

Abstract
The molecular mechanisms linking Aβ to the onset of neurotoxicity are still largely unknown, but several lines of evidence point to reactive oxygen species, which are produced even under the effect of nanomolar concentrations of soluble Aβ-oligomers. The consequent oxidative stress is considered as the mediator of a cascade of degenerative events in many neurological disorders. Epidemiological studies indicate that dietary habits and antioxidants from diet can influence the incidence of neurodegenerative disorders such as Alzheimer's and Parkinson's diseases. In the recent years, a number of reviews have reported on neuroprotective effects of polyphenols in cell and animal models. However, the majority of these studies have focused only on the anti-oxidant properties of these compounds and less on the mechanism/s of action at cellular level. In this work we investigated the effect of cocoa polyphenolic extract on a human AD in vitro model. The results obtained, other than confirming the anti-oxidant properties of cocoa, demonstrate that cocoa polyphenols triggers neuroprotection by activating BDNF survival pathway, both on Aβ plaque treated cells and on Aβ oligomers treated cells, resulting in the counteraction of neurite dystrophy. On the light of the results obtained the use of cocoa powder as preventive agent for neurodegeneration is further supported.
Keywords: Neurodegenerative Diseases, Anti-Oxidant, BDNF Signaling, Neurons, Neurites, Cytoskeletric Proteins
Alzheimer's disease (AD) is a neurodegenerative disorder, characterized by a progressive loss of synapses and neurons, resulting in gradual cognitive impairment and eventually leading to dementia [Selkoe, 2002] The neuropathological features of AD are extracellular deposits of amyloid-β (Aβ) plaques and intracellular neurofibrillary tangles of hyperphosphorylated tau proteins, both caused by the misfolding and gradual conversion of highly soluble proteins into insoluble, filamentous polymers [Selkoe, 2004; Walsh and Selkoe, 2007]. Several events are involved in the pathogenesis of AD, and many studies have focused on the mechanisms underlying the toxicity associated with Aβ plaques. The molecular mechanisms linking Aβ to the onset of neurotoxicity are still largely unknown, but several lines of evidence point to reactive oxygen species (ROS), which are produced even under the effect of nanomolar concentrations of soluble Aβ-oligomers [Varadarajan et al., 2000; Varadarajan et al., 2001; Cimini et al., 2009a]. The consequent oxidative stress is considered as the common effector of the cascade of degenerative events in many neurological disorders [Jenner, 1996]. Studies on post-mortem tissues revealed that the levels of lipid oxidation products, protein oxidation and DNA oxidation are elevated in the brain from Alzheimer's patients [Lovell et al., 1995; Smith et al., 1996; Markesbery, 1997; Sayre et al., 1997; Marcus et al., 1998; Nunomura et al., 1999; Christen, 2000]. Epidemiological studies indicate that dietary habits and antioxidants from diet can influence the incidence of neurodegenerative disorders such as Alzheimer's and Parkinson's diseases [de Rijk et al., 1997; Deschamps et al., 2001; Engelhart et al., 2002]. In a followed-up study for 5 years, between 1991 and 1996 [Commenges et al., 2000] found in a cohort of 1,367 subjects above 65 years of age, a significant inverse association between flavonoids intake and the risk of dementia. The treatment with other sources of flavonoids other than those from tea or wine such as those from the Ginkgo biloba extract has been postulated to improve the cognitive performance of Alzheimer's patients [Oken et al., 1998]. These clinical and epidemiological findings suggesting a protective effect of flavonoids and polyphenols against neurodegenerative disease are supported by data obtained in animal models. In fact, in recent years, a number of reviews have reported on the potential neuroprotective effects of polyphenols in cell and animal models [Dajas et al., 2003; Mandel and Youdim, 2004; Simonyi et al., 2005]. Ad example, in old rats, diet supplemented with fruits and vegetables rich in antioxidants (blueberries, strawberries, and spinaches) can have beneficial effects on age-related decline of cognitive function [Joseph et al., 2005], while in mouse hippocampal cell lines HT-22 and primary neuronal cultures, many flavonoids protect from oxidative injury from glutamate cytotoxicity [Fu and Koo, 2006].
Polyphenols are natural substances that are present in fruits, and vegetables, including olive oil, red wine, and tea. Flavonoids are the largest group of polyphenols and more than 2,000 individual flavonoids are known. They can be divided into various classes on the basis of the molecular structure such as anthocyanins and anthoxanthins. The latter group is divided into flavonols, flavans, flavanols, flavones, and isoflavones, with catechins (CE) being the best described flavanols [Ramassamy, 2006].
The capacity of flavonoids to act as an antioxidant is dependent upon their molecular structure, the position of hydroxyl groups, and other substitutions in the chemical structure of these polyphenols. Numerous exhaustive reviews dealing with their structure, absorption, metabolism, and pharmacokinetics have been published [Bravo, 1998; Ross and Kasum, 2002; Manach and Donovan, 2004]. Besides scavenging free radicals, many phenols also exhibit multiple biological properties, for example, antiinflammatory, anticancer, antiviral, antimicrobial, vasorelaxant, and anticlotting activities [Rahman, 2007]. In general, these phenolic compounds are rapidly converted to their glucuronide derivatives upon ingestion and are transported to the circulatory system and different body organs including the brain. There is evidence that some phenolic compounds exert their mode of action and target different intracellular pathways on a concentration-dependent manner. For example, low dose of red wine polyphenols was shown to promote angiogenesis via activation of the Akt/PI3K/eNOS, p38MAPK pathway but not the NF-KB pathway. However, at high dose, they can be antiangiogenic through inhibition of the Akt/PI3K/eNOS pathway and enhancing the NF-kB pathway [Baron-Menguy et al., 2007]. Another example is epicatechin, which not only exerts antioxidant activity but can also modulate protein kinase signaling pathways, depending on the concentration of the compound administered [Schroeter et al., 2007]. Morever, it has been reported that Epigallocatechin gallate (EGCG), a member of a natural polyphenols family, found in great amount in green tea leaves, is a specific and safe DYRK1A gene (closely associated with Down syndrome phenotypes) inhibitor. The authors suggested that this effect may be due to the modulation of BDNF levels by EGCG [Guedj et al., 2009].
