

Abstract
The cannabinoid receptor 2 (CB2) plays an important role in the immune system. Although a few of GPCRs crystallographic structures have been reported, it is still challenging to obtain functional transmembrane proteins and high resolution X-ray crystal structures, such as for the CB2 receptor. In the present work, we used 10 reported crystal structures of GPCRs which had high sequence identities with CB2 to construct homology-based comparative CB2 models. We applied these 10 models to perform a prescreen by using a training set consisting of 20 CB2 active compounds and 980 compounds randomly selected from the National Cancer Institute (NCI) database. We then utilized the known 170 cannabinoid receptor 1 (CB1) or CB2 selective compounds for further validation. Based on the docking results, we selected one CB2 model (constructed by β1AR) that was most consistent with the known experimental data, revealing that the defined binding pocket in our CB2 model was well-correlated with the training and testing data studies. Importantly, we identified a potential allosteric binding pocket adjacent to the orthosteric ligand-binding site, which is similar to the reported allosteric pocket for sodium ion Na+ in the A2AAR and the δ-opioid receptor. Our studies in correlation of our data with others suggested that sodium may reduce the binding affinities of endogenous agonists or its analogs to CB2. We performed a series of docking studies to compare the important residues in the binding pockets of CB2 with CB1, including antagonist, agonist, and our CB2 neutral compound (neutral antagonist) XIE35-1001. Then, we carried out 50 ns molecular dynamics (MD) simulations for the CB2 docked with SR144528 and CP55940, respectively. We found that the conformational changes of CB2 upon antagonist/agonist binding were congruent with recent reports of those for other GPCRs. Based on these results, we further examined one known residue, Val1133.32, and predicted two new residues, Phe183 in ECL2 and Phe2817.35, that were important for SR144528 and CP55940 binding to CB2. We then performed site-directed mutation experimental study for these residues and validated the predictions by radiometric binding affinity assay.
Introduction
G protein coupled receptors (GPCRs), the largest family of trans-membrane proteins in the human genome, are crucial for many essential physiological processes, including cellular metabolism, immune defense, neurotransmission, cell growth, secretion, and differentiation. It is also known that GPCRs are targeted by 40%–50% of marketed drugs worldwide.1
Cannabinoid receptors2,3 (CB) belong to the members of Rhodopsin-like GPCRs family. Three major groups of ligands can activate the cannabinoid receptors, including endocannabinoids, plant cannabinoids, and synthetic cannabinoids. There are mainly two known subtypes of CB receptors reported, including cannabinoid receptor 1 or CB14 and cannabinoid receptor 2 or CB2,5 which were characterized and cloned in 1990 and 1993, respectively. CB1 can be found to express mainly in the brain, although, it is also found to express in other tissues, including lungs, liver, and kidneys. CB1 plays a fundamental role in the central nervous system (CNS), which has been reported to mitigate numerous pathologies, including Alzheimer’s disease, pain, obesity, and cancer.6 CB2 is predominantly expressed in the peripheral areas of the body, especially in the immune and skeletal systems,7 and it is an important target for the treatment of autoimmune,8 inflamatory neuropathic pain,9 osteoporosis,10 and immune system cancer.11,12
Through Gi/Goα subunits, CB2 and CB1 receptors inhibit the activity of adenylyl cyclase. Moreover, CB2 are also reported to be coupled to the MAPK-ERK pathway13 through their Gβγ subunits. Until now, there are five recognized endocannabinoids, including 2-arachidonoyl glycerol (2-AG), arachidonoylethanolamine (anandamide), virodhamine,14 2-arachidonyl glyceryl ether (noladin ether), and the recently discovered N-arachidonoyl-dopamine (NADA).15 Many of these five ligands preferentially exhibit at the CB2 receptor. 2-Arachidonoyl glycerol (2-AG) selectively activates the pathway of MAPK-ERK. Noladin, as well as another synthetic ligand CP55940, appears to inhibit the activity of adenylyl cyclase through CB2 mediated signaling pathways.14
Many molecular dynamics processes and the underlying mechanisms of CB2 remain unknown due to the lack of high resolution crystal structure. Several research groups tried to construct the 3D model of CB2: Reggio and co-workers16 first reported the 3D homology model for the CB2 in 1999. Their model was based on the α-helical periodicity in the CB2 sequence as detected by Fourier Transform Analysis.17 Gouldson and co-workers18 generated one 3D CB2 model based on their previous rat β2-adrenoceptor model. They used SR144528, a selective CB2 antagonist (inverse agonist), to dock into their model. They also defined a docking position that agreed with the mutation studies. From their model, they observed hydrogen bonds between SR144528 and two residues, including Ser1614.53 and Ser1654.57. In 2000, Palczewski and co-workers19 elucidated the first Class A GPCRs’ crystal structure-bovine rhodopsin.20 Treating this as the starting point, our group21 constructed a comparative 3D CB2 model in 2003. Our model was supported by the results from NMR, X-ray crystallographic studies, and site-directed mutagenesis. For example, a salt bridge was formed between Arg1313.50 and Asp2406.30, an interaction proposed to serve as an ionic lock of rhodopsin-like GPCR activation.22 In 2006, Tuccinardi et al.,23 Stern et al.,24 and Raduner et al.25 generated other rhodopsin-based comparative models of CB2. Their models were generated using the crystal structure of bovine rhodopsin determined at 2.8 Å as a template, which was the only resolved G-protein coupled receptor at that time. Their models represented the inactivated state. Yang et al.26 and Latek et al.27 generated newer 3D CB2 models based on A2AAR in 2012 and 2011, respectively. Specially, a disulfide bond (between residue 174 and residue 179) located in the ECL2 was constructed according to the experiment data. They found that A2AAR had ∼23% sequence identity with CB2, and A2AAR had an entire ECL2 in its crystal structure. All these models above were used for analysis of CB2 ligand binding properties and to explain the effects of individual biological or pharmacological experiments. However, these models may be limited to the few crystal structures available at the time and low sequence identities of their templates, for example, bovine rhodopsin (∼16% sequence identity) and human A2AAR (∼23% sequence identity).
In recent years, significant progress has been made to stabilize GPCRs in order to determine their 3D structure by X-ray crystallography. Since the first crystal structure of GPCR-bovine rhodopsin was reported in 2000, at least 24 crystal structures of GPCRs have been published in the last ten years. By modeling all of the published crystal structures of GPCRs, we found that for most residues in TMs, for example, residue 3.32, its 3D coordinates are almost the same in all GPCRs. The dynamic properties of ligand-induced ionic lock and rotamer toggle molecular switch in the CB2 structural model were also found in congruent with the reported important GPCRs structural functions.28
In the present work, we first built comparative CB2 models based on the structural similar GPCRs. We selected one CB2 model that was supported by the known experimental data for further studies. We found a potential allosteric binding pocket in our CB2 model, which was very similar to the reported allosteric pocket for Na+ in two other GPCRs, e.g. A2AAR and the δ-opioid receptor. The results suggest that sodium may reduce the affinities of endogenous agonists or their analogs. Moreover, we docked CB2 selective antagonist SR144528 for analyzing the similarities and differences of the binding pockets between CB1 and CB2. The dockings of our in-house antagonists and our CB2 neutral compound (neutral antagonist) XIE35-1001 supported the results of SR144528. We also docked the agonist CP55940 into the CB2 model to compare the binding modes between antagonist and agonist. Then we performed 50 ns molecular dynamics (MD) simulations for SR144528 and for CP55940. We compared the conformational changes of CB2 and antagonist/agonist, which was congruent with recent reports for GPCRs. Based on the results from MD simulation and docking studies, we validated one important residue and predicted two residues that may play important roles for SR144528 and CP55940, i.e., Val1133.32, Phe183, and Phe2817.35, respectively. The importance of these residues for ligand binding was further confirmed by site directed mutagenesis and receptor binding assays.
Materials and Methods
Preparation of Crystal Structure of GPCR Proteins
There is no available crystal structure for the CB2 receptor (only one NMR structure for helix VI29). In the present work, we used 10 known GPCRs crystal structures (including Class A and Class B) with different sequence identity to construct the 3D structures of the cannabinoid CB2 receptor. All these 10 GPCRs crystal structures19,30−38 are listed in Table 1.
Table 1. Information of 10 Crystal Structures of GPCRs Used in the Present Worka.
| SMO | bovine rhodopsin | CXCR4 | M2MAR | D3R | β2AR | A2AAR | H1R | S1P | β1AR | |
|---|---|---|---|---|---|---|---|---|---|---|
| PDB entry | 4JKV | 1F88 | 3ODU | 3UON | 3PBL | 2RH1 | 2YDO | 3RZE | 3V2W | 2Y00 |
| resolution (Å) | 2.45 | 2.40 | 2.50 | 3.00 | 2.89 | 2.40 | 3.00 | 3.10 | 3.35 | 2.50 |
| whole sequence identity (±2%) | 14.4 | 17.4 | 19.1 | 20.9 | 23.5 | 24.4 | 25.1 | 25.2 | 28.5 | 28.5 |
| sequence identity in TM (±2%) | 18 | 25 | 21 | 27 | 28 | 28 | 29 | 30 | 34 | 35 |
| proSA-web Z-score | –2.80 | –2.82 | –3.54 | –3.24 | –2.72 | –4.21 | –4.87 | –3.36 | –3.49 | –4.33 |
| ref | (35) | (19) | (37) | (31) | (38) | (30) | (33) | (34) | (32) | (36) |
We listed the information on 10 GPCRs used in the present work, including PDB entry, resolution, sequence identity to CB2, and the related references.
The crystal structures were obtained from the Protein Data Bank (http://www.pdb.org/pdb/). We then prepared these structures using SYBYL-X 1.339 (including repair of residues and minimization of energy). In the present work, the residues in GPCRs are numbered according to the Ballesteros-Weinstin numbering scheme17 in which x.50 denotes the most conserved residue for a particular TM helix.
Homology Modeling
The full sequence of the human CB2 receptor (CNR2_HUMAN or P34972, 360 residues) was obtained from the Web site of UniProtKB/Swiss-Prot (http://www.uniprot.org/uniprot/).
In the present work, we truncated the residues before Ala351.34 for the N terminal and residues after Cys3137.67 for the C terminal. Moreover, the third intracellular loop (ICL3, between TM5 and TM6) had a long-flexible sequence, and we truncated 8 residues from Gln227 (ICL3) to Met234 (ICL3). Therefore, the sequence was from Ala351.34 to His226 (ICL3) and Ala235 to Cys3137.67. We then connected the residues including His226 (ICL3) and Ala235 (ICL3) to generate a continuous polypeptide sequence. The sequence alignments and homology modeling were based on this generated sequence of CB2 using reported protocol.21
Our procedure was chosen for the following three reasons. First, in all crystal structures of GPCRs, ICL3 missed some residues. Second, it had no effect on the binding pocket. Third, it did not make our water–lipid box too big for the MD simulation.
Sequence alignments of CB2 and other known crystal structure of GPCRs revealed a moderate sequence identity between CB2 and these GPCRs, ranging from 14.4% to 28.5% (Table 1). We also compared the sequence identities among the TM regions, as shown in Table 1. We patched a disulfide bridge/bond between Cys1744.66 and Cys1794.71 in ECL2, which was suggested to be present in CB2 according to mutagenesis studies by Gouldson and co-workers.18 After aligning the sequence of CB2 with the templates, we then manually adjusted the alignments from ECL2 to TM5 according to the numbers of residues. For example, we adjusted Ser1935.42 of CB2 to align with the residues-5.42 in other GPCRs. The same operations were performed for other TM5 residues. Moreover, we also made sure that other TMs alignments were reasonable, for which we used conserved motifs in GPCRs. (Examples are “D/ERY” in TM3, “CWxPx″ (or “D/E″6.30) in TM6, and “NPxxY” in TM7.) Last, we checked the alignments of sequences for TMs and made sure that there were no “gaps” in the helical sequences, which were automatically generated by the software upon aligning the TM segment. The detail alignments of sequences between CB2 and 10 templates were shown in Figure S1b. We used all these ten crystal structures of GPCRs to build the CB2 model using Modeller9.12.40
Energy Minimization and Structure Validation
Once the 3D models were generated, energy minimizations were carried out by using SYBYL-X 1.3.38 The parameters defined in the SYBYL were as follows: Gradient was set to 0.5 kcal/mol, Max iterations were set to 5000, force field was set to MMFF94s, and the charges were set to MMFF94.
