

Abstract
Accurate and affordable assessment of ligand–protein affinity for structure-based virtual screening (SB-VS) is a standing challenge. Hence, empirical postdocking filters making use of various types of structure–activity information may prove useful. Here, we introduce one such filter based upon three-dimensional structural protein–ligand interaction fingerprints (SPLIF). SPLIF permits quantitative assessment of whether a docking pose interacts with the protein target similarly to a known ligand and rescues active compounds penalized by poor initial docking scores. An extensive benchmark study on 10 diverse data sets selected from the DUD-E database has been performed in order to evaluate the absolute and relative efficiency of this method. SPLIF demonstrated an overall better performance than relevant standard methods.
Introduction
In structure-based virtual screening (SB-VS), each screened compound is submitted to a two-step process. In the first step, a compound is docked to the putative binding pocket of the protein in a number of energetically acceptable binding modes called poses.1 In the second step, the free energy of binding is assessed for each pose by a scoring function.2 While there is now a general consensus that most of the popular docking algorithms perform fairly well in generating sound poses, scoring functions most often fail to adequately evaluate the binding affinity.3−9 As a result, even the optimiztic success rates that are generally reported in SB-VS benchmark studies8,9 might often be insufficient for true ligand discovery when screening large chemical libraries against a novel target with an objective to experimentally test 50–100 virtual hits. Therefore, all possible means must be employed to improve the odds of obtaining a sizable number of confirmed actives from a small set of designated virtual hits. Scoring approaches that can take advantage of known ligand-bound protein structures (e.g., enzyme-bound substrates) are of special interest. In 2003, Deng et al. introduced structural interaction fingerprints (SIFt),10 with an objective to represent and analyze three-dimensional protein–ligand binding interactions by encoding them into a one-dimensional binary string. Construction of SIFt is a two-step process consisting of (i) identification of residues interacting with the ligand and (ii) classification of ligand–residue interactions into any of seven predetermined types (e.g., whether the protein backbone or side-chain is involved, residue acts as an H-bond donor or acceptor, etc.). Later, similar techniques were proposed by Mpamhanga et al.,11 Pérez-Nueno et al.,12 and Marcou and Rognan13 and implemented in the MOE software suite.14 Although the SIFt-like approaches proved to be useful postdocking analysis techniques, they also have a number of intrinsic limitations. For instance, inferring bond types from quite imperfect binding poses, may lead to frequent bond-type detection mistakes. Moreover, the bond-type categories (e.g., hydrogen bond, polar, nonpolar, and contact10) used in interaction fingerprints do not account for multiple interaction types, such as cation-π, which would be labeled as merely a contact.
Here we introduce a new approach termed structural protein–ligand interaction fingerprints (SPLIF) that also exploits the general idea of quantifying and comparing ligand–protein interactions but does it in a very different way. Particularly, in SPLIF, three-dimensional structures of interacting ligand and protein fragments are explicitly encoded in the fingerprint. Consequently, all possible interaction types that may occur between the fragments (e.g., π–π, CH−π, etc.) are implicitly encoded into SPLIF. The reported fingerprints are wrapped into a normalized quantitative score that expresses the similarity between the interaction profile of a docking pose and that of a reference protein–ligand complex.
In order to quantitatively assess the performance of this new approach, we submitted it to a comparative test using it as a postdocking score against a panel of 10 diverse protein targets. The targets along with the sets of respective actives and decoys were selected from the Database of Useful Decoys: Enhanced (DUD-E).15 The purpose of this evaluation was to ascertain if SPLIF can outperform and/or bring complementary actives compared to standard or analogous approaches.
Materials and Methods
Targets, Ligand Data Sets, and Reference Ligands
The Database of Useful Decoys: Enhanced (DUD-E), a standard test set for virtual screening, was used to validate our fingerprint approach. Ten diverse targets covering six protein classes were randomly selected for this benchmark study (Table 1). The protein structures selected were used as targets for docking of actives and decoys; the resulting poses were processed for subsequent SPLIF generation. All available cocrystallized ligands were retrieved from the Protein Data Bank and used to generate reference SPLIF.