Overall, the antioxidant properties of catechins are more potent than those of α-tocopherol or vitamin C and E [Zhao et al., 1989]. Catechins exert their antioxidative activity by chelating metal ions, such as iron (Fe2+) and copper (Cu2+), and preventing the generation of potentially damaging free radicals [Singh et al., 2008]. Catechins may also transfer an electron to ROS-induced radical sites on DNA and thereby prevent oxidative DNA modifications [Singh et al., 2008]. In addition, Epitachechin (EC) scavenges ROS and inhibits lipid peroxidation [Choi et al., 2001]. It has been suggested that EC chelation reducing the free iron pool, might suppress the translation of APP mRNA and affects the APP levels. In neuronal cells exposed to Aβ, EC decreases malondialdehyde levels and caspase activity, thus protecting against Aβ-induced apoptosis and enhancing hippocampal neuronal survival [Choi et al., 2001]. EC also reduces Aβ-induced oxidative stress in vivo, by decreasing rat hippocampal lipid peroxide [Haque et al., 2008; Kim et al., 2010]. Waterhouse et al. [1996] suggested for the first time that the intake of cocoa and chocolate could contribute to a large proportion of the dietary antioxidants. Since then, several reports have been published demonstrating that cocoa and its products can be sources of flavonoids [Hammerstone et al., 2000; Lamuela-Raventós et al., 2001]. The primary flavonoids in cocoa and chocolate are the flavan-3-ols, epicatechin and catechin (monomeric units), and polymers of these, the proanthocyanidins, also termed procyanidins. Depending on the method used in its production, cocoa powder can contain as much as 10% flavonoids on a dry-weight basis [Steinberg et al., 2003]. The bioavailability of flavonoids in humans ranges from 1% to 26%, depending on the chemical structure and shows individual variability. However, the exact mechanisms of action of the monomeric flavan-3-ols, oligomeric procyanidins, and their metabolites in vivo remain to be elucidated [Steinberg et al., 2003]. In addition to the their free radical scavenging activity, it is likely that these compounds could interact with intracellular signaling mechanisms to elicit a variety of biologic effects [Noé et al., 2004]. In a previous study, the effects of cocoa has been tested in rat pheocromocitoma PC12 cell line treated with Aβ(25–35) demonstrating a neuroprotective effects of cocoa flavonoids in counteracting Aβ-induced cell death [Heo and Lee, 2005].
In the present work, a deeper investigation of cocoa flavonoids effects in AD was performed on a human neuronal model by assaying the BDNF neurotrophin signaling pathway. The results obtained indicate that cocoa polyphenolic extracts other than exerting an aspecific antioxidant action, trigger neuronal survival by increasing the BDNF/TrkB signaling pathway.
Materials and Methods
Materials
Triton X-100, dimethylsulfoxide (DMSO), sodium dodecylsulfate (SDS), Tween20, bovine serum albumine (BSA), l-glutamine, 4′,6 diamino-2-phenylindole dilactate (DAPI), Nonidet P40, sodium deoxycolate, ethylen diamine tetraacetate (EDTA), phenylmethane-sulphonylfluoride (PMSF), sodium fluoride, sodium pyrophosphate, orthovanadate, leupeptin, aprotinin, pepstatin, NaCl, polyvinylidene difluoride (PVDF) sheets, fluorescein-labeled anti-mouse IgG antibodies, rabbit anti-active caspase 3, mouse anti-GAP 43, mouse anti-heavy neurofilament 200 (NF-H), anti P 75 NTR antibodies, Aβ (25–35), and Aβ (1–42) were all purchased from Sigma Chemical Co (St. Louis, CO). Trypsin-EDTA solution, N2 supplement, streptomycin–penicillin were from Gibco Invitrogen GmbH (Austria); Mouse anti p-ERK1/2, rabbit anti-ERK1, rabbit anti-BDNF, rabbit anti-TrkB antibodies were from St. Cruz Biotechnology (Santa Cruz, CA); horseradish peroxidase (HRP)-conjugated anti-rabbit and anti-mouse antibodies were from Vector Laboratories (Burlingame, CA); mouse anti-β-tubulin III antibody was from Promega (Mannheim, Germany); rabbit anti-Pospho Ras/extracellular signal-regulated kinase 5 (pERK5) antibodies was purchased from Upstate, Millipore S.p.A (Milan, Italy). RPMI-1640 medium and fetal bovine serum (FBS) were from Euroclone Ltd (UK); Apoptosis assay kit was from Roche Diagnostic (Indianapolis, IN). Micro BCA protein detection kit was from Pierce (Rockford, IL). Vectashield was purchased from Vector Laboratories (Burlingame, CA). All other chemicals were of the highest analytical grade.
Cell Cultures
SH-SY5Y cells (ATCC) were seeded at 1 × 104 cells/cm2 cultured for 7 DIV in FBS-free RPMI 1640 differentiating medium containing N2 supplement in order to allow the neuronal differentiation.
