cancer in women. The 5-year relative survival rate in patients with local breast cancer is 98%. However, the rate is only about 27% in patients with distant metastases (1). Breast cancer frequently metastasizes to the brain. Specifically, breast cancer is responsible for 16% of all cases of brain metastases (2). Furthermore, clinically overt brain metastases occur in 10–15% of patients with breast cancer, and 20% of patients have brain metastases at autopsy (3, 4). When metastases are present in the brain, the prognosis for breast cancer is consistently poor, with median survival durations less than 1 year in most cases (2, 3). Despite advances in understanding the causes of and treating primary breast cancer, the biological and molecular mechanisms of brain metastases of this malignancy remain understudied.
The oncoprotein signal transducer and activator of transcription 3 (Stat3) is constitutively activated in cancer cells in various types of human cancers (5–12). The Stat3 signaling pathway may impact tumor metastases via regulation of several steps in this process (8, 11, 13). Stat3 is activated by many cytokines and growth factors, including epidermal growth factor and interleukin-6, as well as by oncogenic proteins, such as Src and Ras (14). Conversely, Stat3 activation is negatively regulated by several proteins, including suppressor of cytokine signaling (SOCS)-1 (15, 16). Using our established brain metastases models, we recently described the role and positive regulation of Stat3 activation in melanoma brain metastases (11, 17). However, the underlying mechanisms by which Stat3 promote brain metastases remain unclear.
Caveolin-1 is the principal structural component of caveolar membrane domains in non-muscle cells, including mammary epithelial cells (18, 19). Caveolae are multifunctional organelles in which caveolin-1 plays direct roles in various events, such as membrane trafficking and various cellular signal transductions (20). Interestingly, researchers have observed reduced caveolin-1 expression levels in cases of a number of human cancers (21, 22), suggesting a negative regulatory role for caveolin-1 in tumor development. Although caveolin-1 appears to have both tumor-suppressive and oncogenic activity in different types of cancer, recent studies suggested that caveolin-1 inhibits breast cancer cell migration and metastases (21–26). Specifically, in caveolin-1-null mice carrying the MMTV-PyMT transgene, multifocal dysplastic lesions developed throughout the entire mammary tree, and mammary tumorigenesis and lung metastases were enhanced (27, 28). Furthermore, caveolin-1 expression levels are significantly lower in human breast cancer cells than in their normal mammary epithelial counterparts (24). All of this experimental and clinical evidence supports that caveolin-1 is a tumor suppressor in patients with breast cancer.
These findings underscore the importance of determining the roles and molecular mechanisms by which activated Stat3 and caveolin-1 regulates brain metastases. Therefore, we sought to determine the effect of activated Stat3 on caveolin-1 expression, breast cancer invasion and brain metastases.
Materials and Methods
Cell lines and culture conditions
The NMuMG, MDA-MB-231, BT-474, MCF7 and T-47D and MCF7 cells were obtained from the ATCC. The brain-metastatic variant MDA-MB-231-BR3 (referred to herein as 231-BR), BT-474-BR, and A375Br were generated from their parental cell lines (11, 29). All of the cell lines including the brain-metastatic variants were authenticated by the Authorization for Characterized Cell Line Core of MD. Anderson Cancer Center using the AmpF/STR Identifiler Kit (Applied Biosystems) and the ATCC fingerprint database. The cell lines were cultured in Eagle's minimal essential medium supplemented with 10% FBS, and were not passaged for more than 6 months before bringing new cells out of freeze or purchasing a new cell aliquot from the ATCC. The last cell authentication was tested at February of 2011.
Western blot analysis
Western blotting was performed using whole-cell protein lysates; primary antibodies against phosphorylated Stat3 (pStat3; Tyr 705), Stat3, phosphorylated extracellular signal-regulated kinase1/2 (pERK1/2) or ERK1/2 (Cell Signaling Technology), SOCS-1 (Zymed Laboratories, Inc.), or caveolin-1 (BD Biosciences); and a secondary antibody (anti-rabbit IgG or anti-mouse IgG; Jackson Laboratories). Equal protein-sample loading was monitored using an anti-β-actin antibody (Sigma-Aldrich).
Plasmids and small interfering RNAs
The plasmids pcDNA3.1-SOCS-1, pcDNA3.1-caveolin-1, Stat3C (a constitutively activated mutant of Stat3), Stat3DN (a dominant-negative mutant of Stat3), and control vectors were described previously (14, 16, 17, 30, 31). Small interfering RNA (siRNA) sequences targeting SOCS-1, Stat3, and caveolin-1 were as follows: SOCS-1, 5'-GCAUCCGCGUGCACUUUCAUUtt-3' (32); Stat3, 5'-AACAUCUGCCUAGAUCGGCUAtt-3' (33); and caveolin-1, 5'-AGACGAGCUGAGCGAGAAGCAtt-3' (34).
Transient and stable transfection
Transfection of plasmids and siRNAs into breast cancer cells was performed using Lipofectamine 2000 (Invitrogen). For transient transfection, cells were transfected with siRNA or plasmids at different doses as indicated for 48 h before functional assays were carried out. Stably transfected cell lines were isolated from 231-BR-transfeced with pcDNA3.1-SOCS-1 plasmid via selection with hygromycin (150 µg/mL).
Pharmacologic agents
JSI-124 was purchased from Indofine Chemicals, Inc. (Hillsborough, NJ). STA-21 were obtained from Biomol (Plymouth Meeting, PA). All agents were dissolved in dimethylsulfoxide (DMSO). During the experiment, they were thawed and diluted with culture medium to the appropriate final concentrations.
Promoter reporter, site-specific mutagenesis and dual luciferase assay
The caveolin-1 promoter luciferase reporter pA3Luc-caveolin-1 (pLuc-Cav) was described previously (31). Mutated pLuc-Cav was generated by site-specific mutagenesis using the QuikChange site-directed mutagenesis kit (Stratagene). Cells were transfected with the indicated wild-type or mutated pLuc-Cav, siRNAs or gene-specific expression plasmids. The transfection efficiency was normalized via co-transfection with a β-actin-RL reporter (35). Luciferase activity in the cells was quantified using a dual luciferase assay system (Promega Corp.).
