

Abstract
In drug discovery and development, the conventional “single drug, single target” concept has been shifted to “single drug, multiple targets” – a concept coined as polypharmacology. For studies in this emerging field, dedicated and high-quality databases of multitargeting ligands would be exceedingly beneficial. To this end, we conducted a comprehensive analysis of the structural and chemical/biological profiles of polypharmacological agents and present a Web-based database (Polypharma). All of these compounds curated herein have been cocrystallized with more than one unique protein with intensive reports of their multitargeting activities. The present study provides more insight of drug multitargeting and is particularly useful for polypharmacology modeling. This specialized curation has been made publically available at http:/imdlab.org/polypharma/
1. Introduction
In the past few years, polypharmacology has been recognized as a new avenue for drug discovery and development.1−5 Numerous drugs such as Aspirin,6 topiramate,7 and especially kinase inhibitors are known for their multitarget-directed activities. Along the same lines, drug repurposing/repositioning, which aims to discover new indications for existing approved drugs, has emerged as a critical cost-effective and time-efficient strategy for drug development.8−13 More importantly, the enormous amount of molecular data generated in the postgenomic era will significantly accelerate such polypharmacological research.
Rational design of multitargeting drugs can be challenging with the current drug discovery strategies. Recently we reviewed various polypharmacological approaches14−16 available in the literature. There have been several promising attempts,2,11,17,18 and various methods16,19−23 were developed for associating drugs with their possible unknown off-targets. For instance, Campillos et al. mapped drugs-targets based on their phenotypic side-effect similarities.2 The Shoichet group developed similarity ensemble approach (SEA)24 to relate targets based on the set-wise chemical similarity with their ligands, and it was also applied to a large-scale prediction of drug activity on side-effect targets.11 Several other groups used knowledge-based approaches1,3,25−27 to identify associations among various biomolecules stored in their databases. Recently text mining techniques were also employed to extract ligand-target-disease mapping information from the literature and public databases.28−31 Of course, as the most straightforward methodology in structure-based design, inverse docking has long been used to identify potential targets for a given ligand.21,32−35 Additionally, systems biology/pharmacology approaches have gained more attention recently by integrating experimental and computational approaches to understand drug mechanisms of actions at the systems-level.36−39
During the past decade, numerous databases6,40−45 have been developed such as DrugBank,6 STITCH,40 Supertarget,46 IUPHAR-DB,47 WOMBAT,41 PubChem’s BioAssay Database,48 ChEMBL,49 and so on, which integrate diverse information on molecular pathways, drug targets, crystal structures, etc. There are also a number of small molecule-centric databases including ZINC,50 PubChem,51 Ligand Expo,52 etc. These databases are comprised of enormous information about their disease relevance, chemical properties, and biological activities. Therefore, they could be potentially used for off-target identifications. However, deriving accurate multitargeting information from these databases is not trivial, and, to date, a dedicated, focused polypharmacological database is yet to be developed.
Herein we showcase our implementation of a novel, dedicated database for a unique set of polypharmacological ligands with high-quality, experimentally validated structural and biological activity data. The data was integrated from multiple resources including the following: LigandExpo52 (formerly known as LigandDepot), Protein Data Bank (PDB),53 Universal Protein Resource (UniProt),54 and DrugBank.6 A variety of ligand-protein binding databases such as PDBbind,55 BindingDB,42 and Binding MOAD43 are also taken into consideration in order to extract the available ligand binding affinities. Additionally, literature reports were also mined to obtain as much biological data as possible. By integrating these resources, we have built a novel database, termed Polypharma, specifically for multitargeting ligands, along with their modulated targets and quantitative bioactivities (e.g., binding affinity), in particular for polypharmacology modeling. To date, Polypharma includes 953 ligands that are complexed with two or more protein structures belonging to distinct target families. We also provide other information such as molecular properties of ligands and their targets. A set of query functions has been implemented to search our database, and molecular networks can be constructed to depict ligand-target interactions. The query results can also be visualized with integrated molecular visualization tools. The database is currently accessible at http://imdlab.org/polypharma/.
2. Results
Curation of Polypharmacological Ligands with Unique Targets
The Polypharma database consists of a unique set of multitarget-directed ligands with their high resolution crystal structures and available binding affinities. These data will provide new insights for off-target identification and polypharmacological agent design. A flowchart illustrating the data curation is provided in Figure 1. The curation was started by obtaining ligand data from Ligand Expo,52 and their interactions with targets were analyzed based on their crystal structures in the PDB. As of March 10, 2013, the Ligand Expo contained 15,952 small molecules which were included in 88,714 unique PDB structures. To obtain information on the ligands such as their names, chemical structures, and so on, the mmCIF format dictionary was downloaded from Ligand Expo and analyzed with an in-house program. To make it more applicable for rational drug design, the “filter” module of the OpenEye scientific software was used to keep only the drug-like ligands. To this end, the typical Lipinski’s rule of five56 along with other filtering parameters were applied (Table S1). This process resulted in 8,067 ligands. Finally, several programs were implemented to automatically identify those ligands complexed with more than one protein. This led to 1,674 ligands corresponding to a total of 9,382 unique protein structures (PDB IDs).