Table 1. Targets for SPLIF Benchmarking Collected from DUD-E.
| class | target | description | PDB | activesa | decoysa | Gscore cutoffb | refs |
|---|---|---|---|---|---|---|---|
| kinase | FAK1 | focal adhesion kinase 1 | 3bz3 | 100 (71) | 5350 (2131) | –6.0 | (9) (1mp8, 2etm, 2ijm, 3bz3, 4gu6, 4gu9, 4i4e, 4k8a, 4kab) |
| AKT1 | serine/threonine-protein kinase AKT | 3cqw | 293 (199) | 16450 (6131) | –5.0 | (12) (3cqu, 3cqw, 3mv5, 3mvh, 3ocb, 3ow4, 3qkk, 3cql, 3qkm, 4ekk, 4ekl, 4gv1) | |
| protease | ACE | angiotensin-converting enzyme | 3bkl | 282 (277) | 16900 (16454) | –2.5 | (13) (1o86, 1uze, 1uzf, 2c6n, 2oc2, 2xy9, 2xyd, 2ydm, 3bkk, 3bkl, 3l3n, 3nxq, 4bxk) |
| TRYB1 | tryptase beta-1 | 2zec | 148 (59) | 7650 (1657) | –6.0 | (5) (2f9p, 2f9n, 2zeb, 2zec, 4a6l) | |
| HMDH | HMG-CoA reductase | 3ccw | 170 (170) | 8750 (8456) | –2.5 | 22 (1dq8, 1dq9, 1dqa, 1hw8, 1hw9, 1hwi, 1hwj, 1hwk, 1hwl, 2q1l, 2q6b, 2q6c, 2r4f, 3bgl, 3cct, 3ccw, 3ccz, 3cd0, 3cd5, 3cd7, 3cda, 3cdb) | |
| GPCR | ADRB1 | Beta-1 adrenergic receptor | 2vt4 | 247 (240) | 15842 (13932) | –4.0 | (14) (2vt4, 2y00, 2y01, 2y02, 2y03, 2y04, 2ycw, 2ycx, 2ycy, 2ycz, 3zpq, 3zpr, 4ami, 4amj) |
| nuclear receptor | MCR | mineralocorticoid receptor | 2aa2 | 94 (66) | 5150 (2481) | –6.0 | (13) (2aa2, 1y9r, 1ya3, 2a3i, 2aa5, 2aa6, 2aa7, 2aax, 2ab2, 2abi, 2oax, 3vhu, 3vhv) |
| PRGR | progesterone receptor | 3kba | 293 (222) | 15650 (12914) | –5.0 | (17) (1a28, 1e3k, 1sqn, 1sr7, 1zuc, 2ovh, 2ovm, 2w8y, 3d90, 3g8o, 3hq5, 3kba, 3zr7, 3zra, 3zrb, 4a2j, 4apu) | |
| ion channel | GRIK1 | glutamate receptor ionotropic kainate 1 | 1vso | 101 (96) | 6550 (5980) | –2.5 | (17) (1txf, 1vso, 1ycj, 2f34, 2f35, 2f36, 2pbw, 2qs1, 2qs2, 2qs3, 2qs4, 2wky, 3gba, 3gbb, 3s2v, 4dld, 4e0x) |
| synthase | PGH2 | cyclooxygenase-2 | 3ln1 | 435 (374) | 23150 (17948) | –5.0 | 27 (1cvu, 1cx2, 1ddx, 1pxx, 3hs5, 3hs6, 3hs7, 3krk, 3ln0, 3ln1, 3mdl, 3nt1, 3ntb, 3ntg, 3olt, 3olu, 3pgh, 3q7d, 3qh0, 3qmq, 3rr3, 3tzi, 4cox, 4e1g, 4fm5, 4llz, 6cox) |
Initial numbers of actives and decoys from DUD-E with the numbers after the Gscore filter included in parentheses.
The Gscore cutoffs are set to allow all reference ligands to be retained (in hope to retain the most of actives in the test set as well).
In order to assess the diversity of the actives and decoys, we calculated pairwise Tanimoto similarities for all ligands used in this study. The histograms in Figure S1 (see the Supporting Information) show similarity probability distributions for each combination of target/activity-category (active or decoy). In all histograms, mostly low similarity values are well-populated, meaning that, similar to typical virtual screening libraries, the sets are overall diverse but have clusters of closely related compounds.
The SD files for all data sets including Gscore, SPLIF, 2D similarity and PLIF scores can be obtained on request from the authors.