Electrophoretic mobility shift assay
Electrophoretic mobility shift assay (EMSA) was performed as described previously (35). Double-stranded oligonucleotides of the putative Stat3-binding sites in the caveolin-1 promoter were used as probes. For supershift analysis of EMSA, extracts of tumor cells were preincubated with anti-Stat3 antibodies (Cell Signaling Technology).
Chromatin immunoprecipitation assay
Tumor cells (2×106) were prepared for chromatin immunoprecipitation (ChIP) assay with the ChIP assay kit (Cell Signaling Technology). The resulting precipitated DNA samples were analyzed using PCR to amplify a 212-bp region of the caveolin-1 promoter with the primers 5'-TGGCATAACCTGTTGGCATA-3' (sense) and 5'-GGGTGCCTGTGGTGTACTTT-3' (antisense) (36).
Invasion assay
An invasion assay using growth factor-reduced matrigel coated Invasion Chambers (BD Biosciences) was performed as described previously (11). FBS (10%) containing medium was placed in the lower chambers to act as a chemoattractant. The cells that penetrated through filter were counted at a magnification of x200 in 15 randomly selected fields, and the mean number of cells per field was recorded.
Human tissue samples and immunohistochemical analysis
Human breast ductal carcinoma in situ (DCIS), breast invasive ductal carcinoma (IDC) and breast cancer brain metastases tissue samples were coded without any patient identifiers. The use of the tissue samples was approved by the Institutional Review Board. Immunohistochemical (IHC) was performed using paraffin-embedded sections of the human tissue samples with anti-caveolin-1 or anti-pStat3 antibodies. Staining was scored by two investigators blinded to the clinical data using a four-tier system that incorporated the percentage of cells positive and intensity for staining: negative, weak, moderate positive, and strongly positive.
MTT Assays
Breast cancer cells were seeded at a density of 3X103 cells/ well in 96-well plates. At the end of culture for each indicated time points, 20 µL of MTT (5 mg/mL) (Sigma-Aldrich) was added to each well, and plates were placed at 37°C for 4 hours. DMSO (100 µL) were added to each well to lyse the cells. Absorbance was measured at a wavelength of 570 nm.
Colony formation in soft-agar assay
Cells (5x103 cells/well) were mixed with 0.3% agar solution in MEM containing 10% FBS, and the solution was poured on top of a 0.60% agar layer containing MEM and 10% FBS in 12-well tissue culture plates. After 21 days in culture, colonies were then stained with 0.005% crystal violet and examined microscopically.
Animals
Pathogen-free female athymic BALB/c nude mice were purchased from the National Cancer Institute. The animals were maintained in facilities approved by the Association for Assessment and Accreditation of Laboratory Animal Care International in accordance with the current regulations and standards of the U.S. Department of Agriculture and Department of Health and Human Services.
Human tumor xenografts
Tumor cells (106) in 0.1 mL of Hank’s balanced salt solution were injected into the mammary fat pad of nude mice. The length and width of the resulting tumors were measured using calipers, and the mean tumor diameter was calculated using the equation (L+W)/2, in which L is the tumor length and W is the tumor width.
Brain metastases
Breast tumor cells (3×105) were slowly injected into the intracarotid artery of nude mice. The mice were killed 90 days after tumor-cell inoculation or when they appeared to be moribund or had clinical symptoms of brain metastases. Their brains were then removed, and 10 serial brain sections were cut every 300 µm. Mean number of large metastases and micro metastases was determined by counting the number of metastases in the 10 sections from one hemisphere of each brain as described previously (29, 37).
Statistics
The significance of the data from patient specimens was determined by χ2 test and Spearman rank correlation test. The significance of the in vitro data and in vivo data was determined by Student’s t test (two-tailed) and Mann-Whitney test (two-tailed), respectively. Two-sided P values less than 0.05 were considered statistically significant.
Results
Altered Stat3 signaling impacts the expression of caveolin-1
First, the expression of caveolin-1, pStat3 and Stat3 protein was determined in the normal mammary gland epithelial line NMuMG and various breast cancer cell lines. Substantially higher level of caveolin-1 but lower lever of pStat3 was founded in NMuMG line than in the breast cancer lines (Fig. 1A). Moreover, the decreased level of caveolin-1 protein in brain metastatic cell lines 231-BR and BT-474-BR was directly correlated with increased level of activated Stat3 (Fig. 1B). Transfection of a constitutively activated mutant of Stat3 (Stat3C) plasmid led to decreased caveolin-1 expression in MDA-MB-231 cells, whereas knockdown of Stat3 expression in 231-BR cells using Stat3 siRNA did the opposite at both levels of protein (Fig. 1C) and mRNA expression (Fig. S1). Altered caveolin-1 expression also affected the levels of MMP-9, a known down-stream target gene of caveolin-1 (Fig. 1D) (38). Treatment of 231-BR and BT474-BR cells with STA-21 (inhibiting DNA-binding activity of Stat3) (39) or JSI-124 (Jak2 inhibitor) (40) led to increased caveolin-1 protein expression (Fig. 1E). These data strongly suggested that activated Stat3 and caveolin-1 negatively regulate each other and that activated Stat3 represses caveolin-1 expression in brain-metastatic breast cancer cells.
Figure 1.

Stat3 signaling regulates the expression of caveolin-1. A, Caveolin-1, pStat3 and Stat3 protein expression lever in breast cancer cell lines. B, Stat3 and pStat3 protein expression in breast cancer cell lines and their metastatic variants. C, MDA-MB-231 cells were transfected with pcDNA3.1 vector (Control) or Stat3C, whereas 231-BR cells were transfected with siControl or siStat3 (100 nM) for 48 h. Stat3, caveolin-1, and β-actin protein expressions in the cells were analyzed using immunoblotting. D, 231-BR cells were transfected with pcDNA3.1 (Control; 3 or 6 µg) or pCav1, whereas MDA-MB-231 cells were transfected with 50 or 100 nM of siControl or siCav1 for 48 h. pStat3, caveolin-1, MMP-9 and β-actin protein expressions were analyzed using immunoblotting. E, 231-BR and BT-474-BR cells were pretreated with DMSO (vehicle), 10 or 20 µM STA-21, or 10 or 20 µM JSI-124 for 24 h. pStat3, Stat3, caveolin-1, and β-actin protein expressions were analyzed using immunoblotting.