Figure 1.

Scheme of Polypharma database curation.
During the curation we frequently observed that a ligand can be included in multiple PDB entries which are actually of the same protein. For instance, the drug alitretinoin is complexed with 1FM6, 1FM9, and 1K74, but all belong to the PPAR-γ protein (in a heterodimer with RXR-α), and hence alitretinoin should not be included in Polypharma. To remove cases like alitretinoin, we consider only those ligands complexed with multiple proteins belonging to different families. To this end, we first referred to UniProt identities attempting to obtain unique proteins with an assumption that a protein structure with a unique UniProt identity would represent a unique target. Using our in-house tools, the PDB IDs were mapped to the UniProt identities as annotated in the UniProt database (accessed on March 10, 2013). However, upon analysis, we encountered several problems. First, not all PDB IDs are associated with UniProt IDs. Out of 4,167 PDB IDs, only 4,074 PDB IDs can be mapped to Uniprot IDs. Second, in some cases, the same proteins have different UniProt IDs. For example, HIV-1 protease complexed with the drug darunavir has crystal structures of 3TTP and 3S53, but they have different UniProt IDs as P03367 and Q7SSI0, respectively. The reason is that P03367 corresponds to the gag-pol gene, whereas Q7SSI0 corresponds to the pol gene. Third, sometimes one PDB ID can correspond to multiple UniProt IDs such as 3O3A which is for human Class I MHC HLA-A2 in complex with the Peptidomimetic ELA-1 protein with two UniProt IDs P01892 and P61769. The simple lesson learned from this unsuccessful attempt demonstrated how complicated and difficult it is to perform such data curation (also indicating the urgent need of consistent and clean data integration across different resources).
We also tried other protein classification methods such as CATH/SCOP/EC numbers. Various issues were found, and we conclude that they are not appropriate for our problem here. Therefore, we ventured back to the very basic concept of sequence similarity for identification of unique protein families. All of the proteins bound to the same ligand were compared for their sequence similarity, and the ones with less than 80% similarity were retained. The threshold was determined through a systematic analysis after experimenting with various cutoff values ranging from 70% to 90%. However, we found that we could maintain nonredundant proteins (e.g., some HIV protease mutants have only 80% sequence similarity with the wild type) only when using this 80% sequence similarity cutoff for our data set. The filtering was achieved with the UCLUST program which is a clustering algorithm that employs USEARCH as a subroutine to assign sequences to clusters.57 Since this problem has a significant complexity due to the fact that some PDBs have multiple chains and multiple ligands, the program actually considers each chain separately.57 So for all proteins binding the same ligand, the sequences of their individual chains are compared with each other. The sequences with similarity above a given threshold (80%) will be grouped into one cluster. In each cluster, the chains are sorted (i.e., ranked) according to the following criteria and the order: (a) A quality factor, calculated as ((1/resolution) – R-value); (b) Deposition date (newer structures have higher ranks); (c) Alphabetical order. From each cluster, only the highest ranked chain will be picked as the representative sequence, and this will lead to a set of nonredundant chains for a given ligand.
Database Characterization
Upon the above filtering with the aid of sequence similarity clustering, we obtained 953 multitargeting ligands associated with 4,167 distinct PDBs belonging to various nonredundant proteins. This represents 4,298 positive binding data points as some PDBs have multiple chains with multiple bound ligands. Among the 953 ligands, 550 are crystallized with two unique proteins (1,100 ligand-protein combinations), whereas the other 403 are bound to more than two distinct proteins (3,198 ligand-protein combinations). Figure S10 illustrates the targeting binding profile for each ligand. Of note, it is very critical to understand that there could be various other possible interactions that were not reported yet, as they do not have solved crystal structures of these interactions. This database only outlines the data extracted from the currently available crystal structures and will be updated along the time. Some statistics of our database, including protein families, protein size, structure resolution, etc., are shown in Figures S1–S3. For instance about 10% proteins have below 200 residues, 40% of proteins have 201–500 residues, whereas 31% have residues between 501 and 1000, and the remaining have more than 1000 residues (Figure S3). For ligands, 30% of them have molecular weights between 130 and 200, 27% between 200 and 300, 37% between 300 and 500, and only 6% are above 500 (Figure S4). Similarly, 64% of the ligands have LogP values between 0.0 and 4.0, and 14% with LogP higher than 4.0 (Figure S5), suggesting a high percentage of drug-like ligands. Other characterizations of these multitargeting ligands, as compared to single-targeting agents, are described next.