The CB2 models were also validated by using proSA-web Z-scores41 and PROCHECK Ramachandran plots.42 We also calculated the root mean squared deviation (rmsd), superimposed our model with the templates.43
Conformational Sampling and Secondary Energy Minimization for the CB2 Model
To select the most reasonable conformation of CB2 models, we first performed 10 ns molecular dynamics (MD) simulations for the CB2 candidate with explicit water and lipid. The detailed protocols were described in the section of “Molecular Dynamics Simulation”. During 10 ns MD simulation, we fixed the Cα atoms of seven transmembrane domains of CB2 and made the side chains flexible. After 10 ns MD simulation, we made the protein flexible and performed another 5 ns MD simulation. We selected five conformations with the lowest energy to perform secondary energy minimization by SYBYL, which were described in the previous section (“Energy Minimization and Structure Validation”).
Training Data Set for Model Validation
We first filtered the National Cancer Institute (NCI) Database (NCI2011), eliminating mixtures or metal or isotopes and the compounds with unfavorable molecular weights (lower than 250, higher than 600). Out of the 210,000 remaining compounds, 980 were randomly chosen. The three-dimensional coordinates of these compounds were generated by using SYBYL. Seventeen of our in-house active compounds26,44 and three other active compounds (as shown in Figure S2 in the Supporting Information) were prepared using the same procedure. Special caution was applied to the protonation state of ionizable groups (amines, amidines, carboylic acids) of all 1000 ligands assumed to be ionized at a physiological pH of 7.4. The docked conformations of these 1000 compounds were generated by the the Surflex-Dock program in SYBYL.
Selectivity Training Data Set for Further Validation of the CB2 Model
In our recent publication,44 we retrieved 703 selective compounds of CB1 and CB2 from the public cannabinoid ligand database (http://www.cbligand.org). In the present work, we selected 100 CB2 selective compounds (Ki for CB2 lower than 911 nM) and 70 CB1 selective compounds (all 70 compounds’ Ki for CB1 lower than 555 nM except one with 1120 nM; 24 compounds were with Ki lower than 958 nM for CB2, and another 46 compounds were with Ki higher than 3000 nM for CB2) for validating our binding pocket, which had a high ratio of Ki CB1: Ki CB2. In the present work, all the selected structures did not differ significantly in physicochemical properties.
Molecular Docking for the Study of Ligand/CB Receptors Interaction
We then performed a series of docking experiments for the validation of the CB2 model, comparisons of binding pocket/ligand binding modes, and identification of new binding residues for CB2 ligands. The docking program Surflex-Dock GeomX (SFXC) in SYBYL-X 1.338 was applied to constructed receptor–ligand complexes, in which the Total Score was expressed in −log10(Kd).45
We used the MOLCAD module implemented in SYBYL-X 1.3 to explore the potential binding pocket for our models. The main protocols or parameters of docking can be found in our previous publication.46
Molecular Dynamics Simulation
Following docking, we selected the complexes of CB2 bound with agonist/antagonist to carry out the molecular dynamics (MD) simulations.
Special caution was applied to His residues, because His was ionized at pH 7.40. VEGA ZZ 2.4.047 and PROPKA 3.148 were applied to predict the pK values of His and other residues.
In the CB2 model, all histidines were not protonated, because the calculated pK values ranged from 4.62 to 6.90 (<7.40). Several residues including Asp–, Arg+, Glu–, and Lys+ were charged in our simulations.
The VMD49 program was used to embed the complexes of receptors with ligands into a periodic and pre-equilibrated structure of 1-palmytoyl-2-oleoyl-sn-glycero-3-phosphatidylcholine (POPC). Lipid molecules within 3 Å of the receptor were eliminated. We then inserted these into a water box (TIP3P50 water model) with eliminating the water molecules within 3 Å of the receptor.
The simulation boxes (CB2-SR144528/CB2-CP55940, respectively) contained the CB2 model, 164/165 lipid molecules, 4967/4959 water molecules, 0/0 sodium ions, and 9/9 chloride ions, so the total numbers of per periodic cell were 34313/34295 atoms. The box sizes (Figure S3 in the Supporting Information) were 67 × 67 × 74 Å3/67 × 67 × 74 Å3. Two minimizations were carried out; each minimization was carried out with 50,000 steps. The first minimization was performed with the fixed protein, while the second minimization was carried out with the flexible protein.
Starting from the last frame of the minimization, 50 ns molecular dynamics simulations were carried out. The NAMD package51 (version 2.9b1) with the CHARMM2752,53 force field was applied for the MD simulation. The Particle Mesh Ewald54 (PME) method (with a 12 Å nonbonded cutoff and a grid spacing of 1 Å per grid point in each dimension) was used to calculate the electrostatics. A smooth cutoff (switching radius 10 Å, cutoff radius 12 Å) was used to calculate the van der Waals energies. The temperature and pressure were kept constant using a Langevin thermostat (310 K) and Langevin barostat (1 atm), respectively. The time step of the molecular dynamics (MD) simulation was set to 1 fs (fs). For analysis, we saved the data every 10 picoseconds (ps). VMD software was applied to analyze the trajectory.
Mutagenesis Experiments
Site-directed mutagenesis experiments for different CB2 amino acids mutations were obtained by a site-directed mutagenesis kit according to the manufacturer’s protocol of QuikChange (Stratagene, CA). Briefly, a single amino acid mutation was performed individually to the human CB2 plasmid pcDNA3.1 + 3 × HA-hCB2 (Missouri S&T cDNA Resources Center). Three PCR steps were introduced for mutation of selected amino acid (Val1133.32, Phe183 in ECL2, and Phe2817.35), which can be found in our previous publication.55
Cell Harvesting and Membrane Preparation
The high confluent culture plates were kept on ice prior to harvesting. The plates were rinsed twice with cold (PBS). Then the cells were collected by scraping and centrifuging at 500g for 5 min at 4 °C. The cell pellets were resuspended in 5 mL of membrane preparation buffer (50 mM Tris–HCl pH 7.4, 5 mM MgCl2, 2.5 mM EGTA, and 200 mM sucrose) and homogenized with a Polytron PT1600E Homogenizer (Kinematica, Littau-Lucerne, Switzerland). This step was repeated for three time before the final centrifuge. All supernatants were combined and centrifuged at 68,000g for 90 min at 4 °C. Pellets were then collected and resuspended in membrane preparation buffer for competition binding assays.
Competition Binding Assay
The protein concentration was measured using Pierce BCA Protein Assay (Rockford, IL). Two structurally distinct, representative cannabinoid ligands and amiloride were used in this study. CP55940 (CB agonist) and SR144528 (antagonist/inverse agonist) were obtained from RTI International (Research Triangle Park, NC), while amiloride was obtained from Alfa Aesar. The ligand binding was performed:56 nonradioactive (or cold) ligands were diluted in binding buffer (50 mM Tris-HCl, 5 mM MgCl2, 2.5 mM EGTA, 0.1% (w/v) fatty acid free BSA, pH 7.4), supplemented with 0.4% methyl cellulose and 10% DMSO. Each assay plate well contained a total volume of 200 μL of a mixture of 5 μg of membrane protein, 3 nM of labeled [3H]-CP-55940 (RTI International Research Triangle Park, NC), and concentrations of three unlabeled agonists.55
Plates were incubated at 30 °C for 1 h and then harvested to PerkinElmer 96 well GF/B filter plates using PerkinElmer Filter Mate Harvester (PerkinElmer, NL). GF/B plates were allowed to dry overnight, soaked in liquid microscinct liquid, and read in a PerkinElmer top count reader (PerkinElmer, NL). Binding data were normalized to define 0–100% and analyzed in Graph Pad Prism 5.0 software using one point receptor competitive displacement curve fitting. All assays were performed in duplicate wells, and data were represented as the mean ± SEM. Bound radioactivity data was analyzed for Ki values using the method described in the previous publication.55
Results
Ten CB2 Models
We constructed 10 CB2 models according to ten crystal structures of GPCRs. As shown in Figure S4a, in all our homology models of CB2, residues in 7 TMs constructed by structures of Class A GPCRs were very similar to each other, even the CB2 model constructed by SIP. The main reason was that we adjusted and aligned TMs’ residues/sequences, which helped us to make the alignments more reasonable.
However, residues in the second extracellular loop (ECL2) still seemed different, as the sequences of ECL2 in GPCRs were not conserved. Three CB2 models seemed to be very similar to each other, including the structure of ECL2. These models were constructed by D3R, β1AR, and β2AR. The similarity was obvious when we aligned these three crystal structures, as shown in Figures S5a and S5b. D3R, β1AR, and β2AR shared large similarities. Sequence identities for these three structures were ∼34.4 ± 2% between D3R and β2AR, ∼35.4 ± 2% between D3R and β1AR, and ∼61.9 ± 2% between β1AR and β2AR. Moreover, many key residues involved in the binding pocket were the same. The positions of side chains were almost the same. Examples are Asp3.32 in TM3, Ser5.42, Ser5.43, Ser5.46 in TM5, Trp6.48 in TM6, and Ser7.43 in TM7. The ECL2 superposed very well. Moreover, the detailed Cα rmsd of the TM regions among these 10 models were listed in Table T1.
Conformational Sampling for CB2 Models
For each constructed CB2 model, we first performed 10 ns MD simulations with the fixing backbone, and then we performed 5 ns MD simulations with a flexible protein for sampling conformations.
We selected 5 conformations with the lowest energy during the 5 ns MD simulations with a flexible protein; we then used SYBYL to perform the secondary energy minimization. Our results showed that the energies of five conformations for each model were very similar to each other after minimization (±35 kcal/mol), so we chose the conformation with the lowest energy as the best one for each CB2 model.
CB2 Ligand Data Set for Validating the 3D CB2 Model
Ten CB2 models were utilized to perform the prescreen against the 1000-compounds data set. We then calculated the hit rate (HR) at a given percentage of the data set. The relationship between the number of hit compounds identified and the number of compounds screened were depicted in Figure 1 and Figure S6, apparently, when x%, which was a given percentage of the data set screened, was set to 2% (top 20 compounds), 5% (top 50 compounds), and 10% (top 100 compounds).
Figure 1.
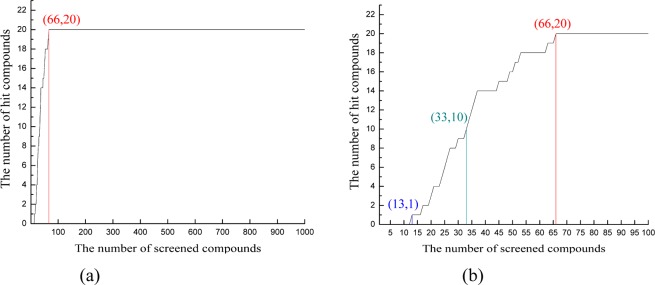
Correlation between the number of hit compounds identified and the number of compounds screened in (a) a 1000-compounds-data set and (b) a top-100-compounds-data set (top 10%).