Docking
Ligands were docked into the active site of the target protein using the Glide program16 in standard docking precision (Glide SP). The binding region was defined by a 20 Å × 20 Å × 20 Å box centered on the reference ligand selected from DUD-E. Default settings were adopted for all the remaining parameters. The top 30 poses were generated for each ligand.
Structural Protein–Ligand Interaction Fingerprints (SPLIF)
Building the Reference Fingerprint
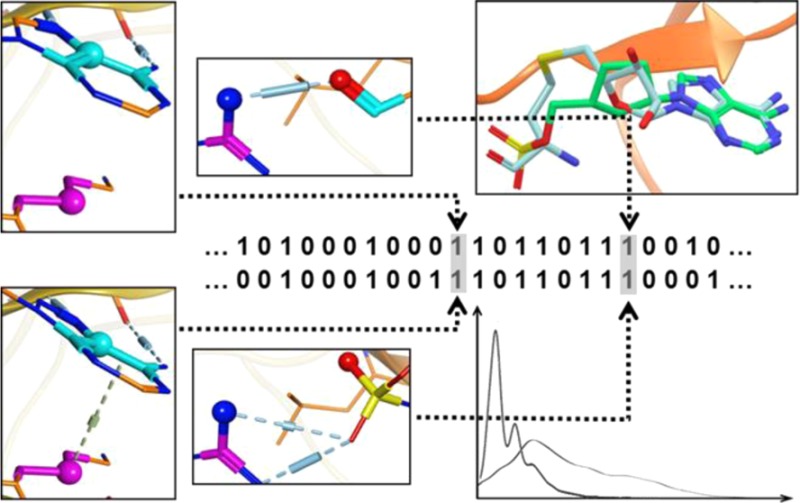
SPLIF-based rescoring consists of calculating SPLIF for each docking pose and comparing it to that of a reference (e.g., experimentally solved) ligand-protein complex. The essential steps of the algorithm for building a reference SPLIF are depicted in Figure 1. In the first step, a reference protein-bound ligand is inspected for protein–ligand contacts. Two atoms are considered being in a contact if the distance between them is within a specified threshold (4.5 Å in this study). In the second step, for each ligand–protein atom pair, the respective ligand and protein atoms are expanded to circular fragments, i.e., fragments that include the atoms in question and their successive neighborhoods up to a certain distance. In Figure 1, the contacting ligand and protein atoms are enclosed in small dotted circles and the respective circular fragments are enclosed in larger concentric circles. Each type of circular fragment is assigned an identifier. Here, we made use of Extended Connectivity Fingerprints up to the first closest neighbor (ECFP2) as defined in the Pipeline Pilot software.17 ECFP retains information about all atom/bond types and uses one unique integer identifier to represent one substructure (i.e., circular fragment). In the third step, 3D coordinates are retrieved for all atoms involved in ligand and protein fragments.
Figure 1.

Essential steps of building a reference SPLIF.
The major difference of SPLIF from earlier—SIFt-type—fingerprints is that in SPLIF the interactions are encoded implicitly, as a result of explicitly encoding ligand and protein fragments, whereas in SIFt-like methods the interaction types need to be encoded explicitly, by means of empirical rules. Consequently, most of current SIFt-like implementations handle only a small number of interaction types. By contrast, SPLIF implicitly accounts for all types of local interactions. For example, two parallel aromatic fragments would imply a π–π interaction; a cation fragment positioned on the axis perpendicular to the aromatic plane of another fragment would imply a cation-π interaction and so forth. For SIFt-like fingerprints, if these two types of interaction are not encoded, they will certainly not be identified. While SPLIF records any contacting fragments from the ligand and the protein, meaning the two aromatic fragments and the cation and the aromatic plane here. If the test ligand presents the same fragments, a match to the reference would be assigned by SPLIF, but not by SIFt-like fingerprints.
Building Fingerprints for Docking Poses of Test Compounds
The interaction fingerprints for the docking poses of a test compound (an active or a decoy from DUD-E) are computed in a similar fashion to that of the reference fingerprints.