Comparison of Multitargeting and Single-Targeting
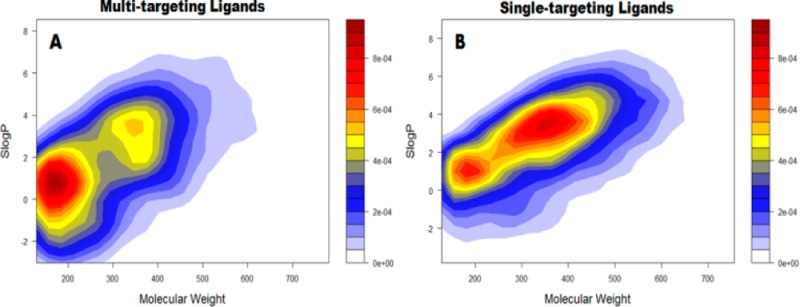
As a dedicated database for multitargeting ligands, it is of great interest to explore whether they are significantly different from single-targeting agents. To this end, typical chemical/physical properties such as molecular weight and hydrophobicity were compared between the two groups of ligands. It is striking to note that, as demonstrated by our 2D plots in Figure 2 and Figure S7B, the 953 multitargeting ligands are on average smaller than the single-targeting ligands. The multitargeting agents mostly have molecular weight below 200 Da, while for single targeting ligands the molecular weight is around 300–420 Da. To some extent, this is not unexpected as lead optimization can improve the selectivity but usually companioned with the increase of molecular size. Accordingly, the hydrophobicity as represented by SLogP here is slightly lower for multitargeting (0–2) than single-targeting ligands (0–4). The similar trend was also observed for the number of rings (Figure S7B) and topological polar surface area (TPSA) (Figure 2). We also performed comparison of other properties such as hydrogen bonding patterns and molecular refractivity, as illustrated in Figure S7. While several properties such as aromatic atoms and molecular refractivity were observed to have more broad range for single targeting ligands (Figures S7B–S7D) since they generally have larger size, it is surprising to see that the hydrogen bonding patterns are similar for multitargeting and single-targeting agents, both have 3–4 hydrogen bond acceptors and 2–4 hydrogen bond donors (Figure S7A).
Figure 2.

Comparison of SLogP, TPSA, and molecular weight in multiple-targeting ligands (the upper panel) vs single-targeting ligands (the lower panel). The plots represent the distribution density of the ligands in the 2D space in terms of the respective chemical/physical properties. The color represents the density as demonstrated by the bar. The color code and scale is the same in each comparison for multitargeting and single-targeting ligands.
Although we the present study is focused on small molecule ligands, we also conducted characterization analysis of protein binding pockets using our in-house programs and a Web server VADAR.58 Of note, in the binding sites, no significant differences were observed between the two groups of proteins, in terms of residue composition, hydrophobicity, and polar surface area (Figure 3). Further case studies with molecular visualization did not identify any unique features of “multitargeting” proteins (Figures S9A,B). This is not uncommon because, as is known, small molecules can be optimized to improve their selectivity toward a specific target. In other words, some ligands can be rather specific, and they are different from other multitargeting molecules, which is the primary point of this manuscript. However, on the other hand, protein targets are a bit different: all proteins are flexible, and each single of them can accommodate quite different small molecules in terms of size, flexibility, and even chemotypes, i.e., they are all always “multitargeting”. Therefore, we do not expect any common features among them or unique features compared to the single-targeting proteins. This is in agreement with our observation here.
Figure 3.

Comparison of residue composition (the upper panel) and chemical/physical properties of protein binding pockets of multitargeting vs single targeting ligands.
Binding Activity Data
For our curated polypharmacological agents, although their binary activities are apparent based on their PDB complexes, the quantitative data of their binding affinities, if available, would be more useful to develop accurate QSAR models or docking/scoring functions for multitargeting predictions. The curation of ligand-protein binding affinities has been conducted by many groups during the past decade, and a variety of databases have been constructed.42,43,49,55 To obtain the binding data for our specific multitargeting agents, we explored these databases along with mining of the published literature. We found that 587 out of 953 ligands (∼61.5%) have available binding data from databases such as PDBbind,55 BindingDB,42 and MOAD.43 As these databases are implemented without a standardized format, it was not trivial to retrieve the activity data automatically from them. To this end, individual programs have been developed to access these databases and extract the activity data in an automated way. Since the data is obtained from multiple databases, the redundancy in the data was eliminated using in-house scripts. Similarly when conflicting data was obtained for the same ligand-protein binding, we double-checked their initial reports to ensure the accuracy of data collections. Eventually we obtained 1,164 quantitative data points for ligand-protein interactions. It is worthy of note that, although the data about the ligands, targets, and their activities are also available elsewhere (e.g., PDB or the databases cited here), Polypharma is a specialized database dedicated to polypharmacological ligands and is uniquely built to perform analysis, visualization, and prediction of multitargeting properties.