Figure 1 and Figure S6 show the results of prescreening for all 10 CB2 models. Our results showed that most CB2 models can distinguish the active compounds from the inactive compounds. CB2 models constructed by β1AR and β2AR had the highest and similar hit rate among all CB2 models. Eight out of ten CB2 models had a 100% hit rate within 15% of the data set screened, except for the models constructed by bovine rhodopsin and A2aAR (had a 100% hit rate within 25% of the data set screened). More details can be found in Figure S6.
The CB2 model constructed by β1AR had the best prediction, as shown in Figures 1a and 1b. All 20 active compounds were detected in the top 66 compounds. The hit rates at 2%, 5%, and 10% of the data set screened were 10% (2 compounds), 80% (16 compounds), and 100% (20 compounds), respectively. It was not surprising that at the hit rate of 2% only 2 active compounds were detected, because we selected compounds from the NCI database randomly. Our data set contained some potential active compounds and flexible compounds that had high docking scores in any pockets.
The results from prescreening showed that the binding pockets of most CB models distinguished the active compounds from the inactive ones.46,57 Moreover, the CB2 model constructed by β1AR seemed more reasonable.
Docking CB Selective Ligands for Further Validation of Our CB2 Models
After prescreening, we used 100 CB2 selective compounds and 70 CB1 selective compounds for further validation. We selected five models with a high hit rate in prescreen docking for further validations, including the CB2 models constructed by β1AR, SIP, H1R, M2MAR, and SMO, which maintained structural diversities.
We chose the docking score (generated by SYBYL-X 1.3) greater than or equal to 7.0 as the cutoff value, which was suggested by SYBLY software.38 Figure 2 and Figure S7 show the predicting results of these five CB2 models. Our results showed that different models had different accuracy for predicting the CB compounds. The predictions of 100 CB2 selective compounds were 44%, 83%, 63%, 77%, and 92% for models constructed by SMO, M2MAR, H1R, S1P, and β1AR. However, the predictions of 70 CB1 selective compounds were 21.43%, 38.57%, 61.43%, 54.29%, and 70% for models constructed by SMO, M2MAR, H1R, S1P, and β1AR. More details can be found in Figure S7.
Figure 2.
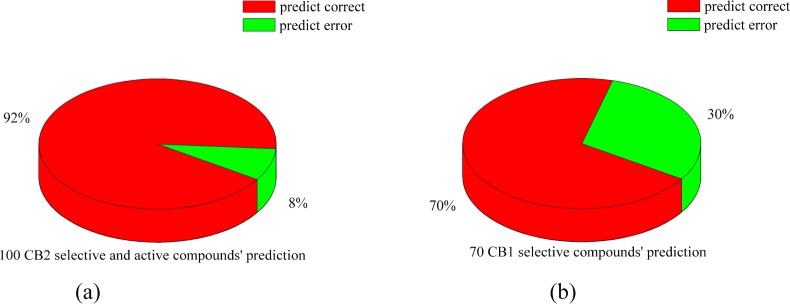
Pie charts showed the prediction for CB1 and CB2 selective compounds in the present work. (a) Pie chart showed that 92 out of 100 CB2 selective compounds predicted correctly (92%); (b) pie chart showed that 49 out of 70 CB1 selective compounds predicted correctly (70%).
The CB2 model constructed by β1AR had the best prediction: out of 170 compounds, 141 (82.9%) were predicted correctly, as shown in Figure 2a and Figure 2b. All the docking results of the CB2 model constructed by β1AR were shown in Tables T2 and T3 in the Supporting Information.
For 70 CB1 selective compounds, 49 compounds (70%) were predicted correctly, as shown in Figure 2b and Table T2. 12 out of 24 active compounds (50%) with Ki lower than 850 nM for CB2 were predicted correctly (docking scores were higher than 7.0). Moreover, 37 out of 46 CB1 selective compounds (80.4%) with Ki higher than 3000 nM for CB2 were also predicted correctly. Our model showed the moderate prediction for CB1-selective-but-CB2-active compounds and had a high prediction for CB1-selective-and-CB2-inactive compounds.
For 100 CB2 selective compounds, 92 compounds (92%) had docking scores higher than 7.0, as shown in Figure 2a and Table T3. The other 8 compounds had the docking scores ranging from 5.87 to 6.99. Only one compound had a docking score lower than 6.0 (docking score: 5.87). Scores of two compounds were higher than 6.90. This model can predict CB2 selective (active) compounds accurately. We tried to find the relationship between the predicted Kd and the Ki value, but there was no direct relationship between them.
By comparing the sequence identity, TM helices, conserved residues, similar motif, hydrogen-bond networks, 3D structures of ten CB2 models, docking results from prescreening, and CB selective compounds, we selected the 3D model of CB2 constructed by β1AR (shown in Figure 3) for further studies in the present work, based on the following reasons. (1) Overall model quality and local model quality of CB2 constructed by β1AR were more reasonable, while they were predicted by ProSA-web, as shown in Table 1 and Figure S8. (2) Ramachandran plots of CB2 constructed by β1AR were more reasonable, as shown in Figure S9. (3) β1AR had the highest sequence identity (28.5 ± 2% for sequence and more than 35% within the TMs regions) to CB2. The structure of β1AR was the fourth highest resolution of GPCRs’ templates in the present work, where the ECL2 in β1AR was also intact. (4) The best docking results from prescreening and from CB selective compounds. The 3D coordinates of our CB2 model based on β1AR can be found in the Supporting Information.
Figure 3.
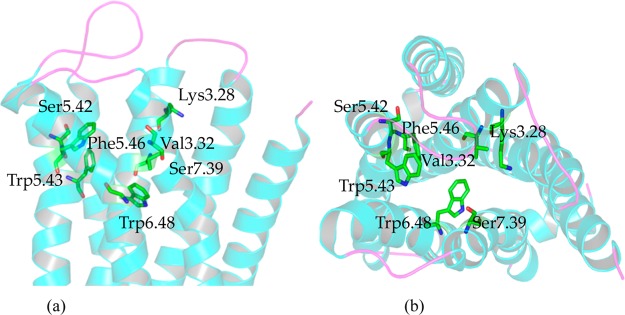
3D structure of CB2 constructed by β1AR after energy minimization (∼28.5 ± 2% sequence identity, PDB entry: 2Y00, resolution: 2.50 Å). Some important residues were highlighted in green sticks: Lys1093.28, Val1133.32, Ser1935.42, Trp1945.43, Phe1965.46, Trp2586.48, and Se285r7.39. (a) Side view from the membrane; (b) top view from the periplasmic side.
Important Residues in the Predicted Binding Pocket of CB2
MOLCAD module implemented in SYBYL-X 1.3 was used to explore the potential binding pocket in the present work.
As shown in Figure 4, the potential binding pocked of CB2 was mainly formed by helices III, V, VI, and VII. The binding pocket of the CB2 model was verified by recent docking results,58−60 which was in agreement with most other GPCRs. Moreover, we found that the binding pocket can be extended into helix IV via a narrow “crevice”. The cavity among TM3, TM4, and TM5 explored by MOLCAD was too narrow for compounds binding. However, some residues in this region showed important roles for antagonists (inverse agonists), which were similar to what Gouldson and co-workers18 found, that SR144528 formed a hydrogen bond with both Ser1614.53 and Ser1654.57.
Figure 4.
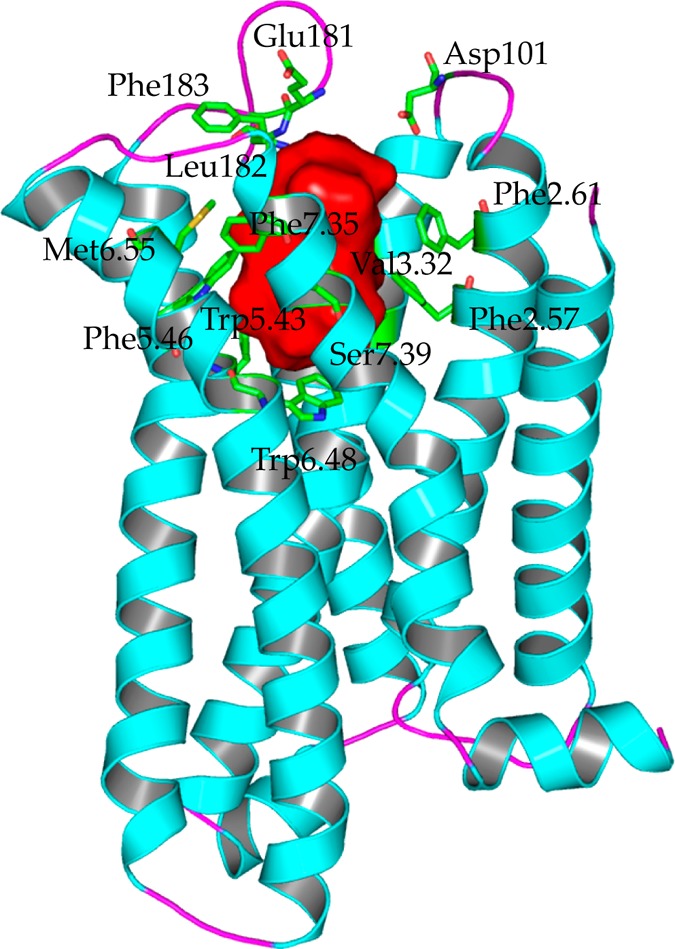
Potential binding pocket and residues involved in the binding pocket of CB2. The potential binding pocket was highlighted in red, which was mainly formed by helices III, V, VI, and VII, but also can extend into helix IV. Important residues involved in the binding pocket were highlighted in green sticks, which included the following: Phe872.57, Phe912.61, Phe942.64 (not shown), Asp101 (ECL1), Phe1063.25 (not shown), Lys1093.28 (not shown), Ile1103.29, Val1133.32, Phe1173.36 (not shown), Glu181 (ECL2), Leu182 (ECL2), Phe183 (ECL2), Trp1945.43, Phe1975.46, Trp2586.48, Val2616.51(not shown), Met2656.55, Phe2817.35, and Ser2857.39.
Moreover, we suggested that this binding pocket was for both agonist and antagonist (inverse agonists). Recently, some GPCRs receptors bound with agonist or bound with antagonist were reported. Figure S10 shows the alignments of agonist (highlighted in green) and antagonist (highlighted in blue) for β2AR and A2AAR, respectively. The binding pocket for agonist or antagonist was formed by helices III, V, VI, and VII both in β2AR and A2AAR. Comparing these crystal structures, many key residues involved in the binding pocket (β2AR or A2AAR) contributed to the recognitions of agonist and also antagonist. For example, for β2AR, residues including Asp3.32, Ser5.42, Ser5.43, Ser5.46, Trp6.48, and Tyr7.43 formed important interactions with agonist BI167107 and antagonist ICI 118,551. Similar results were observed in A2AAR, as shown in Figure S10b, so we concluded that CB2 shared the same binding pocket for agonist and antagonist.
Important residues involved in the binding pocket for CB2 were highlighted in green sticks, as shown in Figure 4, including Phe872.57, Phe912.61, Phe942.64 (not shown), Asp101 (ECL1), Phe1063.25 (not shown), Lys1093.28 (not shown), Ile1103.29, Val1133.32, Phe1173.36 (not shown), Glu181 (ECL2), Leu182 (ECL2), Phe183 (ECL2), Trp1945.43, Phe1975.46, Trp2586.48, Val2616.51 (not shown), Met2656.55, Lys2787.32 (not shown), Lys2797.33 (not shown), Phe2817.35, and Ser2857.39.
Most residues in the CB2 pocket we described were supported by the mutation data and docking data:
Recent docking reports59,61 showed that Phe872.57 and Phe942.64 in TM2 may play roles for the binding of CB2 compounds. Recently, Asp101 in ECL1 was also reported59,61 to play a role for CB2 ligands.