SPLIF-Based Similarity
The calculation of a SPLIF-based similarity score is depicted in Figure 2. In the first step, the ECFP identifiers of a test SPLIF (i.e., the SPLIF of a docking pose to be scored) are compared to ECFP identifiers of the reference fingerprint, which is done to find matching circular fragments between the test ligand and the reference ligand and results in a list of 2D-matching SPLIF-bits. In the second step, 3D coordinates of the matching circular fragments (2D-matching bits) are retrieved and root-mean-square deviations (RMSDs) are calculated in order to assess the 3D overlay. The bits for which RMSDs are within a specific threshold (set to 1 Å in this study) are considered as (fully) matching. Next, all atom lists of all matching bits are fused together and deduplicated to form two consolidated lists: (i) unique matching ligand atoms (UMLA) and (ii) unique matching protein atoms (UMPA). Finally, a SPLIF-based similarity score is calculated as follows:
| 1 |
where NUMLA is the number of unique matching ligand atoms, i.e., atoms constituting the matching circular fragments of the docking pose compared to the reference (on the ligand side); NULA is the number of unique ligand atoms, i.e., atoms constituting all interacting circular fragments of the docking pose (on the ligand side); NUMPA is the number of unique matching protein atoms, i.e., atoms constituting the matching circular fragments of the docking pose compared to the reference (on the protein side); NUPA is the number of unique protein atoms, i.e., atoms constituting all interacting circular fragments of the docking pose (on the protein side).
Figure 2.
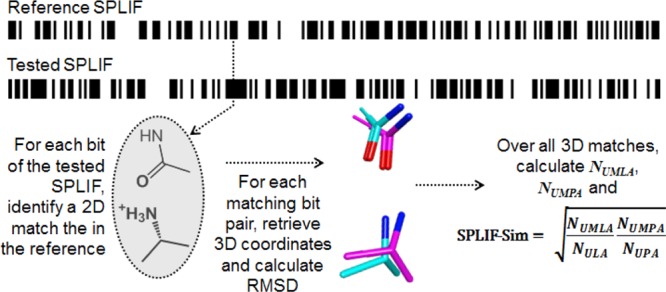
Essential steps of SPLIF-scoring the docking poses.
Implementation
The whole workflow has been implemented in Pipeline Pilot.17 The constituent algorithms were developed in Pipeline Pilot Script. The current implementation allows processing of ∼10 poses per second in screening mode.
Ligand-Based Similarity
Topological fingerprint similarity (expressed as the Tanimoto coefficient) of test vs reference ligands was used as a benchmark showing how efficient VS might be without any knowledge of the protein structure at all. The “Molecular Similarity” component in Pipeline Pilot was used with default settings. Functional Connectivity Fingerprints up to the second closest neighbor (FCFP4) were selected to describe ligand structures. The atomic label in FCFP may be one of the following: (1) hydrogen-bond acceptor; (2) hydrogen-bond donor; (3) positively ionized or positively ionizable; (4) negatively ionized or negatively ionizable; (5) aromatic; and (6) halogen.
PLIF-Based Similarity
The PLIF (protein–ligand interaction fingerprints) descriptors implemented in the MOE suite14 were used as a benchmark with respect to interaction fingerprints. The performance of PLIF would indicate how efficient the first-generation interaction fingerprints might be. Interactions are classified as hydrogen bonds, ionic interactions, and surface contacts according to the residues. We applied it to virtual screening here and compared it to our SPLIF approach. The PLIF descriptors for all protein-bound ligands were generated with the default parameter set in MOE. The PLIF similarity was expressed by means of the Tanimoto similarity coefficient.
Performance Metrics
We made use of enrichment factors (EF) and EF plots to quantitatively assess the performance of the VS scores under study. The EF plot represents the enrichment for a specific top-scoring percentile of the database as a function of the corresponding percentile Px (in log10 scale):
| 2 |
where As is the number of active ligands in the selected top scoring percentile of the database; Ds is the number of decoys in the selected top scoring percentile of the database; At is the total number of active ligands in the whole database; and Dt is the total number of decoys in the whole database. Enrichment factor indicates the ability of a virtual screening score to increase the proportion of true positives in a respective percentile relative to an average random selection. The logarithmic scale for the X-axis is used to accentuate the contribution of the lower percentiles to the plot.