3. Methods, Implementation, and Usage
Polypharma has been designed in a three-tier architecture (Figure 4). The Web user interface (Figure 5) was implemented with HTML/CSS/PHP (version 5.3.27), and the database is managed by MYSQL (version 5.5.32-cll). The Apache HTTP servers (version. 2.2.24) with a HTTP content accelerator are deployed on a Linux operating system (kernel 2.6.32–458). We also implemented many other features. For instance, the JME molecular editor, as a courtesy of Dr. Peter Ertl,59 is integrated to draw ligand molecules for chemical similarity and substructure search. Additionally, MolDB5R,60 a collection of fully functional PHP scripts and Perl scripts, is embedded for search options based on (a) substructure and (b) functional groups. Moreover Jmol61 is used for visualizing ligand-protein interactions directly within an HTML page, and it provides controls of different visualization schemes of the structures in the Jmol applet. Finally we also integrated our molecular network analysis technologies for visualization of multitargeting ligand-target interactions.15
Figure 4.
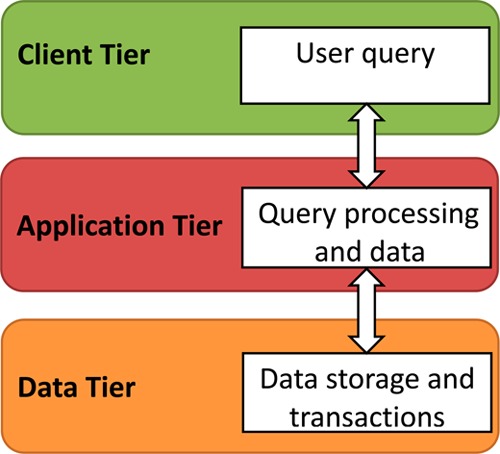
Architecture implemented in the database.
Figure 5.

Some of the screenshots of the graphical user interface of the Polypharma database.
For chemical similarity/substructure search, as a first step all the ligands are encoded into their molecular fingerprints using the checkmol program,60 and they are stored in our MySQL database. To create a query, users can draw a chemical structure using the JME applet embedded into the search page, and the query structure will be converted to a MDL mol file. Based on this file, our backend programs will generate the fingerprints which will be used to search the database for molecules with the similar chemical features (Figure 5). Another useful feature is that the fingerprints of chemical functional groups of all the polypharmacological ligands are stored and used for searching with MolDB5R. Additionally, the user is provided with the option to select multiple functional groups to identify polypharmacological ligands of their interest (Figure S6).
Polypharma is dedicated to multitargeting agents along with their specific targets and biological activities. There are two options for queries. With the “Ligand” option, the queries can be keywords of ligands (e.g., Aspirin) or Ligand Expo IDs (e.g., STI). The results page displays all entries matching the queries, including their Ligand Expo IDs, ligand generic names, etc. The data is linked to a page of the available activity data (Ki, Kd, EC50, IC50, Ka, etc.) of ligands, the target information (e.g., PDB IDs), and so on. As mentioned above, the polypharmacological ligands can also be searched using either substructures or using functional groups. The results are listed along with the 2D structure of each resulted polypharmacological ligand and linked to (a) ligand physicochemical properties, (b) original LigandExpo entry page, and (c) available activity data. With the “Target” option, the queries can be PDB IDs (e.g., 1MLW), disease names (e.g., cancer), target description (e.g., HIV-1), etc. The results page shows the matched protein identities along with their ligands as well as descriptions of the complex structures. Furthermore, the activity data of ligands for different targets is linked to the Ligand Expo IDs. Users can also explore the link-out pages of PDB, DrugBank, and Ligand Expo databases. More documentation with examples and screen shots are available at our Web site http://imdlab.org/polypharma. Users can also communicate with us for further suggestions or questions.
As a unique and interesting feature, the ligand-target relations are depicted and can be used to visualize the multitargeting molecular interaction networks (Figure S8). This was built upon our technology of molecular network analysis as described previously.15 This in-house technique, as the first step, generates the list of all the polypharmacological ligands and, for each ligand, obtains the target information from our curated database through SQL subroutines. Then for each of the targets, all of their complexed ligands’ information is obtained from RCSB and used to construct the graphical network. An open source visualization software Graphviz62 is employed to generate the ligand-protein networks for visualization.