Aβ Fibril Formation
Aβ (25–35) is frequently used in investigating Aβ properties as a less expensive and more easily handled substitute for the native full-length peptide, Aβ (1–42). Indeed, Aβ (25–35) mimics the toxicological and aggregation properties of the full-length peptide, though these characteristics are enhanced; that is, the shorter peptide is more toxic to cultured neurons, exhibits earlier toxicity, causes more severe membrane protein oxidation, and aggregates faster than the native Aβ (1–42) [Santos et al., 2005]. The amyloid fibrils were obtained as previously described [Varadarajan et al., 2001]. Specifically, the Aβ (25–35) stock solution (500 μM) was prepared dissolving Aβ in FBS-free differentiating medium containing N2 supplement (pH 7.4) and stored at −20°C. The amyloid fibrils were obtained incubating Aβ (25–35) stock solution at 37°C for 8 days.
Fluorimetric Assay
The amyloid polymerization status was checked by the thioflavin T (ThT) fluorescence method before each treatment [White et al., 2005]. ThT binds specifically to amyloid fibrils, and such binding produces a shift in its emission spectrum and an increase in the fluorescent signal, which is proportional to the amount of amyloid formed [Naiki et al., 1991; Inestrosa et al., 1996; Munõz and Inestrosa, 1999]. Following incubation, Aβ in 20 mM Tris-HCl Buffer, pH 8.0, and 1.5 μM ThT in a final volume of 2 ml were analyzed. Fluorescence was monitored at excitation wavelength of 450 nm and emission of 485 nm by spectrofluorimetry, as previously described [Munõz and Inestrosa, 1999].
Cocoa Extraction
Commercial cocoa powder was used for this study. Phenols were extracted from 10 g of cocoa as described in [Lee et al., 2003]. The total phenolic content was determined in the extract according to the Folin–Ciocalteu method.
Cocoa powder (10 g) was dissolved in 50 ml of distilled water (ddH2O) at 100°C for 2 min. The cocoa extracts then were centrifuged in a Sorvall RC-5B refrigerated superspeed centrifuge at 12,000g for 5 min, and the resulting supernatants were used as the final samples [Lee et al., 2003]. The water extracts of cocoa was dried and resuspended in deionized distilled water and used at the final concentrations indicated.
Treatments
For acute experiments cells, differentiated for 7 DIV (days in vitro), were treated with aggregates Aβ25–35 (12.5 μM, f.c.) for 24h. For Cocoa treatment, cells, subjected for 4h to acute challenge with Aβ25–35, were treated with different doses (12–120μg/ml EC, 4–40 μg/ml CE, and 70–700 μg/ml total polyphenols) of cocoa extract. To test the protective effects, differentiated cells were treated with Cocoa extract (30 μg/ml EC, 10 μg/ml CE, and 173 μg/ml of total polyphenols) for 4 DIV. After this treatment Cocoa was removed and the cell were incubated with oligomeric Aβ1–42 (10μM, f.c.) at 37°C for 4 DIV.
Cell Viability and Death
Cells, plated on 24 multiwell plates, were incubated, after treatments, for 2 h with CellTiter 96 AQueous one Solution, a colorimetric method based on 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphe-nil)-2-(4-sulfophenyl)-2H-tetrazolium (MTS). The quantity of formazan formed, as a function of viability, was measured at 490 nm using an ELISA plate reader. All MTS assays were performed in triplicate.
For apoptosis detection, cells were seeded in 24-well plates at a density of 1 × 104 cells/cm2. Control and treated cells were analyzed for apoptosis using the Cell death detection ELISA kit for the nucleosome detection. Absorbances at 405 nm with respect to 490 nm were recorded according to manufacturer's directions.
After Aβ exposure, cells on coverslips were fixed in 4% paraformaldehyde at room temperature for 10 min, then stained with DAPI (300ng/ml) for 20 min and examined under UV illumination, using a fluorescence microscope. To quantify the apoptotic process, nuclei with both fragmented or condensed DNA and normal DNA were counted. Five fields/coverslips were counted. Data are expressed as a percentage of the total cells counted.
Morphometry
Control and treated cells, grown on coverslips, were fixed in 4% paraformaldehyde in PBS for 20 min at RT. After washings, coverslips were mounted with Vectashield and phase-contrast observations were performed by an AXIOPHOT Zeiss microscope, equipped with a micrometric ocular lens. The processes longer than the cell body mean diameter (Ø), which should be regarded as neurites, were counted and the results were expressed as neurites number versus the total cell number. The neurite length was determined by comparing the neurite length with the mean diameter (Ø) of cell soma and reported as neurite length/soma (Ø).
Immunofluorescence
Control and treated cells, grown on coverslips, were fixed with absolute methanol for 10 min at −20°C. After that, cells were rehydrated with PBS for 5 min and incubated with mouse anti-GAP 43 (1:300) and mouse anti-NF-H, (1:200) antibodies, overnight at 4°C. After extensive washings with PBS, cells were treated with fluorescein-labeled anti-mouse IgG secondary antibody (1:100 in PBS containing 3% BSA) for 30 min at RT. Nuclei were counterstained with DAPI (300 ng/ml). After extensive washings, coverslips were mounted with Vectashield mounting medium and photographed in a fluorescence microscope (AXIOPHOT, Zeiss).
Western Blot
Cells were washed in ice-cold PBS and homogenizedin ice-cold RIPA buffer (10mM Hepes, pH 7.4, 10mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM dithiothreitol) with a protease inhibitor mixture (100 mg/ml phenylmethylsulfonyl fluoride, 2 mg/ml aprotinin, 2 mM leupeptin, and 1mg/ml pepstatin). The lysate was subjected to centrifugation at 600g for 30 min at 4°C, and the supernatant was collected. Samples (25–50μg/lane) were analyzed by 10% SDS– PAGE, transferred to PVDF membranes, and blocked in Tris–buffered saline containing 5% non fat milk, and 0.1 % Tween 20. Membranes were incubated with different primary antibodies, anti-BDNF (1:200), anti TrkB (1:200) anti p-75 NTR (1: 100), anti p-ERK1,2 (1:200), anti-p-ERK5 (1:200), anti-GAP 43 (1:500), anti-β-TubIII (1:500), anti-Caspase3 (1:500) overnight at 4°C and then probed with horseradish peroxidase-conjugated mouse or rabbit secondary antibodies (1:1,000). Immunoreactive bands were visualized by chemiluminescence. Band relative densities, against most evident band of PVDF membrane Comassie Blu stained, were determined using TotalLab software (ABEL Science-Ware srl, Italy) and values were given as relative units.