Results and Discussion
The generic benchmark workflow for each target included the following steps: (i) collecting reference ligands from crystal structures; (ii) Glide-based docking and scoring of all reference ligands and test ligands (actives and decoys from DUD-E); (iii) selecting test-ligand poses with plausible Gscore values (to leave out the most awkward poses); (iv) generating SPLIF for reference and test ligands, and calculate SPLIF-based similarity scores; (iv) calculating alternative scores (ligand-based similarity and first-generation interaction fingerprints PLIF); and (iv) calculating performance metrics. The targets for the benchmark study represented six protein classes including kinases, proteases, synthases, GPCRs, nuclear receptors, and ion channels (Table 1). In step iii, the Gscore cutoffs were set on per-target basis (see Table 1) to let all reference ligands be selected (in hope to retain most of the actives from the test set as well). The Gscore filter removed substantial numbers of decoys, while sacrificing a relatively small number of actives. However, the enrichment produced by step iii alone was still insufficient, which emphasized the importance of further filtering. The enrichment plots for the 10 targets are shown in Figure 3 and can be used for a quick visual assessment of the VS scores employed. Ideal EF plots (rendered as black solid lines) produced by hypothetic scores that would rank all actives above the decoys are given for reference. As expected, the EFs resulting from the scoring methods under study differed most significantly at relatively small percentages of selected top scoring compounds (<10% of At+ Dt). As can be seen from the plots, the enrichment curves are often entangled, meaning that the relative performance of a method may depend on how many top-scoring compound you choose to select as virtual hits. The predominance of a single method over a broad range of percentile cutoffs has been observed for five targets: ADRB1 and PGH2 (where SPLIF ranked first over the full range of percentile cutoffs); FAK1 and ACE (where ligand-based similarity ranked first); and TRYB1 (where PLIF ranked first). Despite that the performance analysis over a broad percentile range seems to be a frequent practice in VS assessment, we consider that it needs to be complemented by a quantitative analysis of enrichment for low-percentile selections. Indeed, in real-world VS implementations, where millions of compounds are docked to a target, it would be an unaffordable luxury to consider hundreds of thousands of compounds as potential hits. Therefore, in addition to the EF plot analysis, we quantitatively compared the performance of the scores at a 1-percentile cutoff, which was considered realistic for a typical virtual screening campaign (i.e., 10 000 potential hits for a 1 000 000 compound collection). The corresponding enrichment factors are listed in Table 2. SPLIF ranked first for three targets (GRIK1, PGH2, and HMDH), tied for first rank with one other score for three targets (MCR, FAK1, and AKT1) and ranked/tied second for three more targets (ADRB1, PRGR, and ACE). Overall, on the basis of the per-target rankings, SPLIF is definitely the best performing approach in this benchmarking test. The 2D similarity to the reference ligand was the closest follower, which ranked first for two targets and tied for first for two more. Gscore alone ranked first for none of the targets (this result underlines the importance of postscoring functions in SB-VS). Importantly, SPLIF demonstrated a very robust performance on all 10 targets. This result is in a sharp contrast with the performance of PLIF, SPLIF’s closest methodological analog, which failed completely on two tested targets: PGH2, with EF = 0, and PRGR, with EF = 1. For comparison, SPLIF ranked first for PGH2 (with EF = 33) and second for PRGR (with EF = 36).
Figure 3.

EF plots for the performance of benchmarked scores in SB-VS against 10 DUD-E targets: [color legend (by method)] SPLIF red; ligand similarity yellow; Glide-score green; PLIF blue; ideal black.
Table 2. Performance Statistics for the Benchmarked Scores.
| EF (1-percentile) |
actives overlap (%)a |
|||||||
|---|---|---|---|---|---|---|---|---|
| target | SPLIF | ligand similarity | Gscore | PLIF | SPLIF rank | ligand similarity | Gscore | PLIF |
| GRIK1 | 25b | 17 | 18 | 18 | 1 | 25 | 13 | 46 |
| PGH2 | 33 | 17 | 16 | 0 | 1 | 33 | 36 | 0 |
| HMDH | 51 | 50 | 25 | 47 | 1 | 66 | 33 | 72 |
| MCR | 18 | 12 | 9 | 18 | 1–2 | 67 | 25 | 50 |
| FAK1 | 31 | 31 | 23 | 18 | 1–2 | 5 | 68 | 55 |
| AKT1 | 14 | 14 | 8 | 10 | 1–2 | 56 | 26 | 37 |
| ADRB1 | 18 | 17 | 4 | 19 | 2 | 36 | 5 | 46 |
| PRGR | 36 | 40 | 13 | 1 | 2 | 66 | 14 | 14 |
| ACE | 44 | 54 | 5 | 44 | 2–3 | 65 | 1 | 48 |
| TRYB1 | 10 | 7 | 14 | 21 | 3 | 67 | 33 | 83 |
Fraction of the SPLIF true positives also selected by another score.