To assess the mechanism of regulation of caveolin-1 by Stat3, we examined whether inhibition of Stat3 activation suppress caveolin-1 promoter by using the caveolin-1 promoter luciferase reporter pLuc-Cav with a dominant-negative Stat3 (Stat3DN) (41, 42) or the control vector pcDNA3.1 at increasing concentrations. Stat3DN upregulated the activities of pLuc-Cav in 231-BR and BT474-BR cells (Fig. 2A & 2B). Also, the Stat3 inhibitors STA-21 or JSI-124 increased the activities of pLuc-Cav in 231-BR cells (Figs. 2C and 2E) and BT474-BR cells (Figs. 2D & 2F). The inhibition of promoter activities was not due to cytotoxicity caused by the inhibitors (Fig. S2). These results suggest that Stat3 activation negatively regulates caveolin-1 transcription.
Figure 2.

Regulation of Cav-1 promoter activities. pLuc-Cav, pRL, pStat3DN, or pcDNA3.1 were transfected into 231-BR (A) or BT474-BR cells (B) for 48 h; 231-BR (C & E) and BT474-BR cells (D & F) were transfected with pLuc-Cav and pRL for 24 h, and the cells were then treated with DMSO (10 µM), 10 µM STA-21, or 10 µM JSI-124 for 24 h. The caveolin-1 promoter activity was measured and expressed as the fold change relative to that in the control group. Each column indicates the mean± standard error from results of three different experiments. *P < 0.05; **P < 0.01.
Stat3 binds directly to the caveolin-1 promoter
To determine whether caveolin-1 is a direct transcriptional target of Stat3, we analyzed the caveolin-1 promoter sequence using the Stat3-binding consensus sequences TT (N4) AA and TT (N5) AA (43). We identified seven putative Stat3-binding elements in the caveolin-1 promoter (Fig. 3A). To functionally characterize these elements, we initially performed EMSA competition experiments using a Stat3 consensus binding oligonucleotide as a probe. The competitive probes were seven oligonucleotides corresponding to the elements. We found that #2 and #6 probes significantly competed with the Stat3 consensus probe for binding to Stat3 (Fig. 3B).
Figure 3.

Direct binding of Stat3 to the caveolin-1 promoter and inhibitory regulation of caveolin-1 transcription. A, schematic structure of the caveolin-1 promoter with sequences and positions of putative Stat3-binding elements (#1 to #7). B, results of EMSA competition analysis performed with nuclear protein from both 231-BR and A375Br cells using a 32P-labeled Stat3 consensus probe. An unlabeled Stat3 consensus probe (Cold WT) and putative Stat3 binding elements in the caveolin-1 promoter (#1 to # 7) were added to these reactions. Of note is that #2 and #6 significantly competed with the Stat3 consensus probe for binding to Stat3 (*). C, results of EMSA analysis performed with nuclear protein from 231-BR cells. A 32P-labeled #2 Stat3-binding element probe was incubated without nuclear protein (Probe only) or with nuclear protein (Control) in the presence of an unlabeled consensus Stat3 probe (Cold WT) or anti-Stat3 antibody (Stat3 Ab). D, results of a ChIP assay performed with 231-BR, 231-BR-SOCS-1, A375Br, and A375Br-SOCS-1 cells using an anti-Stat3 antibody or normal IgG. E, Activities of wild-type or mutant caveolin-1 promoter in 231-BR and A375Br cells. Bottom panel, the sequence of Stat3-binding site #2 in both wild-type (WT) and mutant (Mut) form. The promoter activity (fold) was calculated relative to that in the wild-type group. Values are mean (± SD) result of two independent experiments. *P<0.05.
To confirm that Stat3 binds to the caveolin-1 promoter, we used the element #2 as a probe for EMSA. As shown in Fig. 3C, incubation of protein extracts from 231-BR cells with the #2 probe resulted in the formation of a major shifted band (DNA-protein complex; lane 2). Moreover, the shifted band was competed out by an unlabeled consensus Stat3 probe (lane 3) and super-shifted by an anti-Stat3 antibody (lane 4), indicating that the shifted band represented a Stat3 DNA-protein complex. These in vitro binding experiments indicated that Stat3-binding elements are present in the caveolin-1 promoter and that #2 sequence in the promoter is a high-affinity Stat3-binding element.
Next, we performed ChIP assays to provide direct evidence that Stat3 is recruited to the endogenous caveolin-1 promoter during transcription in vivo. We used two pairs of primers designed to amplify segments of caveolin-1 promoter encompassing the #2 or #6 elements for the ChIP assays with 231-BR and A375Br cells. Next, we generated 231-BR and A375Br cells with reduced Stat3 activity by transfecting these cells with the SOCS-1 expression vector, as a previous study showed that activation of Stat3 in brain-metastatic cancer cells was caused by decreased SOCS-1 expression and restoration of SOCS-1 expression resulting in inhibition of Stat3 activation (17). ChIP assays were also performed with these cells. We found that the region of the caveolin-1 promoter containing Stat3-binding site #2 bound to endogenous Stat3 protein in above cell lines (Fig. 3D). Moreover, both 231-BR and A375Br exhibited higher levels of binding of Stat3 to the region than did 231-BR-SOCS-1 and A375Br-SOCS-1 cells. However, we did not detect binding of Stat3 to the region of the caveolin-1 promoter containing Stat3-binding site #6 in any of the cell lines tested (data not shown). Collectively, these results showed that Stat3 bound specifically to the caveolin-1 promoter and that TTGGCATAA at positions -571 to -563 in the promoter is a high-affinity Stat3-binding element.
A Stat3-binding site is critical for deactivation of the caveolin-1 promoter in brain-metastatic cancer cells
To assess the functional role of the Stat3-binding site in caveolin-1 gene regulation, we performed site-directed mutagenesis of Stat3-binding site #2 in the caveolin-1 promoter with a base-pair change from TT to CG. We then measured the promoter activity of both wild-type and mutant forms of caveolin-1 promoters in 231-BR and A375Br cells. Mutation of Stat3-binding site #2 significantly increased the promoter activity of caveolin-1, suggesting that this Stat3-binding site acted as a negative regulator of caveolin-1 transcription in these brain-metastatic cancer cells (Fig. 3E).