Statistics
Experiments were performed at least in triplicates. Data were represented as mean ± standard errors. Where appropriate, oneway ANOVA test followed by Scheffe's “post hoc test” analysis was performed using SPSS software. P values less than 0.05 were considered statistically significant.
Results
Cell Viability and Death
In preliminary experiments the effects on cell viability of different concentrations of cocoa phenolyc extracts on N2 differentiated cells were assayed. The final concentrations of cocoa which were no toxic for cells ranged from 12–30μg/ml EC; 4–10μg/ml CE; and 70–170 μg/ml total polyphenols, for these reason these concentrations were used for the apoptotic assay.
Cell viability, evaluated by MTS assay, in control and treated cells is shown in Figure 1A. Aβ treatment leads to significant reduction in cell viability. Aβ-cocoa (12–30 μg/ml EC; 4–10 μg/ml CE, and 70– 170 μg/ml total polyphenols) reverts this effect to the control values. In the same figure apoptotic cell death, evaluated as nucleosome concentration, is shown (Fig. 1B). Aβ treatment induces a significant increase in apoptotic cell death, while after Aβ-cocoa 1 and 2 treatments no significant differences are observed with respect to the control. The dilution containing 30μg/ml EC, 10μg/ml CE, and 170 μg/ml total polyphenols, protected more efficiently cell viability by Aβ-treatment, therefore this concentration (cocoa 2) was used for all subsequent experiments.
Fig. 1.
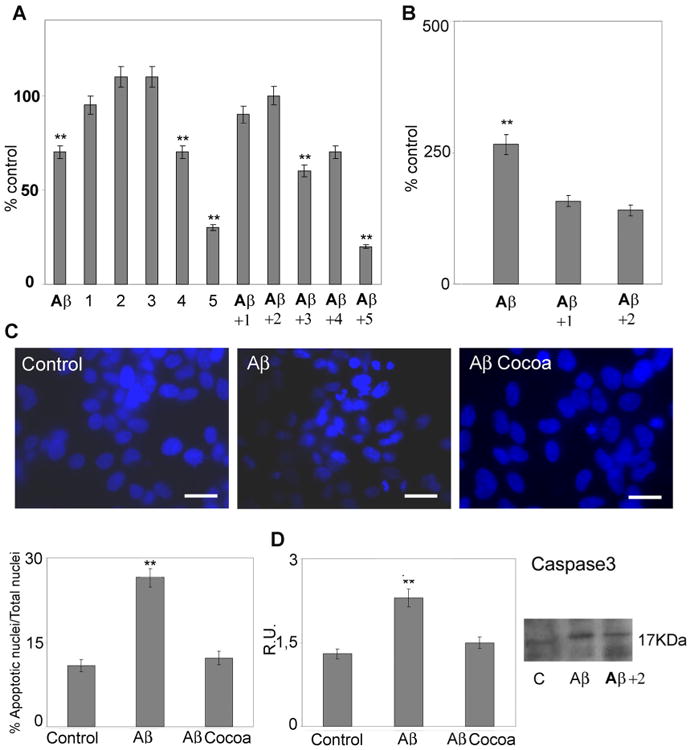
Cell viability and death. Right: A: Cell viability was evaluated by MTS assay in control and treated cells. 1:12 μg/ml EC, 4μg/ml CE, 70 μg/ml total polyphenols; 2:30μg/ml of EC, 10 μg/ml CE, 170 μg/ml total polyphenols; 3:60 μg/ml of EC, 20 μg/ml CE, 340 μg/ml of total polyphenols; 4:120 μg/ml EC, 40μg/ml CE,and 700 μg/ml total polyphenols; 5: 240μg/ml EC, 80 μg/ml CEand 1,400μg/ml total polyphenols. B: Apoptosis was evaluated bycytoplasmaticnucleosome concentration. 1:12 μg/ml EC, 4 μg/ml CE, 70μg/ml total polyphenols; 2: 30μg/ml of EC, 10μg/ml CE, 170μg/ml total polyphenols. Data are mean ± SD of three different experiments run in triplicate. **P < 0.005. C: nuclear fragmentation in control and treated cells, evaluated by DAPI nuclear staining, after Aβ treatment administered alone or with coca 2 (30 μg/ml of EC, 10 μg/ml CE, 170 μg/ml total polyphenols). Five fields/coverslips were counted. Data are mean ± SD of three different experiments. **P < 0.005. Bar = 26 μm. D: Western blotting and relative densitometric analysis of active caspase 3. Data are mean ± SD of three different experiments. **P < 0.005. Cocoa extract was used at the final concentrations of 30μg/ml of EC, 10μg/ml CE, 170μg total polyphenols.
In panel C, the nuclear fragmentation in control and treated cells, evaluated by DAPI nuclear staining, is shown. Consistently with the apoptotic assay, Aβ treatment leads to an increase in apoptotic nuclei, while Aβ-cocoa almost restores the control condition. These results are further confirmed by the Western blotting analysis of active caspase 3 (Fig. 1D). In fact, upon Aβ challenge the active form of caspase 3 is increased, thus indicating apoptosis promotion. Cocoa prevents caspase 3 activation restoring the levels to those of control cells. These results, taken together, indicate that the cocoa plays a protective effect against the Aβ cytotoxic insult.