4. Discussion
Despite their evident applications, polypharmacological studies are attributed with several challenges. The major limitation is that we only partially understand the pathways/mechanisms of many diseases at the molecular level. It is exceedingly difficult to derive the full polypharmacological networks without the complete data. As a critical step of our attempt in this area, Polypharma was built as a dedicated database specifically designed for polypharmacology studies by providing accurate, experimentally validated structural and activity data of multitargeting ligands. We have mined and integrated information from a number of existing databases to extract related data. The database will be updated monthly, and future releases will include new multitargeting ligand molecules together with their targets and biological activities as they become available.
Additional functional features (e.g., searching by properties such as LogP, ChemAxon fingerprints, etc.) will also be added. Similarly we plan to integrate with more databases such as PubChem database,63 ChEMBL database,49 and Community Structure–Activity Resource (CSAR).64 We expect that our dedicated database will lay a foundation for analysis of multitargeting ligand properties and development of novel polypharmacology approaches. In particular, with our accurate activity data (both binary and continuous) along with high resolution structures, investigators can develop various ligand-based (e.g., QSAR) and structure-based (e.g., docking/inverse docking) methods/models to predict off-targets or design polypharmacological agents. Notably, the database would also accelerate other drug development efforts such as drug-repurposing.12,65,66 Therefore, we anticipate that this work will vastly promote polypharmacology studies, and it is significant to propel the field forward.
Moreover, there are a few cases where two different ligands bind to different sites of the same protein, which may not be true examples of polypharmacology but may affect the target functions, e.g., binding of one may affect the binding of the other due to allosteric effects. On the other hand, in many cases, binding of one may have nothing to do with binding of the other–especially if they bind to different domains. This type of situation further makes the study more complicated and needs to be addressed in the future. Last but not least, as we already stated, it is important to realize that the current data collection is far from complete. Absence of a ligand-target data (structure or binding affinity) does not mean they are not really interacting with each other. There could be possible interactions that were not just yet reported. With more data becoming available, we anticipate that our database will be more useful for more accurate polypharmacology modeling.
Acknowledgments
We thank OpenEye Scientific and ChemAxon for providing free academic licenses of their software packages. We acknowledge the use of the following programs: JME molecular editor, Jmol, and MolDB5R. We also thank Dr. Sharangdhar Phatak for his initial development of the database. This work was partially supported by the University Cancer Foundation via the Institutional Research Grant program and NIH 5R01CA138702-03.
Supporting Information Available
Table S1 and Figures S1–S10. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
† These authors contributed equally.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Hopkins A. L. Network pharmacology: the next paradigm in drug discovery. Nat. Chem. Biol. 2008, 411682–690. [DOI] [PubMed] [Google Scholar]
- Campillos M.; Kuhn M.; Gavin A. C.; Jensen L. J.; Bork P. Drug target identification using side-effect similarity. Science 2008, 3215886263–6. [DOI] [PubMed] [Google Scholar]
- Yildirim M. A.; Goh K. I.; Cusick M. E.; Barabasi A. L.; Vidal M. Drug-target network. Nat. Biotechnol. 2007, 25101119–26. [DOI] [PubMed] [Google Scholar]
- Xie L.; Xie L.; Kinnings S. L.; Bourne P. E. Novel Computational Approaches to Polypharmacology as a Means to Define Responses to Individual Drugs. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 361–379. [DOI] [PubMed] [Google Scholar]