Alteration of caveolin-1 expression affects breast cancer cell invasion
We examined the invasive ability of breast cancer cells and found that knockdown of Stat3 expression by Stat3 siRNA attenuated the invasiveness of 231-BR cells (Fig. 4A), whereas Stat3C overexpression promoted the invasiveness of MDA-MB-231 cells (Fig. 4C). We also genetically altered caveolin-1 expression in these two cell types and found that overexpression of caveolin-1 attenuated the invasiveness of 231-BR cells (Fig. 4B), whereas knockdown of expression of caveolin-1 promoted the invasiveness of MDA-MB-231 cells (Fig. 4D). To assess the inhibitory effect of caveolin-1 on Stat3-induced invasion, we co-transfected both Stat3C and caveolin-1 into MDA-MB-231 cells and found that overexpression of caveolin-1 attenuated the invasiveness of Stat3C-overexpressing MDA-MB-231 cells (Fig. 4E).
Figure 4.

Alteration of caveolin-1 expression affects breast cancer cell invasion. A, transfection of siStat3 or siControl in 231-BR cells for 48 h; B, transfection of pCav1 or control pcDNA3.1 in 231-BR cells for 48 h; C, transfection of Stat3C or control pcDNA3.1 in MDA-MB-231 cells for 48 h; D, transfection of siCav1 or siControl in MDA-MB-231 cells for 48 h. E, MDA-MB-231 cells were transfected with pcDNA3.1, Stat3C and pCav1 at the indicated amounts for 48 h. Then, the invasiveness of above cells (in A-E) after the transfection was determined using the invasion assay in 16 h. Representative photomicrographs of tumor cells that invaded through a Matrigel-coated filter were taken and shown. The invasive cells were counted in 15 random fields and expressed as the percentage of the control cells. Values are mean (± SD) results of two independent experiments (left panels). *P<0.001.
Inhibition of Stat3 suppressed the growth and brain metastases of breast cancer cells
Since invasion is a critical step for brain metastasis, we next determined the impact of inhibited Stat3 activation on breast tumor growth and brain metastases and its relationship with caveolin-1 expression. We used the stable SOCS-1-transfected 231-BR cells, because the cells have lower levels of binding of Stat3 to the caveolin-1 promoter than did 231-BR cells (Fig. 3D). SOCS-1 transfection significantly inhibited breast tumor growth (Figs. 5A) and completely abrogated brain metastases of human breast cancer (Figs. 5B) of 231-BR cells in mice. Furthermore, immunohistochemical studies showed that the expression of activated Stat3 decreased, whereas that of caveolin-1 increased in tumors formed by 231-BR-SOCS-1 cells (Fig. 5C). These data indicated that Stat3 inhibition by SOCS-1 suppressed breast cancer metastases at least in part via negative regulation of caveolin-1 expression.
Figure 5.

Inhibition of the growth and brain metastases of 231-BR cells by SOCS-1. 231-BR cells were stable transfected with pcDNA3.1 (Vector) or pSOCS-1 plasmids. To avoid clonal selection, we carried out three independent transfections of pSOCS-1 in the cells and pooled hygromycin-resistant colonies to establish stable transfectants, designated as 231-BR-SOCS-1 #1, #2, and #3. A, 231-BR, 231-BR-Vector, and 231-BR-SOCS-1 (SOCS-1 #1 and #2) cells (106/mouse) were injected into the mammary fat pad of nude mice (n=5). The resulting tumors were measured at the indicated time intervals. The data are the mean tumor diameters observed in five mice per group. B, the above tumor cells (3×105/mouse) also were injected into the intracarotid arteries of nude mice. The mice brains were harvested when they were killed, and metastatic tumor formation was assessed histologically (*P<0.01). Incidence indicates the number of mice with brain metastasis divided by the total number of mice injected with tumor cells. Micro, mean number of micro metastases. Large, mean number of large metastases. C, expression of pStat3 and caveolin-1 in tumors from experiments of (A) was analyzed using IHC staining. Representative photos of the IHC staining are shown. D, SOCS-1 upregulated the expression of caveolin-1. 231-BR or A375Br, vector, or SOCS-1 transfected cells were used for analysis of SOCS-1, pStat3, Stat3, caveolin-1, phosphorylated ERK1/2, ERK1/2, MMP-9 and β-actin protein expression by immunoblotting. E, MDA-MB-231 cells transfected with 100 nM siControl or siSOCS-1 were used for analyses of SOCS-1, pStat3, Stat3, caveolin-1, and β-actin protein expression using immunoblotting.
Inhibition of Stat3 by SOCS-1 upregulates the expression of caveolin-1
In order to provide direct evidence that SOCS-1 inhibited brain metastasis by caveolin-1 upregulation, we determined whether SOCS-1 upregulates caveolin-1 protein expression. We found that the expression level of pStat3, but not phospho-p44/42, was significantly decreased in the 231-BR-SOCS-1 cells (Fig. 5D). Moreover, 231-BR-SOCS-1 cells exhibited a higher level of caveolin-1 than did parental or vector-transfected cells (Fig. 5D). Similar results were found in SOCS-1-transfected A375Br cells (Fig. 5D). In contrast, knockdown of endogenous SOCS-1 expression increased the expression of activated Stat3 and decreased the expression of caveolin-1 protein in MDA-MB-231 cells (Fig. 5E).
Inhibition of Stat3 by SOCS-1 suppressed breast cancer cell invasion and growth
We next determined whether SOCS-1 expression would affect the cell invasion and whether the effect of SOCS-1 on cell invasion is via caveolin-1. First, we found that down-regulation of SOCS-1 by SOCS-1 siRNA enhanced the invasive ability of MDA-MB-231 cells (Fig. 6A and Fig. S3). Conversely, overexpression of SOCS-1 down-regulated the invasive ability of 231-BR cells (Fig. 6B). Overexpression of SOCS-1 also down-regulated the levels of MMP-9 (Fig. 5D), which is consistent with previous studies indicating that MMP-9 is one of the main mechanisms by which caveolin-1 operates in reducing invasion (28, 38). Furthermore, the inhibitory effect of SOCS-1 on cell invasion was rescued by caveolin-1 siRNA (Fig. S4 and Fig. 6C). These data indicated that SOCS-1 inhibition of cell invasion is largely dependent on caveolin-1.