Cell Morphology
Figure 2 shows the contrast phase microscopy and the graphic representation of neurite length and number in control and treated cells. Control cells (N2) show an evident neuronal clustering and neuronal aggregation, Aβ treatment induced an evident neurite loss (Aβ). Aβ-cocoa protects cells from neurite atrophy. The graphical representation of number of neurite, and length shows that Aβ treatment significantly decreases neurite number and length, while cocoa protects the neurites from Aβ-mediated damage.
Fig. 2.
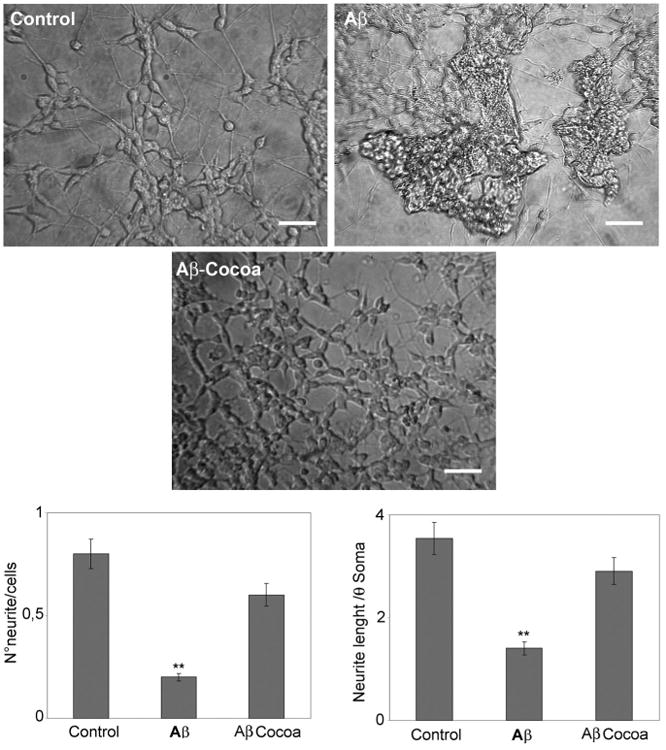
contrast phase microscopy and the graphic representation of neurite length and number in control and treated cells. Data are mean ± SD of three different experiments. **P < 0.005. Bar = 90 μm. Cocoa extract was used at the final concentrations of 30μg/ml of EC, 10μg/ml CE, 170μg total polyphenols.
Neuronal Cytoskeleton Proteins
Since neuronal loss and neuritic/cytoskeletal lesions (synaptic disconnection and proliferation of dystrophic neurites) represent major dementia-associated abnormalities in AD, neuronal differentiation marker localization, such as Neurofilament 200 (NF-H200) and GAP-43 and were investigated in control and treated cells (Figs. 3 and 4). NF-H is a marker of neuronal terminal maturation, while GAP-43 is an axonal marker. In control cells NF-H 200 and GAP-43 localization shows an organized cytoskeletal structure that is lost after Aβ treatment, where a significant decrease of neurite branching is observed. Cocoa preserved cytoskeletal organization, thus confirming the neuroprotective effects by cocoa in counteracting neuronal dystrophy. These findings are further confirmed by the Western blotting analysis for β-tubulin III and GAP-43 shown in Figure 5. Aβ treatment significantly reduces the proteins, cocoa reverts this effect to control levels.
Fig. 3.
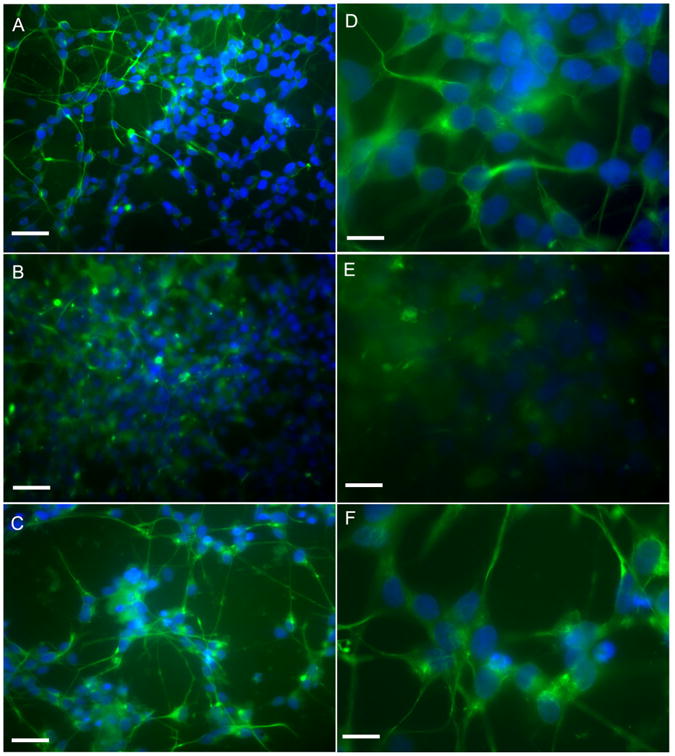
Neurofilament 200 localization in control and treated cell shown at different magnification. Bar A, B, and C = 90μm. Bar D, E, and F = 30 μm. Cocoa extract was used at the final concentrations of 30 μg/ml of EC, 10 μg/ml CE, 170 μg total polyphenols. A and D: control cells; B and E: Aβ-treated cells; C and F: Aβ + cocoa 2 treated cells. Cocoa 2 = 30 μg/ml of EC, 10μg/ml CE, 170μg total polyphenols.
Fig. 4.