The gray background indicates that the respective score ranked or tied first for this target.
It is an interesting result that the least informed method, 2D ligand similarity, was the second best performer in this study. Here, by “least informed” we mean that, unlike other methods on our panel, the ligand similarity does not make use of any protein–structure information. This relative success of SPLIF and ligand similarity, i.e., the two methods on the panel that make explicit use of the chemical structure of known actives, may merely reflect the fact that predicting affinity without a prior knowledge of actives, e.g., by means of docking/scoring, is still in need of substantial improvements. Because the ligand- and the SPLIF-based similarity metrics in our study involve the same type of structural descriptors, one might expect that these two methods should perform with comparable sensitivities and that SPLIF is expected to be more specific due to the 3D information about the protein–ligand interactions embedded in the reference SPLIF (see below a more detailed discussion on the relative SPLIF performance).
We have also analyzed the reasons of the varying SPLIF performance and, more specifically, of its poor performance on TRYB1. A plausible explanation comes from the inspection of the absolute SPLIF-score values. While we did not pay much attention to how high/low the score values are when selecting the top-percentile hits, they display a substantial intertarget variation. For instance, Figure 4 shows the probability density distributions of SPLIF-scores for the most successful SPLIF-screening (HMDH), the least successful one (TRYB1) and the overall distributions for actives and decoys (distributions for all 10 targets can be found in Supporting Information Figure S2). It can be seen from the overall distributions in Figure 4 that a SPLIF-score cutoff of ∼0.5 would allow for a very significant prediction specificity (i.e., low false-positives rate), while lower SPLIF-score values are highly populated by both actives and decoys. Then, it is easy to see that higher SPLIF-score values are significantly populated for HMDH and only marginally for TRYB1. The latter observation simply means that there is a large number of SPLIF-based analogs for HMDH-references among the HMDH-actives and too few analogs for TRYB1-references among TRYB1-actives. That is, in the case of TRYB1, SPLIF underperformed simply because we lacked “right” structural references. Unfortunately, like in the case of most VS scores, this explanation does not offer any means of predicting how SPLIF would perform on a given screening collection against a new target. However, one might suggest general principles of the relative SPLIF performance with respect to other screening approaches. First, the sensitivity of SPLIF is expected to be dictated by the reference ligands (and their respective interactions with the target protein). And, hence, it is restricted compared to docking-based approaches, which potentially can perceive any active ligand in any screening library. Therefore, as in the case of ligand-based similarity, the SPLIF’s sensitivity would grow as the size and diversity of the reference set grow (while its specificity would remain unchanged). Our hypothesis about the SPLIF performance relative to ligand similarity was that SPLIF would be even less sensitive, but more specific, which would result in higher enrichment rates. While a quantitative testing of this hypothesis may need a more substantial effort than the current study, the data presented here corroborate it qualitatively by showing an overall gain in enrichment rate when using SPLIF.
Figure 4.
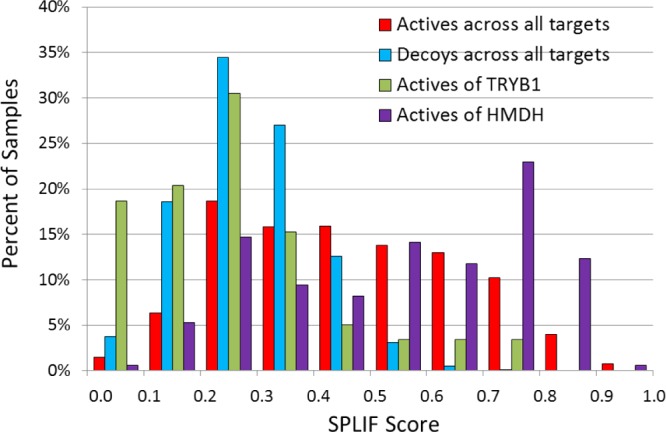
Probability density distributions for SPLIF-scores by category (actives, decoys, HMDH-actives, TRYB1-actives).