Figure 6.

SOCS-1 inhibits the invasive and proliferation abilities of Stat3 and upregulates the expression of caveolin-1 in brain metastatic cells. A, MDA-MB-231 cells were transfected with siControl or siSOCS-1 (100nM) for 48 h. Then the invasive ability of the cells was determined by the invasion assay as described in Figure 4. *, P<0.01. B. The invasive ability of 231-BR, vector and SOCS-1 cells was determined by the invasion assay. *, P<0.01. C. 231-BR-SOCS-1 (#1, #2 and #3) cells were transfected with siControl or siCav-1 (100nM) for 48 h. Then the invasive ability of the cells was determined by the invasion assay. *, P<0.01. D. Cell viability of 231-BR vector and SOCS-1 cells was determined by MTT assay. Values are mean ± S.D. of triplicate samples of two experiments. *, p < 0.05. E. Colony-forming capacity of 231BR vector and 231-BR-SOCS-1 (#1, #2 and #3) cells. F-G. 231-BR-SOCS-1 (#1 and #2) cells were transfected with Stat3CA (0.4µg), pcDNA3.1 vector (0.4µg) or siRNAs (siControl or siCav-1, 100nM) on day 0 and day 3. Then the cell viability at the indicated time points was determined by MTT assay. *, p < 0.05; **, p<0.01.
Additionally, we investigated whether SOCS-1-transfected 231-BR cells also exhibit abnormalities in proliferation. We found the cell viability has no significant difference among 231-BR vector and 231-BR SOCS-1 cells within the five days in MTT assay (Fig. 6C). However, 231-BR SOCS-1 #1 and #2 cells which have relative higher levels SOCS-1 exhibited reduced proliferation in longer time MTT and colony formation in soft agar assays compared to 231-BR vector cell (Fig. 6D-6E). Furthermore, the inhibitory effect of SOCS-1 on the cell proliferation was rescued by Stat3C or caveolin-1 siRNA (Fig. 6F-6G, Fig. S3). Consistently, Stat3C transfection prevented the increase of Cav-1 expression in SOCS-1-transfected 231-BR cells (Fig. S4). These data further support that Stat3 critically regulates breast cancer metastases at least in part via negative regulation of caveolin-1 expression and function.
Increased expression of pStat3 correlates with decreased expression of caveolin-1 in human breast cancer brain metastases
To determine the relationship between Stat3 and caveolin-1 and the clinical significance of this relationship, we performed IHC staining for pStat3 and caveolin-1 in 50 human breast DCIS, 50 human breast IDC and 50 human breast cancer brain metastases samples. We analyzed the data of moderate positive (++) and strongly positive (+++) staining of pStat3 or caveolin-1 by χ2 test. The expression levels of pStat3 were significantly increasing in brain metastases as compared to DCIS or IDC (P<0.001), while the expression levels of caveolin-1 were significantly decreasing in brain metastases as compared to DCIS or IDC (P<0.001) (Table 1, Fig. S5). Moreover, this inverse correlation between pStat3 and caveolin-1 in these three types of samples was statistically significant as determined by Spearman rank correlation test (r = −0.941; P < 0.001) (Table 1). There data further support the critical role of pStat3 in caveolin-1 expression and brain metastasis in human breast cancer.
Table 1.
The expression levels of pStat3 and Caveolin-1 in human DCIS, IDC and Brain metastasis specimens
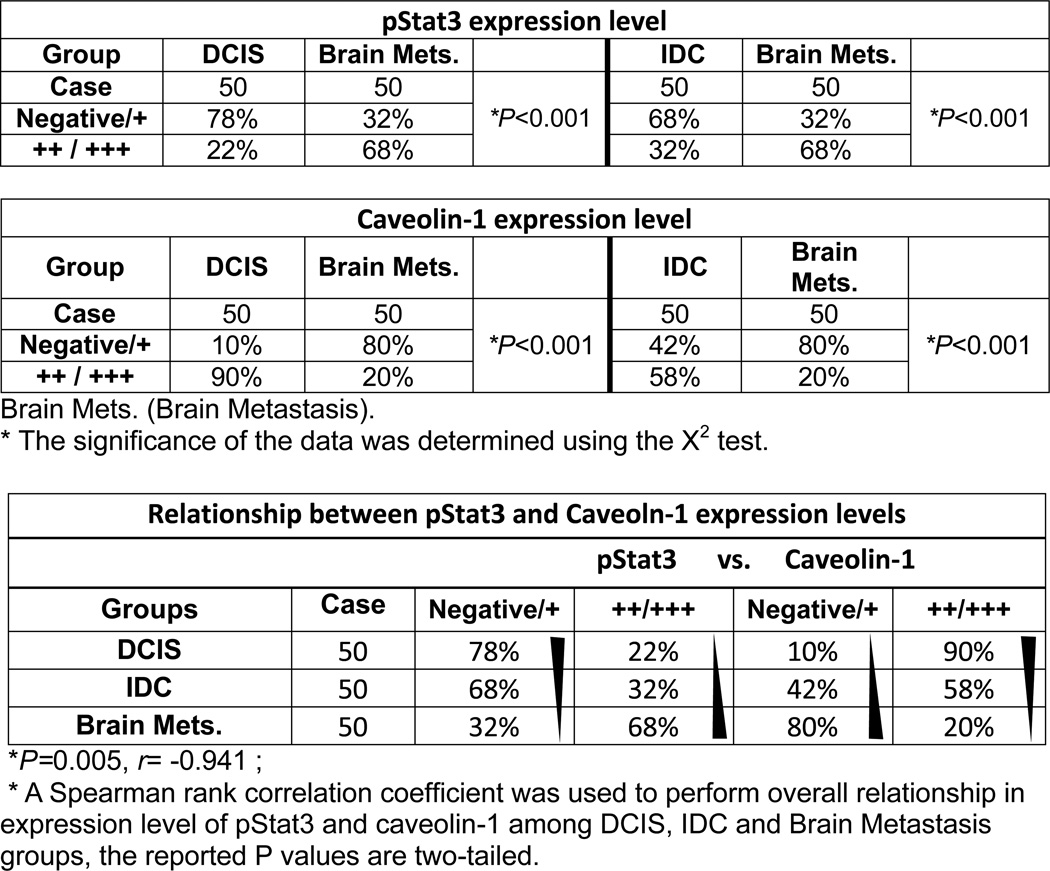 |
Discussion
In the present study, we sought to determine the role of interaction between Stat3 activation and caveolin-1 expression in breast cancer brain metastases. Our novel clinical and mechanistic evidence strongly suggested that activated Stat3 binds directly to the caveolin-1 promoter and inhibits its transcription. Conversely, caveolin-1 negatively regulates activation of Stat3 and invasion of brain-metastatic cancer cells. Moreover, suppression of Stat3 activation inhibited the invasion and brain metastases of breast cancer cells in our animal model.