- Keiser M. J.; Setola V.; Irwin J. J.; Laggner C.; Abbas A. I.; Hufeisen S. J.; Jensen N. H.; Kuijer M. B.; Matos R. C.; Tran T. B.; Whaley R.; Glennon R. A.; Hert J.; Thomas K. L. H.; Edwards D. D.; Shoichet B. K.; Roth B. L. Predicting new molecular targets for known drugs. Nature 2009, 4627270175–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knox C.; Law V.; Jewison T.; Liu P.; Ly S.; Frolkis A.; Pon A.; Banco K.; Mak C.; Neveu V.; Djoumbou Y.; Eisner R.; Guo A. C.; Wishart D. S. DrugBank 3.0: a comprehensive resource for ’omics’ research on drugs. Nucleic Acids Res. 2011, 39Database issueD1035–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley J. T.; Sirota M.; Shenoy M.; Pai R. K.; Roedder S.; Chiang A. P.; Morgan A. A.; Sarwal M. M.; Pasricha P. J.; Butte A. J. Computational repositioning of the anticonvulsant topiramate for inflammatory bowel disease. Sci. Transl. Med. 2011, 39696ra76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekins S.; Williams A. J.; Krasowski M. D.; Freundlich J. S. In silico repositioning of approved drugs for rare and neglected diseases. Drug Discovery Today 2011, 167–8298–310. [DOI] [PubMed] [Google Scholar]
- Schneider G. Designing the molecular future. J. Comput.-Aided Mol. Des 2012, 261115–20. [DOI] [PubMed] [Google Scholar]
- Selassie C. D.; Garg R.; Kapur S.; Kurup A.; Verma R. P.; Mekapati S. B.; Hansch C. Comparative QSAR and the radical toxicity of various functional groups. Chem. Rev. 2002, 10272585–605. [DOI] [PubMed] [Google Scholar]
- Lounkine E.; Keiser M. J.; Whitebread S.; Mikhailov D.; Hamon J.; Jenkins J. L.; Lavan P.; Weber E.; Doak A. K.; Cote S.; Shoichet B. K.; Urban L. Large-scale prediction and testing of drug activity on side-effect targets. Nature 2012, 4867403361–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oprea T. I.; Mestres J. Drug repurposing: far beyond new targets for old drugs. AAPS J. 2012, 144759–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao L.; Zhiyong L. In A new method for computational drug repositioning using drug pairwise similarity; Bioinformatics and Biomedicine (BIBM), 2012 IEEE International Conference on 4–7 Oct. 2012; 2012; pp 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy A. S.; Zhang S. Polypharmacology: drug discovery for the future. Expert Rev. Clin. Pharmacol. 2013, 6141–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phatak S. S.; Zhang S. A novel multi-modal drug repurposing approach for identification of potent ack1 inhibitors. Pac. Symp. Biocomput. 2013, 29–40. [PMC free article] [PubMed] [Google Scholar]
- Morrow J. K.; Tian L.; Zhang S. Molecular networks in drug discovery. Crit. Rev. Biomed. Eng. 2010, 382143–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apsel B.; Blair J. A.; Gonzalez B.; Nazif T. M.; Feldman M. E.; Aizenstein B.; Hoffman R.; Williams R. L.; Shokat K. M.; Knight Z. A. Targeted polypharmacology: discovery of dual inhibitors of tyrosine and phosphoinositide kinases. Nat. Chem. Biol. 2008, 411691–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oprea T. I.; Nielsen S. K.; Ursu O.; Yang J. J.; Taboureau O.; Mathias S. L.; Kouskoumvekaki L.; Sklar L. A.; Bologa C. G. Associating drugs, targets and clinical outcomes into an integrated network affords a new platform for computer-aided drug repurposing. Mol. Inf. 2011, 302–3100–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams J. C.; Keiser M. J.; Basuino L.; Chambers H. F.; Lee D. S.; Wiest O. G.; Babbitt P. C. A mapping of drug space from the viewpoint of small molecule metabolism. PLoS Comput. Biol. 2009, 58e1000474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrant J. D.; Amaro R. E.; Xie L.; Urbaniak M. D.; Ferguson M. A.; Haapalainen A.; Chen Z.; Di Guilmi A. M.; Wunder F.; Bourne P. E.; McCammon J. A. A multidimensional strategy to detect polypharmacological targets in the absence of structural and sequence homology. PLoS Comput. Biol. 2010, 61e1000648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon Z.; Peragovics A.; Vigh-Smeller M.; Csukly G.; Tombor L.; Yang Z.; Zahoranszky-Kohalmi G.; Vegner L.; Jelinek B.; Hari P.; Hetenyi C.; Bitter I.; Czobor P.; Malnasi-Csizmadia A. Drug effect prediction by polypharmacology-based interaction profiling. J. Chem. Inf. Model. 2012, 521134–45. [DOI] [PubMed] [Google Scholar]
- Cheng F.; Zhou Y.; Li W.; Liu G.; Tang Y. Prediction of chemical-protein interactions network with weighted network-based inference method. PLoS One 2012, 77e41064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glick M.; Davies J. W.; Jenkins J. L. Prediction of biological targets for compounds using multiple-category Bayesian models trained on chemogenomics databases. J. Chem. Inf. Model. 2006, 4631124–33. [DOI] [PubMed] [Google Scholar]