Conserved Lys1093.28 was shown to have little effect on known agonist binding or signaling of CB2, even though this particular lysine was crucial for the binding of HU-210 and CP55940 but not WIN55212-2 to the CB1 receptor.62 A double mutation K3.28AS3.31G resulted in a complete loss of affinity for other studied agonists, expected WIN55212-2 and JWH-015. However, downstream signaling by WIN55212-2 was drastically reduced, suggesting an improper coupling of this mutant.63 Moreover, three residues in this helix, including Ile1103.29, Val1133.32, and Phe1173.36, were reported to be involved in ligand binding:58,61 Comparing with the CB1 receptor, Ile1103.29 in CB2, involved in the binding pocket, may have a role in the selectivity for CB2. The corresponding residue in CB1 was Leu3.29. According to the known crystal structure of GPCRs, a residue in position 3.32 was crucial for the ligand recognition. We suggest that Val1133.32 also has an important role for CB2.
Receptor chimera studies by Shire et al.64 showed that ECL2 is important for the binding of CP55940 to CB2. Replacing ECL2 of CB2 with ECL2 of CB1 resulted in a loss of binding of CP55940. Furthermore, Glu181 (ECL2) and Leu182 (ECL2) are unique for CB2 when compared with CB1. These two residues therefore may have roles in the selectivity of CB2. Phe183 in ECL2 was involved in the binding pocket. We conclude that this residue may also have a role in binding some ligands by forming π–π interactions.
Trp1945.43 in TM5 was reported to have an important role in CB2 receptor ligand binding and adenylyl cyclase (AC) activity.65 Moreover, replacing Phe1975.46 with the corresponding Val5.46 of CB1 resulted in a 14-fold decrease of WIN55212-2 affinity to CB2 but had no effect on CP55940, HU-210, or AEA binding.16 An opposite replacement at CB1, V5.46F, enhanced the binding affinity of WIN55212-2. Those data supported the hypothesis16 that an aromatic residue at this position was crucial for the selectivity of WIN55212-2 for CB2.
Song et al.66 used the substituted-cysteine accessibility method (SCAM) to map the residues in the sixth membrane-spanning segment of CB2 that contributed to the surface of the water-accessible binding site crevice. They showed that Val2616.51, Met2656.55, and other residues faced into the CB2 binding pocket. Moreover, Trp2586.48 was highly conserved in GPCRs, supporting a proposal that its rotameric state had a role in CB2 activation.67 Val2616.51 also may have a role in the selectivity for CB2. The corresponding residue in CB1 was Leu6.51. Met2656.55 was also conserved and contributed into the binding for most GPCRs. These residues were reported to have roles in recognition of CB2 by the ligands.22
Ser2857.39 was part of the putative binding pocket in the present model. It had been suggested as a site of interaction61,68,69 for some agonists (for example, HU-243). Binding affinity of HU-243 somewhat decreased with the mutation S7.39A, while this mutation had no signal transduction of HU-210, CP55940, and WIN55212-2.70 However, Ser7.39 in CB1 recently was reported by Kapur and co-workers.71 as having a role in mediating ligand specific interactions for CP55940, HU-210, and AM4056.
Discussion
A Potential Allosteric Pocket at CB2
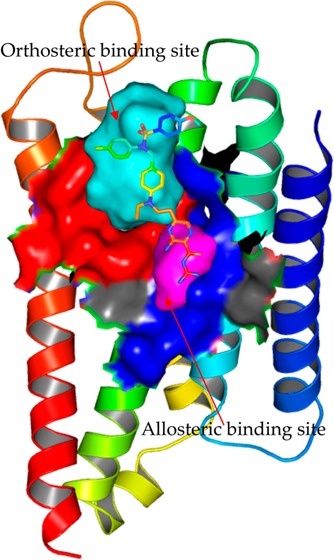
As shown in Figure 5a, beside the orthosteric ligand-binding pocket, we found a small binding pocket, which was closed to the orthosteric one. This allosteric pocket was formed by conserved residues in GPCRs, including Asn511.50, Val541.53, Leu762.46, Ala772.47, Ala792.49, Asp802.50, Ser1203.39, Leu1243.43, Trp2586.48, Asn2917.45, Ser2927.46, Asn2957.49, Asn2967.50, and Tyr2997.53. Moreover, a channel connecting these two pockets together can be observed in our model, as shown in Figure 5a. However, the side chain of Trp2586.48 made the channel between these two pockets narrow. This allosteric pocket seemed highly conserved among Class A of GPCRs.72 Recently, a similar small pocket was also discovered as an allosteric pocket for Na+ ions in A2AAR73 and the δ-opioid receptor.72 However, residue Leu2546.44 at CB2 is unique, while the corresponding residue for other GPCRs is Phe. Moreover, the residue in position 3.35 is also special at CB2 (Thr1163.35). These two residues may differ CB2 from other GPCRs for the bindings of modulators (or ions).
Figure 5.

Potential allosteric binding pocket in the CB2 model. (a) The orthosteric ligand-binding pocket (highlighted in red) and the allosteric pocket (highlighted in magenta), which was formed by Asn511.50, Val541.53, Leu762.46, Ala772.47, Ala792.49, Asp802.50, Ser1203.39, Leu1243.43, Trp2586.48, Asn2917.45, Ser2927.46, Asn2957.49, Asn2967.50, and Tyr2997.53; (b) [3H]CP55940 binding to CB2-WT transiently expressed on cell membrane of Chinese Hamster Ovary (CHO) in the presence of buffer (control) or buffer supplemented with different concentrations of NaCl, the figure represented data combined from two separate experiments performed in duplicate; (c) competition binding of amiloride at CB2 showed that amiloride did not bind to CB2; (d) the binding pose of amiloride in the allosteric binding pocket in the CB2 model.
Howard et al.74 provided evidence that NaCl competed with amiloride, suggesting that Na+ and amiloride shared an allosteric binding site. Liu and co-workers73 performed the binding assays to confirm the allosteric effects of Na+ and amiloride in A2AAR and the role of Na+ in the δ-opioid receptor.
In order to validate further the roles of sodium at CB2, we performed the experiment for the modulation of CB2 by sodium ions: Figure 5b shows [3H]CP55940 binding to CB2-WT transiently expressed on the cell membrane of Chinese Hamster Ovary (CHO) in the presence of buffer (control) or buffer supplemented with different concentrations of NaCl. Our results showed that Na+ did not have any effect on the binding of the CB2 agonist [3H]CP55940 (even with the concentration of 1000 mM), while CP55940 (a synthetic compound) is not an endogenous agonist. Similarly, sodium cannot inhibit the binding of agonist (−)-Bremazocine in the δ-opioid receptor72 (the Ki value was from 35 nM to 47 nM), which (−)-Bremazocine is not an endogenous agonist. However, for agonist [3H]NECA (5′-N-ethylcarboxamidoadenosine, an analog of adenosine) in A2AAR,73 the inhibiting rate (sodium was 150 mM) was up to 75% (only 25% of [3H]NECA binding was detected). Moreover, for peptide agonist [3H]DADLE ([d-Ala2, d-Leu5]-Enkephalin, an analog of Enkephalin) in the δ-opioid receptor,72 140 mM sodium reduced the affinity of this agonist, the Ki value from 280 nM (with 0 mM NaCl) to 2800 nM (with 140 mM NaCl).
Until now, there have been no endogenous ligands tested for the effect of sodium ions for CB2. More experiments on endogenous agonists for CB2 are being performed in our lab, including AEA (arachidonoyl ethanolamide) and 2-AG (2-arachidonoylglycerol). We hope these can help us learn more about the role of sodium at GPCRs. However, based on the current data, NECA and DADLE were related to endogenous agonists, while CP55940 and Bremazocine were not. We think that sodium may affect endogenous agonists or their analogs more than the chemical compounds.
We also explored the role of amiloride for CB2. Figure 5c shows the competition binding curve of amiloride at CB2. Our results showed that the Ki value of amiloride at CB2 is only 495 μM (the Ki value of amiloride with 150 mM NaCl at CB2 was 310 μM), indicating that amiloride has very low (or no) binding affinity at CB2. Our docking results supported this result, because the docking score is only ∼4.0 in both the orthosteric ligand-binding pocket and the allosteric pocket.
As shown in Figure 5d, we docked amiloride into the allosteric binding pocket of CB2. Our results showed only minor changes in the conformations of the surrounding side chain for the CB2 model. Furthermore, the charged guanidinium group of amiloride interacted tightly with Asp802.50 in the CB2 model. Asn2957.49 also formed a hydrogen bond with amiloride. The docking results of amiloride at CB2 shared large similarities with that from A2AAR.73
However, there were also some differences between CB2 and A2AAR. For A2AAR,73 Thr883.36 formed a hydrogen bond with the benzene ring of amiloride; but for CB2, the residue was Phe1173.36 in this same position, and Phe1173.36 formed a strong hydrophobic interaction with amiloride. Moreover, for CB2, our results showed that Ser1203.39 formed the hydrogen bond with amiloride, while there was no apparent hydrophilic interaction with Ser913.39 at A2AAR. Subtle but important differences of the allosteric binding pocket between CB2 and A2AAR caused the binding pose of amiloride to differ from that of A2AAR. Also, the distances of Trp2586.48 (5.1 Å), Asn2917.45 (5.3 Å), and amiloride from CB2 were greater than from A2AAR. All these differences helped us to understand the diversities of GPCRs.
Based on the high conservations of the residues involved in this allosteric pocket, we suggest that CB2 also has this potential allosteric pocket for Na+ ions. The sodium may affect the affinities of endogenous agonists or their analogs at CB2 more than the chemical compounds. The roles of other ions (including Ca2+ and Zn2+) and amiloride are still being explored. They may contribute to the further understanding of GPCRs.
This potential allosteric binding pocket will help us to develop new allosteric modulators for CB2 or other GPCRs. The new potential binding pocket formed by an orthosteric ligand-binding pocket and an allosteric binding pocket may help us to develop new drugs for GPCRs.
Comparisons of the Binding Pocket between CB1 and CB2
CB1 and CB2 belong to class A of a G-protein coupled receptor (GPCR). They share large similarities with each other, especially among TMs, which have more than 48% sequence identity throughout the whole protein and 68% identity within the trans-membrane regions.
Using the same methods, we constructed the homology model of CB1. Then we docked SR144528 into CB1 and CB2 for exploring the similarities and differences between their binding pockets. SR14452818 is a compound that acts as a potent and highly selective CB2 receptor inverse agonist, with a Ki of 0.6 nM to CB2 and 400 nM to the CB1 receptor.
We docked SR144528 into the model of CB2 and CB1. As shown in Figure 6, the docking scores (predicted Kd) of SR144528 were 7.21 in the CB1 receptor and 9.56 in the CB2 receptor, which agreed with the Ki values.
Figure 6.
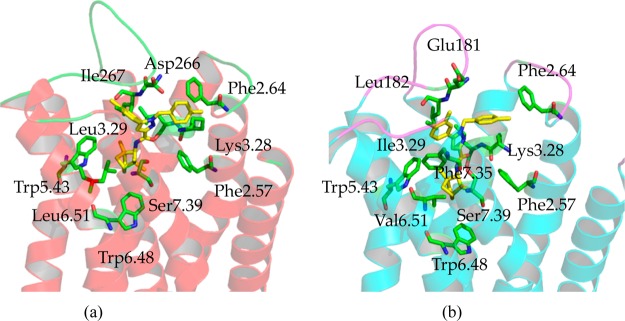
Comparing the differences and similarities of binding pockets between CB1 and CB2 by docking with SR144528. (a) The binding mode of SR144528 within the CB1 model; (b) the binding mode of SR144528 within the CB2 model. SR144528 is a drug that acts as a potent and highly selective CB2 receptor inverse agonist, with a Ki of 0.6 nM at CB2 and 400 nM at the CB1 receptor.