Brain metastasis is one of the major causes of morbidity and mortality in patients with breast cancer. The underlying mechanisms of breast cancer brain metastases remain unclear; however, a full understanding of these mechanisms is essential to controlling brain metastases. In the present study, we found that activated Stat3 expression is higher in breast cancer brain metastases than in breast DCIS and IDC tumors. Consistently, several studies have linked activated Stat3 protein expression with metastases of various tumor types (12), and activated Stat3 promotes liver and lung metastases in mouse models (8, 44). Also, recent studies by our group indicated that alteration of Stat3 activation directly impacts melanoma brain metastases (11, 17). These previous findings are substantially extended by those of our present study showing that Stat3 regulates the expression and function of caveolin-1 and the invasive and metastatic properties of breast cancer. Mechanistically, activated Stat3 binds to the promoter of and inactivates the transcription of caveolin-1.
In the present study, we also observed that SOCS-1, a negative regulator of Stat3, enhances caveolin-1 expression in brain metastatic breast cancer cells and inhibits breast cancer invasion and metastases. Interestingly, authors have reported that the caveolin-1 scaffolding domain shares primary sequence similarities with the SOCS-1 pseudosubstrate domain (45); thus, caveolin-1 may also negatively regulate the activation of Stat3 via interactions with members of the Janus kinase family (46). Thus, activation of Stat3 in brain-metastatic cancer cells probably shut down the negative feedback of caveolin-1 by downregulating its expression. This may be one of the mechanisms responsible for sustained Stat3 activation in brain metastases.
Studies have demonstrated that caveolin-1 acts as a tumor suppressor protein in human breast cancer (17–28, 46-48). The mechanisms by which caveolin-1 exerts its inhibitory role in transformation, tumor growth, and metastases include cell proliferation and invasion. Caveolin-1 functions as an endogenous inhibitor in the p42/44 mitogen-activated protein kinase cascade and as a transcriptional repressor of cyclin D1 (46). Caveolin-1 also inhibits secretion and expression of MMP-9 and MMP-2 (28, 38). Consistently, in the present study, we found that elevation of caveolin-1 expression by SOCS-1 suppressed tumor growth in vitro and in nude mice and that increased caveolin-1 expression attenuated the invasiveness of breast cancer, whereas decreased caveolin-1 expression promoted the cell growth and invasiveness of breast cancer. Moreover, we observed that caveolin-1 overexpression inhibited Stat3 activation, an important pathway in breast tumor growth and metastases. Thus, caveolin-1 expression may affect brain metastases of breast cancer via multiple mechanisms.
Prior studies have suggested that activated Stat3 promotes tumor growth and metastases, presumably through its critical role in the expression of many genes key to regulation of multiple aspects of tumor-cell survival, growth, angiogenesis and evasion of immune surveillance (7, 49), such as cyclin D1, MMP-2, vascular endothelial growth factor, and 10-kDa interferon-γ–induced protein (7, 14, 49). In the present study, we obtained evidence that Stat3 activation transcriptionally represses caveolin-1 expression and promotes breast cancer invasion and brain metastases. Our results strongly suggest that Stat3 has an important role in brain metastases of breast cancer through the regulation of the expression of caveolin-1 and could be a potential therapeutic target for brain metastases of breast cancer. It would be of great interest to test the efficacy of Stat3 inhibitors on brain metastases of breast cancer.
Supplementary Material
Acknowledgments
Grant support: U.S. Department of Defense Breast Cancer Research Program Center of Excellence Award Grant W81XWH-06-2-0033, American Cancer Society Research Scholar Grant, and National Cancer Institute Cancer Center Support Grant CA 16672.
We thank Don Norwood for editorial comments.