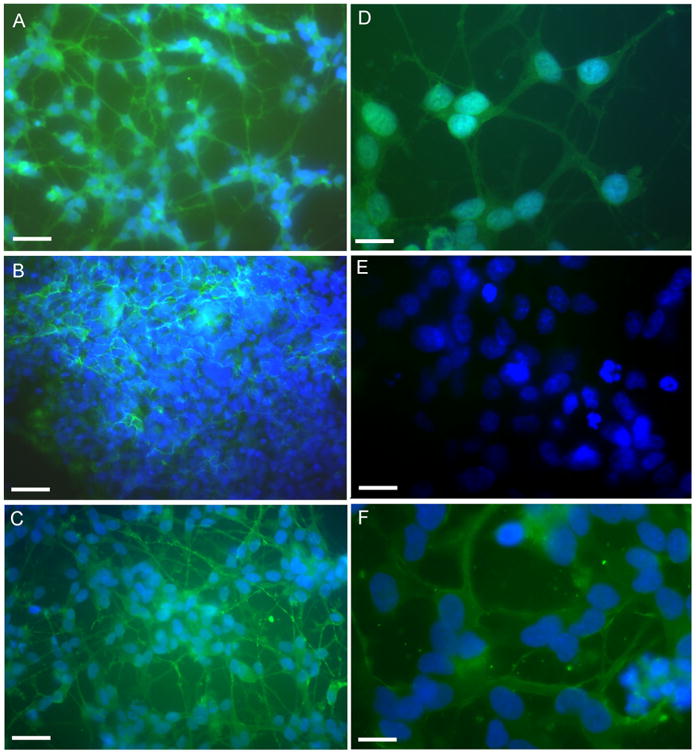
GAP-43 immuno localization in control and treated cell shown at different magnification. Bar A, B, and C = 90μm. Bar D, E, and F = 30 μm. Cocoa extract was used at the final concentrations of 30μg/ml of EC, 10μg/ml CE, 170μg total polyphenols. A and D: control cells; B and E: Aβ-treated cells; C and F: Aβ + cocoa 2 treated cells. Cocoa 2=30μg/ml of EC, 10μg/ml CE, 170μg total polyphenols.
Fig. 5.
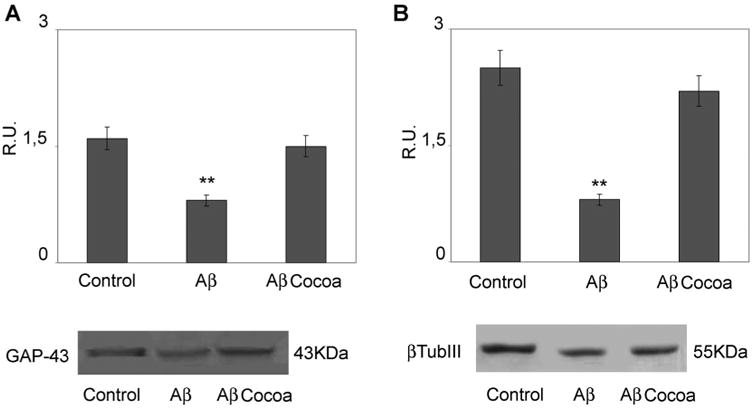
Western blotting and relative densitometric analysis of GAP43 and β-tubulin III in control and treated cells. Data are mean ± SD of three different experiments. **P < 0.005. Cocoa extract was used at the final concentrations of 30 μg/ml of EC, 10 μg/ml CE, 170μg total polyphenols.
Signal Transduction Pathways
The signal transduction pathways involved in neuronal survival, neurotrophins modulation, and neuronal death were investigated in control and treated cells (Figs. 6 and 7).
Fig. 6.
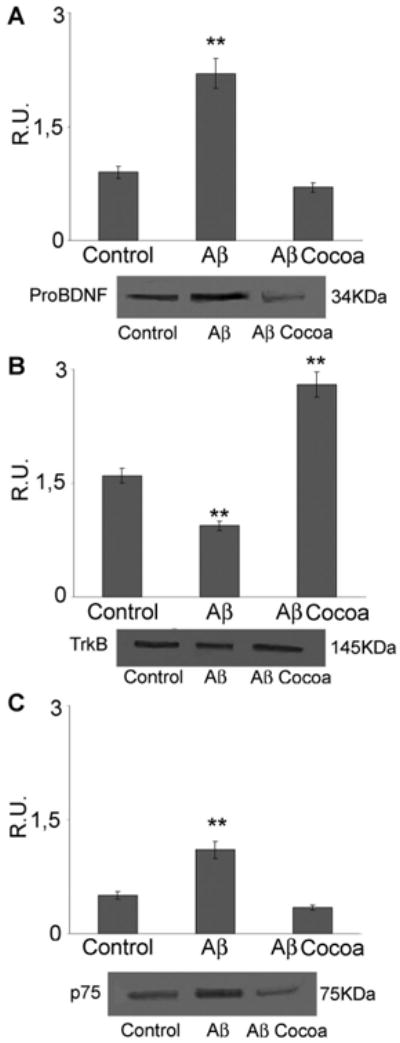
Western blotting and relative densitometric analysis of pro-BDNF, TrkB, and p75NTR in control and treated cells. Data are mean ± SD of three different experiments. **P < 0.005. Cocoa extract was used at the final concentrations of 30μg/ml of EC, 10μg/ml CE, 170μg total polyphenols.
Fig. 7.