We had the similar binding mode between SR144528 and CB2 as that from Reggio’s group,59 which the group 2,2,4-trimethyl-3-bicyclo[2.2.1]heptanyl of SR144528 was on the top of the binding pocket (interacted with the ECL1 and extracellular side of TM1 and TM2). However, we chose the different binding mode in the present work for two reasons, which 2,2,4-trimethyl-3-bicyclo[2.2.1]heptanyl of SR144528 was on the bottom of the binding pocket (interacted with Trp1945.43 and Trp2586.48). First, three residues including Lys1133.28, Ser1163.31, and Ser2857.39 pointed to the middle part of the binding pocket. These residues can provide the polar environment for amide in SR144528; the role of these three corresponding residues71 in CB1 had been reported. Second, the binding mode of SR144528 at CB2 was supported by our other docking results, as shown in Figure S11 and Table T3: three other CB2 selective compounds sharing very similar scaffolds with SR144528 had almost the same binding modes with SR144528.
Many similarities can be found in the SR144528 binding pockets of CB1 and CB2. On the bottom of the binding pocket, SR144528 formed hydrophobic interactions with several important residues, including Phe3.36 (not shown in Figure 6), Val/Leu6.51, and Trp6.48. In the middle of the binding pocket, SR144528 formed hydrophobic interactions with Val3.32 (not shown in Figure 6), and it also formed a π–π interaction with Phe2.57 and Trp5.43. Moreover, the amide group of SR144528 faced the polar environment formed by Lys3.28, Ser/Gly3.31, and Ser7.39. On the top of the binding pocket, SR144528 formed a hydrophobic and a π–π interaction with Phe2.64 and Phe3.25 (not shown in Figure 6).
Subtle but important information was that the distance between Lys1133.28 in CB2 and SR144528 was more than 5.7 Å, implying that there was no hydrogen bond between them. However, there is a hydrogen bond (∼3.9 Å) between Lys3.28 in CB1 and SR144528, showing that Lys3.28 had a key role for CB1 ligands recognition. Moreover, Ser7.39 in CB1 also formed a hydrogen bond with SR144528, and the distance was 4.2 Å. The role of Ser7.39 in CB1 was recently reported by Kapur and co-workers.71 Furthermore, Leu3.29, Asp266, Ile267, Phe5.42, and Leu6.51 in CB1 interacted directly with SR144528. The corresponding residues in CB2 were Ile1133.29, Glu181, Leu182, Phe1975.46, and Val2656.51, which also interacted tightly with SR144528. Some of these residues may have key roles in the selectivity for CB1 and CB2. Also, residues in positions 5.42 and 5.46 were different: Phe2785.42 and Val2825.46 in CB1, Ser1935.42 and Phe1975.46 in CB2. However, both Phe2785.42 in CB1 and Phe1975.46 in CB2 may contribute to the selectivity.
Although SR144528 is an active compound for CB1 and CB2, it is a highly selective CB2 receptor inverse agonist. Our docking results indicate at least two reasons. One is that SR144528 preferred more hydrophobic interactions. For example, Met2656.55 and Phe2817.35 in CB2 formed stronger hydrophobic interactions and a π–π interaction with SR144528. The other reason is that different residues involved in the binding pocket contributed to the selectivity, SR144528 preferring Ile1133.29, Ser1153.31, Glu181, Leu182, Phe1975.46, and Val2616.51 in CB2.
Binding Modes of our In-House Compounds with CB2
We docked our in-house antagonists (inverse agonists) and CB2 neutral compound (neutral antagonist) XIE35-1001 into the CB2 model to find the similarities of binding for antagonists and neutral compound (neutral antagonist). Moreover, we selected and docked 11 in-house inverse agonists into the CB2 model and then aligned their binding poses with SR144528, which is highlighted in yellow in Figure 7a.
Figure 7.

Analyzing the binding modes of our in-house antagonists (inverse agonists) and our CB2 neutral compound (neutral antagonist) XIE35-1001. (a) Alignments the binding poses between SR144528 (highlighted in yellow) and our 11 in-house antagonists (inverse agonists); (b) the interactions between CB2 and our in-house antagonist (inverse agonist) XIE95-1171 (0.5 nM for CB2); (c) structure and bioactivity information for our in-house CB2 neutral compound (neutral antagonist) XIE35-1001; (d) the interactions between CB2 and our in-house neutral compound (neutral antagonist) XIE35-1001.
We selected one of our compounds XIE95-1171 (0.5 nM for CB2 and 514 nM for CB1) to analyze further, as shown in Figure S12 and Figure 7b. The interactions between our compound XIE95-1171 (docking score: 9.29) and CB2 were almost the same as that of SR144528 (Figure 6b). Our compound XIE95-1171 formed a strong hydrophobic interaction with Val1133.32 (not shown in Figure 7b), Phe872.57, and Trp1945.43; our compound interacted strongly with Phe942.64 (hydrophobic interaction), Asp101 (not shown in Figure 7, hydrophobic interaction), and Phe1063.25 (not shown in Figure 7, π–π interactions) on the top of the binding pocket. The distance between Lys1093.28 in the CB2 receptor and our compound was more than 5.1 Å. Moreover, XIE95-1171 formed a strong hydrophobic interaction with Met2656.55 (not shown in Figure 7b) and formed a π–π interaction with Phe2817.35. It also had a hydrophobic interaction with Ile1133.29, Glu181, Leu182, Phe1975.46, and Val2616.51 in CB2. These residues therefore may contribute to the selectivity for CB2. One item that was different was the distance of ∼4.6 Å between Ser2857.39 and our compound XIE95-1171.
We then docked our CB2 neutral compound (neutral antagonist) Xie35-1001 (US 20110118214 A1, Figure 7c) into our CB2 model, which has a Ki of 37 nM at CB2 and 4082 nM at the CB1 receptor. Figure 7d shows the binding mode of our compound XIE35-1001, with the docking score of 8.67. The interactions of this compound with CB2 were almost the same as those of SR144528. The compound formed strong interactions with Phe942.64, Ile1103.29, Val1133.32, Phe1173.36, Glu181 (ECL2), Leu182 (ECL2), Trp1945.43, Val2616.51, and Met2656.55. The π–π interactions also were observed between compound XIE35-1001 and several residues, including Phe1063.25 and Phe2817.35. The distances between compound XIE35-1001 and Lys1093.28, Ser2857.39 were larger than 5.0 Å.
The docking results of our in-house compounds further supported the binding mode of SR144528. The docking results for antagonists (inverse agonists) and the neutral compound (neutral antagonist) showed that residues in TM3, TM5, TM6, and TM7 had important roles in the recognition for CB2 by the ligands.
Molecular Dynamics Study of the Complex of CB2 with SR144528
To validate further the binding mode of SR144528 within CB2, we performed 50 ns molecular dynamics simulation with lipid and water. For the MD simulations from this section and the following, all our CB2 models were flexible (that meant the backbones of CB2 were not fixed).
During 50 ns MD simulation, we found that the rmsd of CB2 was equilibrated after 5 ns. The time scale of 50 ns is reasonable for our purpose to validate the binding mode of SR144528.
Figure 8a shows the rmsd of CB2 and SR144528, respectively. Our results showed that the deviations of CB2 kept stable at 3.5 Å after system equilibration (after 5 ns), while the deviation of SR144528 was stable at 1 Å during the simulation. We aligned the conformations between before MD simulation and after 50 ns MD simulation. As shown in Figure 8d, the structure of CB2 kept stable during the MD simulation, especially the binding pocket and the loops, which agreed with the recent report for CB2 by Reggio and co-workers.59
Figure 8.

Time evolutions for (a) the deviation of CB2 protein (highlighted in black color) and SR144528 (highlighted in red color); (b) the distances between different residues and SR144528, including Phe1063.25, Lys1093.28, Ile1103.29, and Val1133.32; (c) the distances between Leu182/Phe183/Trp1945.43/Val2616.51/Phe2817.35 and SR144528; (d) comparing conformations of CB2 with SR144528 before MD simulation (CB2 highlighted in blue and SR144528 highlighted in yellow) and after 50 ns MD simulation (CB2 highlighted in orange and SR144528 highlighted in green).
However, our results showed that SR144528 moved upward (ECL2) about 0.7 Å after MD simulation. We found that SR144528 was moving to form a π–π interaction with Phe183 in ECL2. From Figure 8c, our results showed that the distance (highlighted in wine color) between Phe183 and SR144528 was ∼7.5 Å initially. After 5 ns MD simulation, the distance was stable within 4.3 Å. We infer that this residue may play an important role in SR144528 binding.
Three important residues in TM3 formed strong interactions with SR144528, including Phe1063.25 (Figure 8b, top graph, π–π interaction), Ile1133.29 (third graph, hydrophobic interaction), and Val1133.32 (fourth graph, hydrophobic interaction). The distances between these three residues and SR144528 were stable within 4.5 Å during the MD simulation. We suggest that Val1133.32 is important for the ligand recognition of CB2. Ile1133.29 may have a role in the selectivity for CB2 according to its stable interaction. The distance (second graph) between Lys1093.28 and SR144528 ranged from 5.3 to 6 Å. The weak interaction showed that Lys1093.28 may not be important for the binding of SR144528. During the MD simulation of SR144528 and CB2, we did not observe any water mediated interactions between Lys1093.28 and SR144528, which may be due to the short time of our simulation.
Some important residues involved in the binding pocket also formed strong interactions with SR144528 during the MD simulation, including Trp1945.43 (hydrophobic interaction) and Phe2817.35 (Figure 8c, π–π interaction). The role of Trp1945.43 had been reported.65 In the present work, we suggest that Phe2817.35 may have an important role for SR144528 binding.
Some residues that contributed to the selectivity also interacted tightly with SR144528, including Leu182 (Figure 8c, hydrophobic interaction), Val2616.51 (Figure 8c, hydrophobic interaction), and Glu181 (not shown the data, hydrophobic interaction).
Docking and Molecular Dynamics Study of CP55940 in CB2
CP55940 is considered as a full agonist to the CB2 receptor and has a Ki of 0.68 nM on CB2. We docked CP55940 into our CB2 model and tried to compare the similarities and differences between CB2 bound with agonist and antagonist (inverse agonist).
Figure 9a shows the binding mode of CP55940 in CB2. The polar parts of CP55940 formed hydrogen bonds with the extracellular part, while the flexible and hydrophobic part of CP55940 mainly interacted with Phe1173.36 (not shown in Figure 9a), Phe1975.46 (not shown in Figure 9a), and Trp2586.48 initially.
Figure 9.

(a) Binding mode of CP55940 within CB2; (b) binding mode of CP55940 within CB2 after 50 ns MD simulation; (c) the deviation of CP55940, and the distances between Lys1093.28/Ile1103.29/Val1133.32/Leu182 and CP55940; (d) the distances between Phe183/Trp1945.43/Trp1975.436/Val2616.51/Phe2817.35/Ser2857.39 and CP55940.
Several residues from the extracellular part of the pocket formed hydrogen bonds with CP55940 including Asp101 in ECL1, Glu181 (hydrogen donor), Leu182 (hydrogen acceptor) in ECL2, Lys1093.28 (hydrogen donor) in TM3, and Ser2857.39 (hydrogen acceptor) in TM7. Several residues in TM3 interacted tightly with CP55940: Ile1133.29 and Val1133.32 mainly forming hydrophobic interactions with CP55940. However, Phe1063.25 formed a weak hydrophobic interaction (larger than 6 Å initially) with CP55940, a residue that formed a π–π interaction with SR144528. Val2616.51 in TM6 also formed a hydrophobic interaction with CP55940 (a weak interaction, the distance was 6 Å), where it had a very strong interaction with SR144528 (within 4 Å). Moreover, for the binding of CP55940, many residues in CB2 formed hydrophobic interactions instead of π–π interactions, including Phe872.57 (not shown in Figure 9a), Phe912.61 (not shown in Figure 9a), Trp1945.43 (not shown in Figure 9a, also a weak interaction), Phe1975.46, Trp2586.48, and Phe2817.35.