References
- 1.Chang EL, Lo S. Diagnosis and management of central nervous system metastases from breast cancer. Oncologist. 2003;8:398–410. doi: 10.1634/theoncologist.8-5-398. [DOI] [PubMed] [Google Scholar]
- 2.Lagerwarrd FJ, Levendag PC, Nowak PJ, Eijkenboom WM, Hanssens PE, Schmitz PI. Identification of prognostic factors in patients with brain metastases: a review of 1292 patients. Int J Radiat Oncol Biol Phys. 1999;43:795–803. doi: 10.1016/s0360-3016(98)00442-8. [DOI] [PubMed] [Google Scholar]
- 3.Boogerd W. Central nervous system metastasis in breast cancer. Radiother Oncol Biol. 1996;40:5–22. doi: 10.1016/0167-8140(96)01766-5. [DOI] [PubMed] [Google Scholar]
- 4.Lin NU, Bellon JR, Winer EP. CNS metastases in breast cancer. J Clin Oncol. 2004;22:3608–3617. doi: 10.1200/JCO.2004.01.175. [DOI] [PubMed] [Google Scholar]
- 5.Kortylewski M, Jove R, Yu H. Targeting STAT3 affects melanoma on multiple fronts. Cancer Metastasis Rev. 2005;24:315–327. doi: 10.1007/s10555-005-1580-1. [DOI] [PubMed] [Google Scholar]
- 6.Kujawski M, Kortylewski M, Lee H, Herrmann A, Kay H, Yu H. Stat3 mediates myeloid cell-dependent tumor angiogenesis in mice. J Clin Invest. 2008;118:3367–3377. doi: 10.1172/JCI35213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bromberg JF. Stat proteins and oncogenesis. J Clin Invest. 2002;109:1139–1142. doi: 10.1172/JCI15617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xie TX, Wei D, Liu M, et al. Stat3 activation regulates the expression of matrix metalloproteinase-2 and tumor invasion and metastasis. Oncogene. 2004;23:3550–3560. doi: 10.1038/sj.onc.1207383. [DOI] [PubMed] [Google Scholar]
- 9.Ling X, Arlinghaus RB. Knockdown of STAT3 expression by RNA interference inhibits the induction of breast tumors in immunocompetent mice. Cancer Res. 2005;65:2532–2536. doi: 10.1158/0008-5472.CAN-04-2425. [DOI] [PubMed] [Google Scholar]
- 10.Chang KC, Wu MH, Jones D, Chen FF, Tseng YL. Activation of STAT3 in thymic epithelial tumours correlates with tumour type and clinical behavior. J Pathol. 2006;210:224–233. doi: 10.1002/path.2041. [DOI] [PubMed] [Google Scholar]
- 11.Xie TX, Huang FJ, Aldape KD, et al. Activation of stat3 in human melanoma promotes brain metastasis. Cancer Res. 2006;66:3188–3196. doi: 10.1158/0008-5472.CAN-05-2674. [DOI] [PubMed] [Google Scholar]
- 12.Yuan ZL, Guan YJ, Chatterjee D, Chin YE. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science. 2005;307:269–273. doi: 10.1126/science.1105166. [DOI] [PubMed] [Google Scholar]
- 13.Palmieri D, Chambers AF, Felding-Habermann B, Huang S, Steeg PS. The biology of metastasis to a sanctuary site. Clin Cancer Res. 2007;13:1656–1662. doi: 10.1158/1078-0432.CCR-06-2659. [DOI] [PubMed] [Google Scholar]
- 14.Bromberg JF, Wrzeszczynska MH, Devgan G, et al. Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 15.Endo TA, Masuhara M, Yokouchi M, et al. A new protein containing an SH2 domain that inhibits JAK kinases. Nature. 1997;387:921–924. doi: 10.1038/43213. [DOI] [PubMed] [Google Scholar]
- 16.Starr R, Willson TA, Viney EM, et al. A family of cytokine-inducible inhibitors of signaling. Nature. 1997;387:917–921. doi: 10.1038/43206. [DOI] [PubMed] [Google Scholar]
- 17.Huang FJ, Steeg PS, Price JE, et al. Molecular basis for the critical role of suppressor of cytokine signaling-1 in melanoma brain metastasis. Cancer Res. 2008;68:9634–9642. doi: 10.1158/0008-5472.CAN-08-1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Drab M, Verkade P, Elger M, et al. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 genedisrupted mice. Science. 2001;293:2449–2452. doi: 10.1126/science.1062688. [DOI] [PubMed] [Google Scholar]
- 19.Fra AM, Williamson E, Simons K, Parton RG. De novo formation of caveolae in lymphocytes by expression of VIP21-caveolin. Proc Natl Acad Sci U S A. 1995;92:8655–8659. doi: 10.1073/pnas.92.19.8655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krajewska WM, Maslowska I. Caveolins: structure and function in signal transduction. Cell Mol Biol Lett. 2004;9:195–220. [PubMed] [Google Scholar]
- 21.Koleske AJ, Baltimore D, Lisanti MP. Reduction of caveolin and caveolae in oncogenically transformed cells. Proc Natl Acad Sci U S A. 1995;92:1381–1385. doi: 10.1073/pnas.92.5.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Williams TM, Lisanti MP. Caveolin-1 in oncogenic transformation, cancer, and metastasis. Am J Physiol Cell Physiol. 2005;288:494–506. doi: 10.1152/ajpcell.00458.2004. [DOI] [PubMed] [Google Scholar]
- 23.Sloan EK, Stanley KL, Anderson RL. Caveolin-1 inhibits breast cancer growth and metastasis. Oncogene. 1998;23:7893–7897. doi: 10.1038/sj.onc.1208062. [DOI] [PubMed] [Google Scholar]
- 24.Lee SW, Reimer CL, Oh P, Campbell DB, Schnitzer JE. Tumor cell growth inhibition by caveolin re-expression in human breast cancer cells. Oncogene. 1998;16:1391–1397. doi: 10.1038/sj.onc.1201661. [DOI] [PubMed] [Google Scholar]
- 25.Fiucci G, Ravid D, Reich R, Liscovitch M. Caveolin-1 inhibits anchorage-independent growth, anoikis and invasiveness in MCF-7 human breast cancer cells. Oncogene. 2002;21:2365–2375. doi: 10.1038/sj.onc.1205300. [DOI] [PubMed] [Google Scholar]
- 26.Bouras T, Lisanti MP, Pestell RG. Caveolin-1 in breast cancer. Cancer Biol Ther. 2004;3:931–941. doi: 10.4161/cbt.3.10.1147. [DOI] [PubMed] [Google Scholar]
- 27.Williams TM, Cheung MW, Park DS, et al. Loss of caveolin-1 gene expression accelerates the development of dysplastic mammary lesions in tumor-prone transgenic mice. Mol Biol Cell. 2003;14:1027–1042. doi: 10.1091/mbc.E02-08-0503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Williams TM, Medina F, Badano I, et al. Caveolin-1 gene disruption promotes mammary tumorigenesis and dramatically enhances lung metastasis in vivo. Role of Cav-1 in cell invasiveness and matrix metalloproteinase (MMP-2/9) secretion. J Biol Chem. 2004;279:51630–61646. doi: 10.1074/jbc.M409214200. [DOI] [PubMed] [Google Scholar]