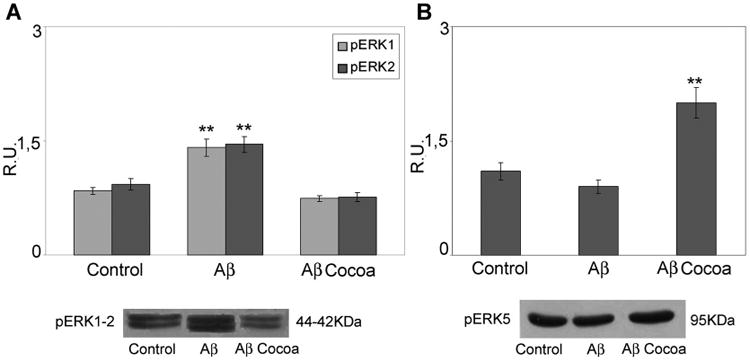
Western blotting and relative densitometric analysis of ERK1, 2 and E RK5 in control and treated cells. Data are mean ± SD of three different experiments. **P < 0.005. Cocoa extract was used at the final concentrations of 30 μg/ml of EC, 10 μg/ml CE, 170 μg total polyphenols.
Neuronal morphology and plasticity are related to the brain derived neurotrophic factor (BDNF) signal transduction pathway, therefore we assayed the BDNF, its receptors such as TrkB and p75, and the extracellular signal regulated kinases such as ERK1,2 and ERK5. Brain derived neurotrophic factor (BDNF) is a neurotrophin involved in neuronal survival and differentiation. Its activity is mediated by the high affinity receptor TrkB and by the low affinity receptor p75NTR. Upon Aβ challenge, the cytoplasmic levels of BDNF immature form (pro-BDNF) appear upregulated (Fig. 6). This strong increase of pro-BDNF may be responsible for the promotion of the neuronal death and atrophy, as it is known that the immature form of BDNF induces neuronal apoptosis via activation of a receptor complex p75NTR-sortilin [Teng et al., 2005]. This notion is supported by the results obtained, in our experimental condition, for p75NTR protein (Fig. 6). In fact, as observed for pro-BDNF, Aβ increases p75NTR protein levels, while concomitantly triggers a decrease of the specific receptor TrkB involved in the action of mature and cleaved form of BDNF. Moreover, Aβ induces the active form of ERK1,2 (p-ERK1,2; Fig. 7), known to be involved in apoptosis promotion. In Aβ-treated cells, following cocoa, a complete reversion of pro-BDNF levels is observed. In the same time, cocoa, significantly increase TrkB as well as the p-ERK5 (Fig. 7), involved in neuronal survival, with the concomitant decrease of ERK1,2, thus suggesting an activation of the neuronal survival pathway BDNF/TrkB/ERK5.
Preventive Neuroprotective Effects of Cocoa
Finally, in order to assess if cocoa may exerts preventive effects, cells were pre-treated for 4 days with cocoa extract (30 μg/ml EC, 10 μg/ml CE, and 170 μg/ml total polyphenols) and then challenged with oligomeric Aβ (1–42) for further 4 days. The viability assay (A) shows that, indeed, cocoa exerts a strong neuroprotective effect on cell survival with respect to Aβ-treated cells (Fig. 8). Moreover, the same figure shows that cocoa pre-treatment prevents apoptosis promotion in Aβ-treated cells (B).
Fig. 8.
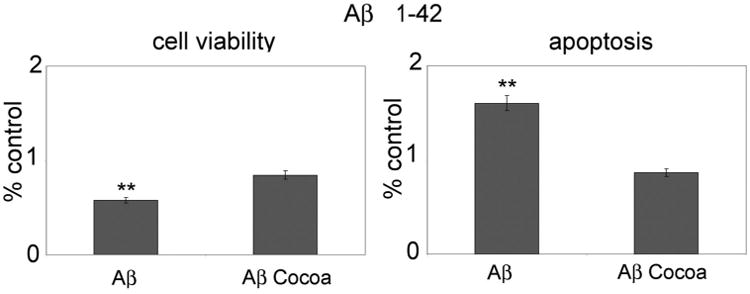
Cell viability (A) and cell death (B) upon oligomeric Aβ (1–42) challenge. Data are mean ± SD of three different experiments run in triplicate. **P < 0.005. Cocoa extract was used at the final concentrations of 30 μg/ml of EC, 10 μg/ml CE, 170μg total polyphenols.
Discussion
Oxidative stress is considered as a risk factor in the incidence and progression of cognitive declines occurring during normal cerebral aging and in dementia and playing a critical role in many neurodegenerative processes, such as AD and Parkinson's disease [Jenner, 1996; Selkoe, 2002]. Recently, food-derived antioxidants such as vitamins and phytochemicals have received growing attention, as chemopreventive agents against oxidative damage. Many of the biological actions of phytochemicals including flavonoids have been attributed to their antioxidant properties [Rice-Evans, 2001]. Aβ 25–35 has neurotoxicity comparable to that of Aβ (1–42) and produces free radical adducts in aqueous solutions. It sensitizes neurons to injury resulting from oxidative stress-induced neurotoxicity induced by glutamate or free radicals [Cimini et al., 2009a]. In a previous study, Aβ (25–35) has been shown to induce cytotoxicity in a rat PC12 neuronal model where cocoa extracts were able to protect PC12 cells by apoptotic death [Heo and Lee, 2005]. To get more insight into the mechanisms triggered by cocoa in counteracting Aβ cytotoxicity, in the present study, the neuroprotective and preventive role of cocoa from Aβ-induced cytotoxicity, has been investigated particularly focusing on parameters involved in neuronal survival and in the maintenance of neuronal network, such as BDNF signaling.
In our experimental conditions, in agreement with previous studies [D'Angelo et al., 2009], the acute treatment with Aβ was neurotoxic to cells, as demonstrated by the loss of neurite branching and the decrease of cell viability. In fact, control cells, which under control conditions form extensive networks of healthy and long neurites, after Aβ treatment show evident morphological alterations and axonal and dendritic dystrophy. Cocoa extract protects cell viability and cell morphology from Aβ injury suggesting a neurotrophic role for this compound.