Another goal in this study was to assess how complementary SPLIF is to the other benchmarked scores. To this end, for each target we calculated the overlap between the true-positives identified by SPLIF and each other benchmarked score (see Table 2). The overlaps range from 0% to 83% with a median of 36%. The highest overlaps (83% and 72%) are with PLIF, followed by the overlaps with Gscore (68%) and ligand-based similarity (67%). Interestingly, and contrary to expectations, the overlaps with PLIF are low (<50%) for most targets. The latter observation means that the explicit encoding of the chemical structure (which constitutes the novelty of SPLIF compared to PLIF and other first-generation interaction fingerprints) does indeed result in better VS performance and complementary actives. Overall, the data show that in terms of true positives identified SPLIF is highly complementary to the other benchmarked scores.
When it comes to a general assessment of whether a new scoring method may be recommended for broad use, there are three key questions to be answered: Does the new method show any improvement compared to its close analogs and popular standard tools? Does the new method show robust performance across a broad range of targets? Does the new method bring additional actives compared to similarly performing techniques? While the importance of the first question is evident, the second and third questions are equally important for the following reasons. It has been demonstrated in multiple benchmark studies that the relative performance of any scoring method varies with protein target and that in general it is impossible to predict, which function would perform best on a given target.9,18−21 A solution to address this uncertainty would be to use a panel of scoring functions and merge the respective hit lists using either AND or OR logic. However, this still requires all methods on the panel to perform reasonably well. Otherwise, a single strongly underperforming (as happened twice to PLIF and twice to Gscore in this study) would significantly undermine the overall performance. We believe that the presented data on SPLIF performance supports an affirmative response to all three questions. Indeed, although it did not rank first for all protein targets, it did rank first more frequently than any other benchmarked method, hence showing a tangible improvement. Also, SPLIF did not rank last for any of the targets and showed significant enrichment for all of them. Finally, for 9 out of 10 targets SPLIF brought 30% or more additional true positives compared to the method that ranked higher or tied with SPLIF (or was one rank below when SPLIF ranked first).
Conclusions
SPLIF is an interaction fingerprint that explicitly encodes structural information and hence implicitly captures all types of ligand–protein interactions (e.g., stacking, polarization, cation-π, CH-π). Here, we demonstrated that SPLIF-based similarity of ligand-protein interaction motifs is a valuable metric that helps to improve the odds of finding true hits via structure-based virtual screening. SPLIF-score showed a robust performance over a broad panel of diverse protein targets. The SPLIF-based hit lists featured true positives, which (i) cannot be obtained by a docking score alone, (ii) are not structurally similar to reference ligands, and (iii) are complementary to the hits that might be obtained using analogous interaction fingerprints. We therefore recommend the use of SPLIF either as a single rescoring technique to increase the probability of success or as a complement to other scoring functions in order to increase the chemical diversity of resulting hits.
Acknowledgments
This work was supported by the University Cancer Research Fund (University of North Carolina at Chapel Hill) and by Federal Funds from the National Cancer institute, National Institute of Health, under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. We thank Stephen Frye for helpful discussions and critical reading of the manuscript.
Glossary
Abbreviations:
- SB-VS
structure-based virtual screening
- SPLIF
structural protein–ligand interaction fingerprints
- ECFP
extended connectivity fingerprint
- DUD-E
Database of Useful Decoys–Enhanced
Supporting Information Available