- 29.Kim LS, Huang S, Lu W, Lev DC, Price JE. Vascular endothelial growth factor expression promotes the growth of breast cancer brain metastasis in nude mice. Clin. Exp. Metastasis. 2004;21(2):107–118. doi: 10.1023/b:clin.0000024761.00373.55. [DOI] [PubMed] [Google Scholar]
- 30.Fujimoto T, Kogo H, Nomura R, Une T. Isoforms of caveolin-1 and caveolar structure. J Cell Sci. 2000;113:3509–3517. doi: 10.1242/jcs.113.19.3509. [DOI] [PubMed] [Google Scholar]
- 31.Engelman JA, Zhang XL, Razani B, Pestell RG, Lisanti MP. p42/44 MAP kinase-dependent and -independent signaling pathways regulate caveolin-1 gene expression. Activation of Ras-MAP kinase and protein kinase A signaling cascades transcriptionally down-regulates caveolin-1 promoter activity. J Biol Chem. 1999;274:32333–32341. doi: 10.1074/jbc.274.45.32333. [DOI] [PubMed] [Google Scholar]
- 32.Neuwirt H, Puhr M, Santer FR, et al. Suppressor of cytokine signaling (SOCS)-1 is expressed in human prostate cancer and exerts growth-inhibitory function through down-regulation of cyclins and cyclin-dependent kinases. Am J Pathol. 2009;174:1921–1930. doi: 10.2353/ajpath.2009.080751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Konnikova L, Kotecki M, Kruger MM, Cochran BH. Knockdown of STAT3 expression by RNAi induces apoptosis in astrocytoma cells. BMC Cancer. 2003;3:23–31. doi: 10.1186/1471-2407-3-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sunaga N, Miyajima K, Suzuki M, et al. Different roles for caveolin-1 in the development of non-small cell lung cancer versus small cell lung cancer. Cancer Res. 2004;64:4277–4285. doi: 10.1158/0008-5472.CAN-03-3941. [DOI] [PubMed] [Google Scholar]
- 35.Huang S, Jean D, Luca M, Tainsky M, Bar-Eli M. Loss of AP-2 results in downregulation of c-KIT and enhancement of melanoma tumorigenicity and metastasis. EMBO J. 1998;17:4358–4369. doi: 10.1093/emboj/17.15.4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abulrob A, Giuseppin S, Andrade MF, McDermid A, Moreno M, Stanimirovic D. Interactions of EGRF and caveolin-1 in human glioblastoma cells: evidence that tyrosine phosphorylation regulates EGFR association with caveolae. Oncogene. 2004;23:6967–6979. doi: 10.1038/sj.onc.1207911. [DOI] [PubMed] [Google Scholar]
- 37.Palmieri D, Bronder JL, Herring JM, et al. Her-2 overexpression increases the metastatic outgrowth of breast cancer cells in the brain. Cancer Res. 2007;67:4190–4198. doi: 10.1158/0008-5472.CAN-06-3316. [DOI] [PubMed] [Google Scholar]
- 38.Fei Han, Hong-Guang Zhu. Caveolin-1 Regulating the Invasion and Expression of Matrix Metalloproteinase (MMPs) in Pancreatic Carcinoma Cells. Journal of Surgical Research. 2010;159:443–450. doi: 10.1016/j.jss.2009.03.079. [DOI] [PubMed] [Google Scholar]
- 39.Song H, Wang R, Wang S, Lin J. A low-molecular-weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cell. Proc Natl Acad Sci U S A. 2005;102:4700–4705. doi: 10.1073/pnas.0409894102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blaskovich MA, Sun J, Cantor A, Turkson J, Jove R, Sebti SM. Discovery of JSI-124 (cucurbitacin I), a selective Janus kinase/signal transducer and activator of transcription 3 signaling pathway inhibitor with potent antitumor activity against human and murine cancer cells in mice. Cancer Res. 2003;63:1270–1279. [PubMed] [Google Scholar]
- 41.Kaptein A, Paillard V, Saunders M. Dominant negative Stat3 mutant inhibits interleukin-6-induced Jak-STAT signal transduction. J Biol Chem. 1996;271:5961–5964. doi: 10.1074/jbc.271.11.5961. [DOI] [PubMed] [Google Scholar]
- 42.Nakajima K, Yamanaja Y, Nakae K, et al. A central role for Stat3 in IL-6-induced regulation of growth and differentiation in M1 leukemia cells. EMBO J. 1996;15:3651–3658. [PMC free article] [PubMed] [Google Scholar]
- 43.Seidel HM, Milocco LH, Lamb P, Darnell JE, Jr, Stein RB, Rosen J. Spacing of palindromic half sites as a determinant of selective STAT (signal transducers and activators of transcription) DNA binding and transcriptional activity. Proc Natl Acad Sci U S A. 1995;92:3041–3045. doi: 10.1073/pnas.92.7.3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li WC, Ye SL, Sun RX, et al. Inhibition of growth and metastasis of human hepatocellular carcinoma by antisense oligonucleotide targeting signal transducer and activator of transcription 3. Clin Cancer Res. 2006;12:7140–7148. doi: 10.1158/1078-0432.CCR-06-0484. [DOI] [PubMed] [Google Scholar]
- 45.Jasmin JF, Mercier I, Sotgia F, Lisanti MP. SOCS proteins and caveolin-1 as negative regulators of endocrine signaling. Trends Endocrinol Metab. 2006;17:150–158. doi: 10.1016/j.tem.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 46.Engelman JA, Chu C, Lin A, et al. Caveolin-mediated regulation of signaling along the p42/44 MAP kinase cascade in vivo. A role for the caveolin-scaffolding domain. FEBS Lett. 1998;428:205–211. doi: 10.1016/s0014-5793(98)00470-0. [DOI] [PubMed] [Google Scholar]
- 47.Galbiati F, Volonte D, Engelman JA, et al. Targeted downregulation of caveolin-1 is sufficient to drive cell transformation and hyperactivate the p42/44 MAP kinase cascade. EMBO J. 1998;17:6633–6648. doi: 10.1093/emboj/17.22.6633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hayashi K, Matsuda S, Machida K, et al. Invasion activating caveolin-1 mutation in human scirrhous breast cancers. Cancer Res. 2001;61:2361–2364. [PubMed] [Google Scholar]
- 49.Yu H, Jove R. The STATs of cancer--new molecular targets come of age. Nat Rev Cancer. 2004;4:97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.