CP55940 is also considered as a full agonist to CB1, has a Ki of 0.58 nM on CB1, but is an antagonist to GPR55, the putative “CB3″ receptor.75 The model of CB1 bound with CP55940 was reported by Kapur and co-workers.71 Comparing the binding mode between CB1/CB2 and CP55940, we found that Lys3.28 and Ser7.39 in CB1/CB2 should be involved in the recognition of CP55940. The differences included the following. For CB1, Lys3.28 and Ser7.39 had important roles for ligand binding. Moreover, Ser1.39 and Lys373 in ECL3 also had important roles for CP55940s binding. However, Asp101 in ECL1, Glu181, and Leu182 in ECL2 may contribute to the binding of CP55940 in our CB2 model.
We then performed 50 ns MD simulation for the complex of CP55940 within CB2. Figure 9c shows the rmsd of CP55940 during the MD simulation. Our results showed that the deviations of CB2 kept stable (∼4 Å during the MD) after system equilibration (after 5 ns MD simulation). TM3, ECL2, TM6, and TM7 endured larger conformational changes, while the deviation of CP55940 (highlighted in red) was stable at 2 Å during the simulation.
Comparing the conformations of CP55940 during the MD simulations, our results showed that CP55940 endured large conformational changes, as shown in Figures 9b, 9c, and 9d. First, the flexible and hydrophobic part of CP55940 moved from the “crevice” between TM3 and TM4 to the “crevice” between TM5 and TM6. As a result, the distance (highlighted in orange in Figure 9d) between Phe1975.46 and CP55940 became larger (from 3 Å initially to 6.5 Å after equilibration), while the distances between CP55940 and Trp1945.43 (highlighted in olive in Figure 9d, from 5.5 to4 Å) and Val2616.51 (highlighted in violet in Figure 9d, from 6 to 4 Å) became smaller.
Moreover, CP55940 moved ∼2.0 Å toward intracellular side. Consequently, Lys1093.28 (highlighted in green in Figure 9c) and Ser2857.39 (highlighted in gray in Figure 9d) were far away from CP55940, respectively. Our results showed that the distance between Lys1093.28 and CP55940 was 3.8 Å initially and extended to 6.0 Å, while the distance between Ser2857.39 and CP55940 was 3.7 Å initially and extended to 5.2 Å.
Moreover, the situation of Asp101 in ECL1 was similar to Lys1093.28 and Ser2857.39. The distance between Asp101 and CP55940 was larger than 6.5 Å after MD simulation. However, Ile1103.29, Val1133.32, and Phe2817.35 formed stronger hydrophobic interactions with CP55940 after MD simulation, as shown in Figures 9c and 9d.
Residues in ECL2 contributed important roles to CP55940 binding. Our results showed that Leu182 (highlighted in purple in Figure 9d) and Glu181 formed hydrogen bonds with CP55940. The distance of hydrogen bonds were within 4–5 Å, as shown in Figure 9c. Phe183 (ECL2, Figure 9d) did not interact tightly with CP55940. The distance between Phe183 and CP55940 was larger than 6 Å. However, Phe183 in ECL2 covered the binding pocket, which may have an important role for the conformation of the binding pocket.
These results indicate that the agonist CP55940 has the flexibility to interact CB2 with different H-bond partners or hydrophobic partners. The flexibility can facilitate the role of CP55940 in the activation of CB2, which agrees with the recent report.76
Comparison of Conformations of CB2 between Agonist-Bound and Antagonist-Bound Status
We compared the conformations of CB2 between agonist-bound (CP55940) and antagonist-bound (SR144528) to illustrate the structural information during the activation of CB2, as shown in Figures S13a and S13b:
1. Large conformational changes upon CB2 bound with agonist CP55940. Our results showed that CB2 had the deviation of ∼4.5 Å from its original conformation; moreover, TM3, ECL2, TM6, and TM7 fluctuated greatly when bound with CP55940.
2. “Ionic lock” for CB2 bound with SR144528 was more stable than that for CB2 bound with CP55940. “Ionic lock” motif was as follows: Arg3.50 of the conserved D/ERY in TM3 and highly conserved residue Glu/Asp6.30 in TM6. Our results showed that the “ionic lock” for CB2 bound with SR144528 was more stable, as shown in Figure S13b.
3. Agonist CP55940 endured large conformational changes to facilitate its role for the activation of CB2; CP55940 moved toward the intracellular side, and the distance between CP55940 and Trp2586.48 was almost ∼3.0 Å during the MD simulation. However, the antagonist SR144528 moved toward the extracellular side, and the distance between antagonist SR144528 and Trp2586.48 was larger than ∼5.0 Å during the MD simulation, as shown in Figure S13b.
4. Conformational change of Tyr2997.53 can be observed for CB2 bound with agonist CP55940: Tyr7.53, which belongs to the highly conserved NPxxY motif, endured a conformational change (∼1.3 Å), accompanying the intracellular side of TM7 moved inward.
5. Phe872.57, Phe912.61, Phe942.64, Lys1093.28, Ile1103.29, Val1133.32, Phe1173.36, Trp1945.43, Val2616.51, Trp2586.48, Phe2817.35, and Ser2857.39 had roles in the recognition of CP55940 and SR144528. However, Phe183 (ECL2) may have an important role for SR144528 and CP55940, due to its π–π interactions or changing the conformation of binding pocket. Some important residues in ECL2 may have the similar functions of Phe183, for example, Glu181 (ECL2) and Leu182 (ECL2).
Last but not least, we compared the structure of our CB2 model after MD simulation with agonist and the crystal structure of β2AR bound with Gα subunit (PDBID:3SN6),77 as shown in Figure S13c. Our results showed that almost all TMs aligned well, except TM6. Figure S13c shows that TM6 in our CB2 model had a “crash” with the Gα subunit when we aligned CB2 and β2AR. The intracellular side of TM6 in our CB2 did not endure enough conformational changes for fitting the Gα subunit. We think the only plausible method is to construct a homology model based on the beta-2 active structure (PDBID:3SN6)77 and relaxing it a little bit before using for docking and other studies.
Mutation Data of CB2 Val1133.32, Phe183, and Phe2817.35
Based on our docking and MD results, we performed a mutation study for several residues, including Val1133.32, Phe183, and Phe2817.35of CB2. We performed V113E, V113L, F183Q, F183V, and F281K mutations and tested the binding affinity of SR144528 and CP55940. The mutations described in the present work were based on our in silico docking results.
The results showed that mutating the valine to a glutamic acid residue (V113E), which has a negatively charged side chain, led to the complete loss of CB2 ligand binding, as shown in Table 2. The mutation for V113E agreed with the computation in silico: the mutation from valine to a leucine (V113L) allowed CB2 to retain its ligand binding, because the leucine also provided hydrophobic interactions with SR144528 or CP55940 and also affected the conformation of the binding pocket (Table 2). Residue 3.32 is occupied by a negatively charged amino acid in biogenic amine receptors (D2 dopamine, β2 adrenergic, and M3 muscaranic) and that it is essential for recognition and binding of endogenous amines. In an attempt to resemble the functionality of this residue in amine receptors, Li et al.78 substituted endogenous V843.32 with aspartic acid (D) in the A2aAR. The V84D mutant in A2aAR was unable to bind to any ligand tested. Interestingly, substitution of the same valine residue with leucine (V84L) did not significantly affect binding with any ligand with respect to the wild-type A2aAR, which agreed with our mutation for Val1133.32.
Table 2. Binding Affinities (Ki, nM) of Ligands to Cannabinoid Receptors of Wild-Type and Mutant CB2 Stably Expressed in CHO Cellsa.
| receptors | WT | V113E | V113L | F183Q | F183V | F281K |
|---|---|---|---|---|---|---|
| SR144528 | 3.2 (1.1–8.8) | ND | 6.1 (2.8–12) | ND | ND | ND |
| CP55940 | 2.7 (1.5–4.8) | ND | 3.8 (0.3–46) | ND | ND | ND |
Data are the means and corresponding 95% confidence intervals of two independent experiments each performed in duplicate. WT: wild-type; V113E, V113L, F183Q, F183V, and F281K: mutant huCB2.ND: binding not detected.
Our studies also demonstrate the role of ECL2 in the ligands-binding recognition. A prior report18 affirmed that this loop dipped down toward the binding site of SR144528 to make a π-sulfur interaction with the Cys1754.67. Therefore, we investigated the effect of the aromatic side chain of Phe183 in ECL2 for SR144528 binding. Mutation of Phe183 in silico, CB2 did not form strong hydrophobic interactions with SR144528 and CP55940. Mutation of Phe183 to the polar uncharged amino acid such as glutamine (F183Q) or to a hydrophobic side chain such as valine (F183V) had a significant effect on the binding affinity of SR144528 to CB2, as shown in Figure 10 and Table 2. CP55940 binding affinity to Phe183 mutated CB2 were also tested, and the similar results can be found in Figure 10 and Table 2. As we discussed in the previous section, Phe183 did not form a π–π interaction with CP55940 and maybe did not form a hydrophobic interaction (or a very weak hydrophobic interaction), but the mutations of Phe183 showed a significant effect for CP55940 binding.
Figure 10.
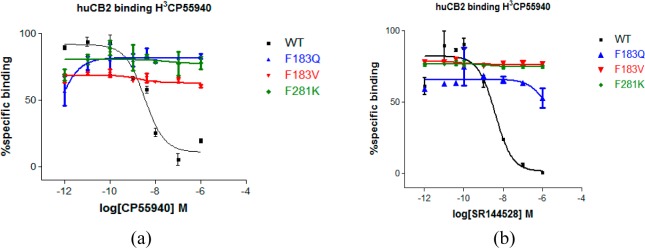
Competitive ligand displacement assay. For mutant CB2 for agonist and antagonist (inverse agonist). Binding profile of (a) CP55940; (b) SR144528 for WT, F183Q/V, and F281K. Competitive displacement of the [3H]CP55940 was obtained by using an increased amount of unlabeled ligands. Assay was performed in duplicate (n = 2). Data represented as mean ± SEM.
We therefore suggested that the mutations of Phe183 significantly affected both SR144528 and CP55940. Moreover, some residues in ECL2 may have similar effects, including Glu181 (ECL2) and Leu182 (ECL2). This agreed with the finding64 that ECL2 is important for the binding of CP55940 to CB2, where replacing of ECL2 of CB2 with ECL2 of CB1 resulted in a loss of binding for CP55940.
In our model, Phe2817.35 formed a strong π–π interaction with SR144528, but it formed a hydrophobic interaction with CP55940. We therefore mutated it to a charged chain such as lysine (F281K) to study its influence on CB2 ligand binding. As shown in Figure 10 and Table 2, our results showed that this kind of mutation led to the complete loss of CB2 ligand binding. Phe2817.35 also had an important role in the binding of SR144528 and CP55940 to CB2 through a π–π interaction or a hydrophobic interaction.
In our model, we found that Phe183 in ECL2, Met2656.55, and Phe2817.35 formed a strong hydrophobic pocket in the extracellular side of CB2. Mutations of F183Q and F281K will change the charge environment, while the mutation of F183V will affect the hydrophobic pocket. We suggest that these three mutations will reduce the affinities of ligands on CB2.
All our mutation results agreed with our docking analysis and MD simulation, which can help us to learn more about structure and function of CB2.