- Keiser M. J.; Roth B. L.; Armbruster B. N.; Ernsberger P.; Irwin J. J.; Shoichet B. K. Relating protein pharmacology by ligand chemistry. Nat. Biotechnol. 2007, 252197–206. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L. Network pharmacology. Nat. Biotechnol. 2007, 25101110–1111. [DOI] [PubMed] [Google Scholar]
- Davis A. P.; Murphy C. G.; Rosenstein M. C.; Wiegers T. C.; Mattingly C. J. The Comparative Toxicogenomics Database facilitates identification and understanding of chemical-gene-disease associations: arsenic as a case study. BMC Med. Genomics 2008, 1, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanishi Y.; Araki M.; Gutteridge A.; Honda W.; Kanehisa M. Prediction of drug-target interaction networks from the integration of chemical and genomic spaces. Bioinformatics 2008, 2413i232–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschman L.; Burns G. A.; Krallinger M.; Arighi C.; Cohen K. B.; Valencia A.; Wu C. H.; Chatr-Aryamontri A.; Dowell K. G.; Huala E.; Lourenco A.; Nash R.; Veuthey A. L.; Wiegers T.; Winter A. G. Text mining for the biocuration workflow. Database (Oxford) 2012, 2012, bas020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krallinger M.; Leitner F.; Vazquez M.; Salgado D.; Marcelle C.; Tyers M.; Valencia A.; Chatr-aryamontri A. How to link ontologies and protein-protein interactions to literature: text-mining approaches and the BioCreative experience. Database (Oxford) 2012, 2012, bas017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowell K. G.; McAndrews-Hill M. S.; Hill D. P.; Drabkin H. J.; Blake J. A. Integrating text mining into the MGI biocuration workflow. Database (Oxford) 2009, 2009, bap019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegers T. C.; Davis A. P.; Cohen K. B.; Hirschman L.; Mattingly C. J. Text mining and manual curation of chemical-gene-disease networks for the comparative toxicogenomics database (CTD). BMC Bioinf. 2009, 10, 326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinter S. Z.; Liang Y.; Huang S. Y.; Hyder S. M.; Zou X. An inverse docking approach for identifying new potential anti-cancer targets. J. Mol. Graphics Modell. 2011, 296795–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui-fang L.; Qing S.; Jian Z.; Wei F. Evaluation of various inverse docking schemes in multiple targets identification. J. Mol. Graphics Modell. 2010, 293326–30. [DOI] [PubMed] [Google Scholar]
- Chen Y. Z.; Zhi D. G. Ligand-protein inverse docking and its potential use in the computer search of protein targets of a small molecule. Proteins 2001, 432217–26. [DOI] [PubMed] [Google Scholar]
- Kolb P.; Ferreira R. S.; Irwin J. J.; Shoichet B. K. Docking and chemoinformatic screens for new ligands and targets. Curr. Opin. Biotechnol. 2009, 204429–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boran A. D. W.; Iyengar R. Systems approaches to polypharmacology and drug discovery. Curr. Opin. Drug Discovery Dev. 2010, 133297–309. [PMC free article] [PubMed] [Google Scholar]
- Boran A. D. W.; Iyengar R. Systems pharmacology. Mt. Sinai J. Med. 2010, 774333–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S.; Iyengar R. Systems pharmacology: network analysis to identify multiscale mechanisms of drug action. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 505–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dar A. C.; Das T. K.; Shokat K. M.; Cagan R. L. Chemical genetic discovery of targets and anti-targets for cancer polypharmacology. Nature 2012, 486740180–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn M.; von Mering C.; Campillos M.; Jensen L. J.; Bork P. STITCH: interaction networks of chemicals and proteins. Nucleic Acids Res. 2008, 36Database issueD684–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olah M.; Mracec M.; Ostopovici L.; Rad R.; Bora A.; Hadaruga N.; Olah I.; Banda M.; Simon Z.; Mracec M.; Oprea T. I.. WOMBAT: World of molecular bioactivity. In Chemoinformatics in Drug Discovery; Oprea T. I., Ed.; Wiley-VCH: New York, 2004; pp 223–239. [Google Scholar]
- Liu T.; Lin Y.; Wen X.; Jorissen R. N.; Gilson M. K. BindingDB: a web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res. 2007, 35Database issueD198–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson M. L.; Smith R. D.; Khazanov N. A.; Dimcheff B.; Beaver J.; Dresslar P.; Nerothin J.; Carlson H. A. Binding MOAD, a high-quality protein-ligand database. Nucleic Acids Res. 2008, 36Database issueD674–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid A.; Blank L. M. Hypothesis-driven omics integration. Nat. Chem. Biol. 2010, 67485–487. [DOI] [PubMed] [Google Scholar]
- Joyce A. R.; Palsson B. O. The model organism as a system: integrating ’omics’ data sets. Nat. Rev. Mol. Cell Biol. 2006, 73198–210. [DOI] [PubMed] [Google Scholar]