Figures S1 and S2 as mentioned in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Brooijmans N.; Kuntz I. D. Molecular Recognition and Docking Algorithms. Annu. Rev. Biophys. Biomol. Struct. 2003, 321335–373. [DOI] [PubMed] [Google Scholar]
- Jain A. N. Scoring functions for protein-ligand docking. Curr. Protein Pept Sci. 2006, 75407–420. [DOI] [PubMed] [Google Scholar]
- Kirchmair J.; Markt P.; Distinto S.; Wolber G.; Langer T. Evaluation of the performance of 3D virtual screening protocols: RMSD comparisons, enrichment assessments, and decoy selection—What can we learn from earlier mistakes?. J. CAMD 2008, 223213–228. [DOI] [PubMed] [Google Scholar]
- Scior T.; Bender A.; Tresadern G.; Medina-Franco J. L.; Martínez-Mayorga K.; Langer T.; Cuanalo-Contreras K.; Agrafiotis D. K. Recognizing Pitfalls in Virtual Screening: A Critical Review. J. Chem. Inf Model 2012, 524867–881. [DOI] [PubMed] [Google Scholar]
- Warren G.; Andrews C.; Capelli A.; Clarke B.; LaLonde J. A critical assessment of docking programs and scoring functions. J. Med. Chem. 2005, 49, 5912–5931. [DOI] [PubMed] [Google Scholar]
- Kitchen D. B.; Decornez H.; Furr J. R.; Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat. Rev. Drug Discov 2004, 311935–949. [DOI] [PubMed] [Google Scholar]
- Ferrara P.; Gohlke H.; Price D.; Klebe G.; Brooks C. I. Assessing scoring functions for protein-ligand interactions. J. Med. Chem. 2004, 47, 3032–3047. [DOI] [PubMed] [Google Scholar]
- Cross J. B.; Thompson D. C.; Rai B. K.; Baber J. C.; Fan K. Y.; Hu Y.; Humblet C. Comparison of several molecular docking programs: pose prediction and virtual screening accuracy. J. Chem. Inf Model 2009, 4961455–1474. [DOI] [PubMed] [Google Scholar]
- Kellenberger E.; Rodrigo J.; Muller P.; Rognan D. Comparative evaluation of eight docking tools for docking and virtual screening accuracy. Proteins: Struct., Funct., Bioinf. 2004, 572225–242. [DOI] [PubMed] [Google Scholar]
- Deng Z.; Chuaqui C.; Singh J. Structural Interaction Fingerprint (SIFt): A Novel Method for Analyzing Three-Dimensional Protein-Ligand Binding Interactions. J. Med. Chem. 2003, 472337–344. [DOI] [PubMed] [Google Scholar]
- Mpamhanga C. P.; Chen B.; McLay I. M.; Willett P. Knowledge-Based Interaction Fingerprint Scoring: A Simple Method for Improving the Effectiveness of Fast Scoring Functions. J. Chem. Inf Model 2006, 462686–698. [DOI] [PubMed] [Google Scholar]
- Pérez-Nueno V. I.; Rabal O.; Borrell J. I.; Teixidó J. APIF: A New Interaction Fingerprint Based on Atom Pairs and Its Application to Virtual Screening. J. Chem. Inf Model 2009, 4951245–1260. [DOI] [PubMed] [Google Scholar]
- Marcou G.; Rognan D. Optimizing Fragment and Scaffold Docking by Use of Molecular Interaction Fingerprints. J. Chem. Inf. Model. 2006, 471195–207. [DOI] [PubMed] [Google Scholar]
- Molecular Operating Environment (MOE), v.2010.10; Chemical Computing Group Inc.2010. [Google Scholar]
- Mysinger M. M.; Carchia M.; Irwin J. J.; Shoichet B. K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55146582–6594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesner R. A.; Banks J. L.; Murphy R. B.; Halgren T. A.; Klicic J. J.; Mainz D. T.; Repasky M. P.; Knoll E. H.; Shelley M.; Perry J. K.; Shaw D. E.; Francis P.; Shenkin P. S. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 4771739–1749. [DOI] [PubMed] [Google Scholar]
- Pipeline Pilot, ver. 8.5, Accelrys Software Inc. 2009. [Google Scholar]
- Venkatraman V.; Pérez-Nueno V. I.; Mavridis L.; Ritchie D. W. Comprehensive Comparison of Ligand-Based Virtual Screening Tools Against the DUD Data set Reveals Limitations of Current 3D Methods. J. Chem. Inf. Model. 2010, 50122079–2093. [DOI] [PubMed] [Google Scholar]
- Oda A.; Tsuchida K.; Takakura T.; Yamaotsu N.; Hirono S. Comparison of consensus scoring strategies for evaluating computational models of protein-ligand complexes. J. Chem. Inf. Model. 2006, 46, 380. [DOI] [PubMed] [Google Scholar]
- Hawkins P. C.; Skillman A. G.; Nicholls A. Comparison of shape-matching and docking as virtual screening tools. J. Med. Chem. 2007, 50174–82. [DOI] [PubMed] [Google Scholar]
- McGaughey G. B.; Sheridan R. P.; Bayly C. I.; Culberson J. C.; Kreatsoulas C.; Lindsley S.; Maiorov V.; Truchon J. F.; Cornell W. D. Comparison of topological, shape, and docking methods in virtual screening. J. Chem. Inf Model 2007, 4741504–1519. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.