Conclusion
In the present work, we used 10 GPCRs to construct 10 comparative CB2 models. We selected one model for further studies that was shown to be consistent with the known experimental data. Importantly, we found a potential allosteric binding pocket and suggest that sodium may reduce the binding affinity of endogenous agonists or its analogs. Subsequently, the similarities and differences of the binding pockets between CB1 and CB2 were analyzed in comparison of the binding modes between antagonist and agonist. We also performed 50 ns molecular dynamics simulations for CB2 bound with agonist and antagonist. The conformational changes of CB2 and antagonist/agonist were congruent with recent reports for GPCRs. Finally, we validated the roles of Val1133.32 and two newly predicted residues for CB2 binding, i.e., Phe183 in ECL2 and Phe2817.35 by radiometric binding affinity assay experiment. Overall, the new 3D CB2 structural model based on recent GPCRs crystal data as well as the in silico and in vitro experimental studies performed here provide new insight into better understanding of the structural and functional roles of the CB2 receptor and facilitated the future structure-based design novel CB2 ligands with therapeutic potential.
Acknowledgments
The project is supported by funding from the NIH R01 DA025612 (Xie) and NIDA P30 DA035778A1 (Xie).
Supporting Information Available
Figure S1 shows the alignments of all known GPCR crystal structures. Figure S2 shows 20 active compounds for CB2 prescreen. Figure S3 shows the molecular dynamics simulation box of CB2 bound with agonist or antagonist. Figure S4 shows the alignments of constructed CB2 models. Figure S5 shows the alignments of three crystal structures, including D3R, β1AR, and β2AR. Figure S6 shows the hit rates of prescreen for the other nine CB2 models. Figure S7 shows the prediction of CB1 and CB2 selective compounds for the other four CB2 models. Figure S8 shows the overall model quality and local model quality of CB2 constructed by β1AR, predicted by ProSA-web. Figure S9 shows the Ramachandran plots of the CB2 model constructed by β1AR. Figure S10 shows the alignments between β2AR/A2AAR bound with agonist and with antagonist. Figure S11 shows three CB2 selective compounds sharing the very similar binding poses with SR144528. Figure S12 shows the alignments between SR144528 and our antagonist (inverse agonist) XIE95-1171. Figure S13 shows the comparison of conformations between CB2 bound with CP5940 and with SR144528. Table T1 shows the rmsd of Cα in the TMs regions among our 10 CB2 models. Table T2 shows the structure, bioactivity information, and docking score of 70 CB1 selective compounds. Table T3 shows the structure, bioactivity information, and docking score of 100 CB2 selective compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Overington J. P.; Al-Lazikani B.; Hopkins A. L. How many drug targets are there?. Nat. Rev. Drug Discovery 2006, 5, 993–996. [DOI] [PubMed] [Google Scholar]
- Howlett A. C. The cannabinoid receptors. Prostaglandins Other Lipid Mediators 2002, 68, 619–631. [DOI] [PubMed] [Google Scholar]
- Mackie K. Cannabinoid receptors: where they are and what they do. J. Neuroendocrinol. 2008, 20, 10–14. [DOI] [PubMed] [Google Scholar]
- Matsuda L. A.; Lolait S. J.; Brownstein M. J.; Young A. C.; Bonner T. I. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 1990, 346, 561–564. [DOI] [PubMed] [Google Scholar]
- Gerard C. M.; Mollereau C.; Vassart G.; Parmentier M. Molecular cloning of a human cannabinoid receptor which is also expressed in testis. Biochem. J. 1991, 279, 129–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee R. G. Emerging strategies for exploiting cannabinoid receptor agonists as medicines. Br. J. Pharmacol. 2009, 156, 397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng R.; Milcarek C. A.; Xie X.-Q. Antagonism of cannabinoid receptor 2 pathway suppresses IL-6-induced immunoglobulin IgM secretion. BMC Pharmacol. Toxicol. 2014, 15, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabral G. A.; Griffin-Thomas L. Emerging role of the cannabinoid receptor CB2 in immune regulation: therapeutic prospects for neuroinflammation. Expert Rev. Mol. Med. 2009, 11, e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon J.; Hohmann A. Cannabinoid CB2 receptors: a therapeutic target for the treatment of inflammatory and neuropathic pain. Br. J. Pharmacol. 2008, 153, 319–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karsak M.; Cohen-Solal M.; Freudenberg J.; Ostertag A.; Morieux C.; Kornak U.; Essig J.; Erxlebe E.; Bab I.; Kubisch C.; de Vernejoul M. C.; Zimmer A. Cannabinoid receptor type 2 gene is associated with human osteoporosis. Hum. Mol. Genet. 2005, 14, 3389–3396. [DOI] [PubMed] [Google Scholar]
- Beltramo M. Cannabinoid type 2 receptor as a target for chronic - pain. Mini-Rev. Med. Chem. 2009, 9, 11–25. [DOI] [PubMed] [Google Scholar]
- Mbvundula E. C.; Rainsford K. D.; Bunning R. A. Cannabinoids in pain and inflammation. Inflammopharmacology 2004, 12, 99–114. [DOI] [PubMed] [Google Scholar]
- Bouaboula M.; Poinot-Chazel C.; Marchand J.; Canat X.; Bourrié B.; Rinaldi-Carmona M.; Calandra B.; Fur G.; Casellas P. Signaling pathway associated with stimulation of CB2 peripheral cannabinoid receptor. Eur. J. Biochem. 1996, 237, 704–711. [DOI] [PubMed] [Google Scholar]
- Shoemaker J. L.; Ruckle M. B.; Mayeux P. R.; Prather P. L. Agonist-directed trafficking of response by endocannabinoids acting at CB2 receptors. J. Pharmacol. Exp. Ther. 2005, 315, 828–838. [DOI] [PubMed] [Google Scholar]
- Bisogno T.; Melck D.; Bobrov M.; Gretskaya N.; Bezuglov V.; De Petrocellis L.; Di Marzo V. N-Acyl-dopamines: novel synthetic CB1 cannabinoid-receptor ligands and inhibitors of anandamide inactivation with cannabimimetic activity in vitro and in vivo. Biochem. J. 2000, 351, 817–824. [PMC free article] [PubMed] [Google Scholar]
- Song Z.-H.; Slowey C.-A.; Hurst D. P.; Reggio P. H. The difference between the CB1 and CB2 cannabinoid receptors at position 5.46 is crucial for the selectivity of WIN55212-2 for CB2. Mol. Pharmacol. 1999, 56, 834–840. [PubMed] [Google Scholar]
- Bramblett R.; Panu A. M.; Ballesteros J. A.; Reggio P. H. Construction of a 3D model of the cannabinoid CB1 receptor: determination of helix ends and helix orientation. Life Sci. 1995, 56, 1971–1982. [DOI] [PubMed] [Google Scholar]
- Gouldson P.; Calandra B.; Legoux P.; Kernéis A.; Rinaldi-Carmona M.; Barth F.; Le Fur G.; Ferrara P.; Shire D. Mutational analysis and molecular modelling of the antagonist SR 144528 binding site on the human cannabinoid CB2 receptor. Eur. J. Pharmacol. 2000, 401, 17–25. [DOI] [PubMed] [Google Scholar]
- Palczewski K.; Kumasaka T.; Hori T.; Behnke C. A.; Motoshima H.; Fox B. A.; Trong I. L.; Teller D. C.; Okada T.; Stenkamp R. E. Crystal structure of rhodopsin: AG protein-coupled receptor. Science 2000, 289, 739–745. [DOI] [PubMed] [Google Scholar]
- Krebs A.; Villa C.; Edwards P. C.; Schertler G. F. Characterisation of an improved two-dimensional p22121 crystal from bovine rhodopsin. J. Mol. Biol. 1998, 282, 991–1003. [DOI] [PubMed] [Google Scholar]
- Xie X. Q.; Chen J. Z.; Billings E. M. 3D structural model of the G-protein-coupled cannabinoid CB2 receptor. Proteins: Struct., Funct., Bioinf. 2003, 53, 307–319. [DOI] [PubMed] [Google Scholar]
- Ballesteros J. A.; Jensen A. D.; Liapakis G.; Rasmussen S. G.; Shi L.; Gether U.; Javitch J. A. Activation of the β2-adrenergic receptor involves disruption of an ionic lock between the cytoplasmic ends of transmembrane segments 3 and 6. J. Biol. Chem. 2001, 276, 29171–29177. [DOI] [PubMed] [Google Scholar]
- Tuccinardi T.; Ferrarini P. L.; Manera C.; Ortore G.; Saccomanni G.; Martinelli A. Cannabinoid CB2/CB1 selectivity. Receptor modeling and automated docking analysis. J. Med. Chem. 2006, 49, 984–994. [DOI] [PubMed] [Google Scholar]
- Stern E.; Muccioli G. G.; Millet R.; Goossens J.-F.; Farce A.; Chavatte P.; Poupaert J. H.; Lambert D. M.; Depreux P.; Hénichart J.-P. Novel 4-oxo-1,4-dihydroquinoline-3-carboxamide derivatives as new CB2 cannabinoid receptors agonists: synthesis, pharmacological properties and molecular modeling. J. Med. Chem. 2006, 49, 70–79. [DOI] [PubMed] [Google Scholar]
- Raduner S.; Majewska A.; Chen J.-Z.; Xie X.-Q.; Hamon J.; Faller B.; Altmann K.-H.; Gertsch J. Alkylamides from Echinacea are a new class of cannabinomimetics cannabinoid type 2 receptor-dependent and-independent immunomodulatory effects. J. Biol. Chem. 2006, 281, 14192–14206. [DOI] [PubMed] [Google Scholar]
- Yang P.; Myint K.-Z.; Tong Q.; Feng R.; Cao H.; Almehizia A. A.; Alqarni M. H.; Wang L.; Bartlow P.; Gao Y. Lead discovery, chemistry optimization, and biological evaluation studies of novel biamide derivatives as CB2 receptor inverse agonists and osteoclast inhibitors. J. Med. Chem. 2012, 55, 9973–9987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latek D.; Kolinski M.; Ghoshdastider U.; Debinski A.; Bombolewski R.; Plazinska A.; Jozwiak K.; Filipek S. Modeling of ligand binding to G protein coupled receptors: cannabinoid CB1, CB2 and adrenergic β2AR. J. Mol. Model. 2011, 17, 2353–2366. [DOI] [PubMed] [Google Scholar]
- Xie X.-Q.; Chowdhury A. Advances in methods to characterize ligand-induced ionic lock and rotamer toggle molecular switch in G protein-coupled receptors. Method. Enzymol. 2012, 520, 153–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiburu E. K.; Tyukhtenko S.; Deshmukh L.; Vinogradova O.; Janero D. R.; Makriyannis A. Structural biology of human cannabinoid receptor-2 helix 6 in membrane-mimetic environments. Biochem. Biophy. Res. Commun. 2009, 384, 243–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherezov V.; Rosenbaum D. M.; Hanson M. A.; Rasmussen S. G.; Thian F. S.; Kobilka T. S.; Choi H.-J.; Kuhn P.; Weis W. I.; Kobilka B. K. High-resolution crystal structure of an engineered human β2-adrenergic G protein–coupled receptor. Science 2007, 318, 1258–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haga K.; Kruse A. C.; Asada H.; Yurugi-Kobayashi T.; Shiroishi M.; Zhang C.; Weis W. I.; Okada T.; Kobilka B. K.; Haga T. Structure of the human M2 muscarinic acetylcholine receptor bound to an antagonist. Nature 2012, 482, 547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson M. A.; Roth C. B.; Jo E.; Griffith M. T.; Scott F. L.; Reinhart G.; Desale H.; Clemons B.; Cahalan S. M.; Schuerer S. C. Crystal structure of a lipid G protein–coupled receptor. Science 2012, 335, 851–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebon G.; Warne T.; Edwards P. C.; Bennett K.; Langmead C. J.; Leslie A. G.; Tate C. G. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature 2011, 474, 521–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamura T.; Shiroishi M.; Weyand S.; Tsujimoto H.; Winter G.; Katritch V.; Abagyan R.; Cherezov V.; Liu W.; Han G. W. Structure of the human histamine H1 receptor complex with doxepin. Nature 2011, 475, 65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.; Wu H.; Katritch V.; Han G. W.; Huang X.-P.; Liu W.; Siu F. Y.; Roth B. L.; Cherezov V.; Stevens R. C. Structure of the human smoothened receptor bound to an antitumour agent. Nature 2013, 497, 338–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warne T.; Moukhametzianov R.; Baker J. G.; Nehmé R.; Edwards P. C.; Leslie A. G.; Schertler G. F.; Tate C. G. The structural basis for agonist and partial agonist action on a β 1-adrenergic receptor. Nature 2011, 469, 241–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B.; Chien E. Y.; Mol C. D.; Fenalti G.; Liu W.; Katritch V.; Abagyan R.; Brooun A.; Wells P.; Bi F. C. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 2010, 330, 1066–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien E. Y.; Liu W.; Zhao Q.; Katritch V.; Han G. W.; Hanson M. A.; Shi L.; Newman A. H.; Javitch J. A.; Cherezov V. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 2010, 330, 1091–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SYBYL-X 1.3; Tripos International, 1699 South Hanley Rd., St. Louis, Missouri 63144, USA. 2010.