- Hecker N.; Ahmed J.; von Eichborn J.; Dunkel M.; Macha K.; Eckert A.; Gilson M. K.; Bourne P. E.; Preissner R. SuperTarget goes quantitative: update on drug-target interactions. Nucleic Acids Res. 2012, 40Database issueD1113–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharman J. L.; Mpamhanga C. P.; Spedding M.; Germain P.; Staels B.; Dacquet C.; Laudet V.; Harmar A. J. IUPHAR-DB: new receptors and tools for easy searching and visualization of pharmacological data. Nucleic Acids Res. 2011, 39Database issueD534–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. L.; Xiao J. W.; Suzek T. O.; Zhang J.; Wang J. Y.; Zhou Z. G.; Han L. Y.; Karapetyan K.; Dracheva S.; Shoemaker B. A.; Bolton E.; Gindulyte A.; Bryant S. H. PubChem’s BioAssay Database. Nucleic Acids Res. 2012, 40D1D400–D412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaulton A.; Bellis L. J.; Bento A. P.; Chambers J.; Davies M.; Hersey A.; Light Y.; McGlinchey S.; Michalovich D.; Al-Lazikani B.; Overington J. P. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40Database issueD1100–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin J. J.; Shoichet B. K. ZINC--a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 451177–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler D. L.; Barrett T.; Benson D. A.; Bryant S. H.; Canese K.; Chetvernin V.; Church D. M.; Dicuccio M.; Edgar R.; Federhen S.; Feolo M.; Geer L. Y.; Helmberg W.; Kapustin Y.; Khovayko O.; Landsman D.; Lipman D. J.; Madden T. L.; Maglott D. R.; Miller V.; Ostell J.; Pruitt K. D.; Schuler G. D.; Shumway M.; Sequeira E.; Sherry S. T.; Sirotkin K.; Souvorov A.; Starchenko G.; Tatusov R. L.; Tatusova T. A.; Wagner L.; Yaschenko E. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2008, 36Database issueD13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z.; Chen L.; Maddula H.; Akcan O.; Oughtred R.; Berman H. M.; Westbrook J. Ligand Depot: a data warehouse for ligands bound to macromolecules. Bioinformatics 2004, 20132153–5. [DOI] [PubMed] [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. The Protein Data Bank. Nucleic Acids Res. 2000, 281235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium U. Reorganizing the protein space at the Universal Protein Resource (UniProt). Nucleic Acids Res. 2012, 40Database issueD71–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R.; Fang X.; Lu Y.; Wang S. The PDBbind database: collection of binding affinities for protein-ligand complexes with known three-dimensional structures. J. Med. Chem. 2004, 47122977–80. [DOI] [PubMed] [Google Scholar]
- Lipinski C.; Hopkins A. Navigating chemical space for biology and medicine. Nature 2004, 4327019855–61. [DOI] [PubMed] [Google Scholar]
- Edgar R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26192460–1. [DOI] [PubMed] [Google Scholar]
- Willard L.; Ranjan A.; Zhang H.; Monzavi H.; Boyko R. F.; Sykes B. D.; Wishart D. S. VADAR: a web server for quantitative evaluation of protein structure quality. Nucleic Acids Res. 2003, 31133316–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ertl P. Molecular structure input on the web. J. Cheminf. 2010, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haider N. Functionality pattern matching as an efficient complementary structure/reaction search tool: an open-source approach. Molecules 2010, 1585079–5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herraez A. Biomolecules in the computer - Jmol to the rescue. Biochem. Mol. Biol. Educ. 2006, 344255–261. [DOI] [PubMed] [Google Scholar]
- Ellson J.; Gansner E. R.; Koutsofios E.; North S. C.; Woodhull G. Graphviz and dynagraph - Static and dynamic graph drawing tools. Graph Drawing Software 2004, 127–148. [Google Scholar]
- Bryant S. H. PubChem. Abstr. Pap.–Am. Chem. Soc. 2005, 230, U1008–U1009. [Google Scholar]
- Dunbar J. B. Jr.; Smith R. D.; Damm-Ganamet K. L.; Ahmed A.; Esposito E. X.; Delproposto J.; Chinnaswamy K.; Kang Y. N.; Kubish G.; Gestwicki J. E.; Stuckey J. A.; Carlson H. A. CSAR data set release 2012: ligands, affinities, complexes, and docking decoys. J. Chem. Inf. Model. 2013, 5381842–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Achenbach J.; Tiikkainen P.; Franke L.; Proschak E. Computational tools for polypharmacology and repurposing. Future Med. Chem. 2011, 38961–8. [DOI] [PubMed] [Google Scholar]
- Allison M. NCATS launches drug repurposing program. Nat. Biotechnol. 2012, 307571–2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.