- Martí-Renom M. A.; Stuart A. C.; Fiser A.; Sánchez R.; Melo F.; Šali A. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 291–325. [DOI] [PubMed] [Google Scholar]
- Wiederstein M.; Sippl M. J. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski R. A.; MacArthur M. W.; Moss D. S.; Thornton J. M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Yang P.; Wang L.; Feng R.; Almehizia A. A.; Tong Q.; Myint K.-Z.; Ouyang Q.; Alqarni M. H.; Wang L.; Xie X.-Q. Novel triaryl sulfonamide derivatives as selective cannabinoid receptor 2 inverse agonists and osteoclast inhibitors: discovery, optimization, and biological evaluation. J. Med. Chem. 2013, 56, 2045–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain A. N. Scoring noncovalent protein-ligand interactions: a continuous differentiable function tuned to compute binding affinities. J. Comput. Aided-Mol. Des. 1996, 10, 427–440. [DOI] [PubMed] [Google Scholar]
- Chen J.-Z.; Wang J.; Xie X.-Q. GPCR structure-based virtual screening approach for CB2 antagonist search. J. Chem. Inf. Model. 2007, 47, 1626–1637. [DOI] [PubMed] [Google Scholar]
- Pedretti A.; Villa L.; Vistoli G. VEGA: a versatile program to convert, handle and visualize molecular structure on Windows-based PCs. J. Mol. Graphics Modell. 2002, 21, 47–49. [DOI] [PubMed] [Google Scholar]
- Søndergaard C. R.; Olsson M. H.; Rostkowski M.; Jensen J. H. Improved treatment of ligands and coupling effects in empirical calculation and rationalization of pKa values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [DOI] [PubMed] [Google Scholar]
- Hsin J.; Arkhipov A.; Yin Y.; Stone J. E.; Schulten K. Using VMD: an introductory tutorial. Curr. Protoc. Bioinf. 2008, 5.7. 1–5.7. 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926. [Google Scholar]
- Kalé L.; Skeel R.; Bhandarkar M.; Brunner R.; Gursoy A.; Krawetz N.; Phillips J.; Shinozaki A.; Varadarajan K.; Schulten K. NAMD2: greater scalability for parallel molecular dynamics. J. Comput. Phys. 1999, 151, 283–312. [Google Scholar]
- Brooks B. R.; Bruccoleri R. E.; Olafson B. D. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983, 4, 187–217. [Google Scholar]
- MacKerell A. D.; Bashford D.; Bellott M.; Dunbrack R. L.; Evanseck J. D.; Field M. J.; Fischer S.; Gao J.; Guo H.; Ha S.; Joseph-McCarthy D.; Kuchnir L.; Kuczera K.; Lau F. T. K.; Mattos C.; Michnick S.; Ngo T.; Nguyen D. T.; Prodhom B.; Reiher W. E.; Roux B.; Schlenkrich M.; Smith J. C.; Stote R.; Straub J.; Watanabe M.; Wiorkiewicz-Kuczera J.; Yin D.; Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- Essmann U.; Perera L.; Berkowitz M. L.; Darden T.; Lee H.; Pedersen L. G. A smooth Particle Mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar]
- Prism, G, version 5.01; GraphPad Software Inc.: San Diego, CA, USA, 2007.
- Lunn C. A.; Fine J. S.; Rojas-Triana A.; Jackson J. V.; Fan X.; Kung T. T.; Gonsiorek W.; Schwarz M. A.; Lavey B.; Kozlowski J. A. A novel cannabinoid peripheral cannabinoid receptor-selective inverse agonist blocks leukocyte recruitment in vivo. J. Pharmacol. Exp. Ther. 2006, 316, 780–788. [DOI] [PubMed] [Google Scholar]
- Tandon M.; Wang L.; Xu Q.; Xie X.; Wipf P.; Wang Q. J. A targeted library screen reveals a new inhibitor scaffold for protein kinase d. PloS One 2012, 7, e44653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aghazadeh Tabrizi M.; Baraldi P. G.; Saponaro G.; Moorman A.; Romagnoli R.; Preti D.; Baraldi S.; Ruggiero E.; Tintori C.; Tuccinardi T. Discovery of 7-oxo-pyrazolo[1,5-a]pyrimidine-6-carboxamides as potent and selective CB2 cannabinoid receptor inverse agonists. J. Med. Chem. 2013, 56, 4482–4496. [DOI] [PubMed] [Google Scholar]
- Kotsikorou E.; Navas F. III; Roche M. J.; Gilliam A. F.; Thomas B. F.; Seltzman H. H.; Kumar P.; Song Z.-H.; Hurst D. P.; Lynch D. L.; Reggio P. H. The importance of hydrogen bonding and aromatic stacking to the affinity and efficacy of cannabinoid receptor CB2 antagonist, 5-(4-chloro-3-methylphenyl)-1-[(4-methylphenyl)methyl]-N-[(1S,2S,4R)-1,3,3-trimethylbicyclo[2.2.1]hept-2-yl]-1H-pyrazole-3-carboxamide (SR144528). J. Med. Chem. 2013, 56, 6593–6612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusakabe K.-i.; Tada Y.; Iso Y.; Sakagami M.; Morioka Y.; Chomei N.; Shinonome S.; Kawamoto K.; Takenaka H.; Yasui K. Design, synthesis, and binding mode prediction of 2-pyridone-based selective CB2 receptor agonists. Bioorg. Med. Chem. 2013, 21, 2045–2055. [DOI] [PubMed] [Google Scholar]
- Ashton J. C.; Wright J. L.; McPartland J. M.; Tyndall J. D. Cannabinoid CB1 and CB2 receptor ligand specificity and the development of CB2-selective agonists. Curr. Med. Chem. 2008, 15, 1428–1443. [DOI] [PubMed] [Google Scholar]
- Raitio K.; Salo O.; Nevalainen T.; Poso A.; Jarvinen T. Targeting the cannabinoid CB2 receptor: mutations, modeling and development of CB2 selective ligands. Curr. Med. Chem. 2005, 12, 1217–1237. [DOI] [PubMed] [Google Scholar]
- Tao Q.; McAllister S. D.; Andreassi J.; Nowell K. W.; Cabral G. A.; Hurst D. P.; Bachtel K.; Ekman M. C.; Reggio P. H.; Abood M. E. Role of a conserved lysine residue in the peripheral cannabinoid receptor (CB2): evidence for subtype specificity. Mol. Pharmacol. 1999, 55, 605–613. [PubMed] [Google Scholar]
- Shire D.; Calandra B.; Delpech M.; Dumont X.; Kaghad M.; Le Fur G.; Caput D.; Ferrara P. Structural features of the central cannabinoid CB1 receptor involved in the binding of the specific CB1 antagonist SR 141716A. J. Biol. Chem. 1996, 271, 6941–6946. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Xie Z.; Wang L.; Schreiter B.; Lazo J. S.; Gertsch J.; Xie X.-Q. Mutagenesis and computer modeling studies of a GPCR conserved residue W5.43(194) in ligand recognition and signal transduction for CB2 receptor. Int. Immunopharmacol. 2011, 11, 1303–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebane N. M.; Hurst D. P.; Carrasquer C. A.; Qiao Z.; Reggio P. H.; Song Z.-H. Residues accessible in the binding-site crevice of transmembrane helix 6 of the CB2 cannabinoid receptor. Biochemistry 2008, 47, 13811–13821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katritch V.; Abagyan R. GPCR agonist binding revealed by modeling and crystallography. Trends Pharmacol. Sci. 2011, 32, 637–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salo O. M.; Raitio K. H.; Savinainen J. R.; Nevalainen T.; Lahtela-Kakkonen M.; Laitinen J. T.; Järvinen T.; Poso A. Virtual screening of novel CB2 ligands using a comparative model of the human cannabinoid CB2 receptor. J. Med. Chem. 2005, 48, 7166–7171. [DOI] [PubMed] [Google Scholar]
- Rhee M. H. Functional role of serine residues of transmembrane dopamin VII in signal transduction of CB2 cannabinoid receptor. J. Vet. Sci. 2002, 3, 185–191. [PubMed] [Google Scholar]
- Poso A.; Huffman J. Targeting the cannabinoid CB2 receptor: modelling and structural determinants of CB2 selective ligands. Br. J. Pharmacol. 2008, 153, 335–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur A.; Hurst D. P.; Fleischer D.; Whitnell R.; Thakur G. A.; Makriyannis A.; Reggio P. H.; Abood M. E. Mutation studies of Ser7.39 and Ser2.60 in the human CB1 cannabinoid receptor: evidence for a serine-induced bend in CB1 transmembrane helix 7. Mol. Pharmacol. 2007, 71, 1512–1524. [DOI] [PubMed] [Google Scholar]
- Fenalti G.; Giguere P. M.; Katritch V.; Huang X.-P.; Thompson A. A.; Cherezov V.; Roth B. L.; Stevens R. C. Molecular control of [dgr]-opioid receptor signalling. Nature 2014, 506, 191–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W.; Chun E.; Thompson A. A.; Chubukov P.; Xu F.; Katritch V.; Han G. W.; Roth C. B.; Heitman L. H.; IJzerman A. P. Structural basis for allosteric regulation of GPCRs by sodium ions. Science 2012, 337, 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard M. J.; Hughes R. J.; Motulsky H. J.; Mullen M. D.; Insel P. A. Interactions of amiloride with alpha-and beta-adrenergic receptors: amiloride reveals an allosteric site on alpha 2-adrenergic receptors. Mol. Pharmacol. 1987, 32, 53–58. [PubMed] [Google Scholar]
- Kapur A.; Zhao P.; Sharir H.; Bai Y.; Caron M. G.; Barak L. S.; Abood M. E. Atypical responsiveness of the orphan receptor GPR55 to cannabinoid ligands. J. Biol. Chem. 2009, 284, 29817–29827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z.; Hou T.; Li Y. Studies on the interactions between β2 adrenergic receptor and Gs protein by molecular dynamics simulations. J. Chem. Inf. Model. 2012, 52, 1005–1014. [DOI] [PubMed] [Google Scholar]
- Rasmussen S. G.; DeVree B. T.; Zou Y.; Kruse A. C.; Chung K. Y.; Kobilka T. S.; Thian F. S.; Chae P. S.; Pardon E.; Calinski D. Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 2011, 477, 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Jonsson A. L.; Beuming T.; Shelley J. C.; Voth G. A. Ligand-dependent activation and deactivation of the human adenosine A2A receptor. J. Am. Chem. Soc. 2013, 135, 8749–8759. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.