The increase of survival triggered by cocoa was paralleled by the maintenance of neuronal network, as evaluated by neurites length and number and by the expression and localization of the cytoskeleton proteins such as NF200, β-tubulin III, and GAP-43.
Aβ-treated cells showed an interesting picture as to regard BDNF, TrkB, p75NTR, ERK1,2 and ERK5. In our experimental conditions, upon Aβ challenge, pro-BDNF and p-ERK1,2 appear all increased and this effect may be related to the known “dark side” effect of pro-BDNF under oxidative stress conditions [Teng et al., 2005]: pro-BDNF may activate, through p-75 and sortilin, ERK1,2, which, in AD, has been involved in apoptotic neuronal death [White et al., 2005]. Moreover, Aβ slightly decreased also the active form of ERK5, known to be involved in neuronal survival. On the other hand, BDNF is known to be effective in the promotion of neuronal survival in the presence of anti-oxidants [Teng et al., 2005]. In this view, cocoa, by decreasing pro-BDNF and p75NTR and increasing TrkB and ERK5 levels, tries to counteract neuronal death and to promote cell survival, since Aβ plaques are however present under cocoa treatment.
It has been previously reported that oligomeric Aβ is more toxic than fibrils, especially concerning the induction of neuroinflammation and of subtle and early neuronal damage [Chong et al., 2006; Cimini et al., 2009a,b]. In this study, we show that cocoa is also able to exert a preventive effect on the progression of AD, since, when administered before the chronic treatment with oligomeric Aβ (1–42), it promotes neuronal survival with respect to cells treated only with oligomeric Aβ (1–42).
On the basis of the results obtained it is possible to speculate that under oxidative injury the survival pathways depending on BDNF are downregulated with concomitant increase of the pro-BDNF pathway, as suggested by Teng et al. [2005]. The antioxidants, like cocoa extracts, are known to counteract oxidative stress and this action is, in this model, closed related to an increase of the specific TrkB receptor and a decrease of pro-BDNF. This suggests that TrkB expression and BDNF cleavage may be ROS-dependent events and that polyphenols may be involved in this process for their anti-oxidant properties.
Conclusions
The results obtained appear original since demonstrate that cocoa seems to affect, directly or indirectly, signal transduction pathways involved in neuronal death and neuroprotection, supporting the possibility of its potential use as preventive agent for neurodegenerative diseases characterized by oxidative stress. Although additional confirmatory studies are warranted, these findings suggest that the regular dietary inclusion of flavanols could be one element of a dietary approach for the maintaining and improving brain health, as previously suggested [Desideri et al., 2012; Williams and Spencer, 2012].
Supplementary Material
Supplementary Fig. 1 is the total gel staining for Western blotting presented in Figs. 1 and 5.
Supplementary Fig. 2 is the total gel staining for Western blotting presented in Figs. 6 and 7.
Acknowledgments
This work has been supported by RIA 2010 (Prof. Cimini and Desideri).
Grant sponsor: RIA 2010; Grant sponsor: Human Health Foundation.
Footnotes
Supporting Information: Additional supporting information may be found in the online version of this article at the publisher's web-site.
References
- Baron-Menguy C, Bocquet A, Guihot AL, Chappard D, Amiot MJ, Andriantsitohaina R, Loufrani L, Henrion D. Effects of red wine polyphenols on postischemic neovascularization model in rats: Low doses are proangiogenic, high doses anti-angiogenic. FASEB J. 2007;21:3511–3521. doi: 10.1096/fj.06-7782com. [DOI] [PubMed] [Google Scholar]
- Bravo L. Polyphenols: Chemistry, dietary sources, metabolism, and nutritional significance. Nutr Rev. 1998;56:317–333. doi: 10.1111/j.1753-4887.1998.tb01670.x. [DOI] [PubMed] [Google Scholar]
- Choi YT, Jung CH, Lee SR, Bae JH, Baek WK, Suh MH, Park J, Park CW, Suh SI. The green tea polyphenol (—)-epigallocatechin gallate attenuates beta-amyloid-induced neurotoxicity in cultured hippocampal neurons. Life Sci. 2001;70:603–614. doi: 10.1016/s0024-3205(01)01438-2. [DOI] [PubMed] [Google Scholar]
- Chong YH, Shin YJ, Lee EO, Kayed R, Glabe CG, Tenner AJ. ERK1/2 activation mediates Abeta oligomer-induced neurotoxicity via caspase-3 activation and tau cleavage in rat organotypic hippocampal slice cultures. J Biol Chem. 2006;281:20315–20325. doi: 10.1074/jbc.M601016200. [DOI] [PubMed] [Google Scholar]
- Christen Y. Oxidative stress and Alzheimer disease. Am J Clin Nutr. 2000;71:621S–629S. doi: 10.1093/ajcn/71.2.621s. [DOI] [PubMed] [Google Scholar]
- Cimini A, Moreno S, D'Amelio M, Cristiano L, D'Angelo B, Falone S, Benedetti E, Carrara P, Fanelli F, Cecconi F, Amicarelli F, Cerù MP. Early biochemical and morphological modifications in the brain of a transgenic mouse model of Alzheimer disease. A role for peroxisomes. J Alzheimers Dis. 2009a;18:935–952. doi: 10.3233/JAD-2009-1199. [DOI] [PubMed] [Google Scholar]
- Cimini A, Benedetti E, D'Angelo B, Cristiano L, Falone S, Di Loreto S, Amicarelli F, Cerù MP. Neuronal response of peroxisomal and peroxisome-related proteins to chronic and acute Aβ injury. Curr Alzh Res. 2009b;6:238–250. doi: 10.2174/156720509788486518. [DOI] [PubMed] [Google Scholar]
- Commenges D, Scotet V, Renaud S, Jacqmin-Gadda H, Barberger-Gateau P, Dartigues JF. Intake of flavonoids and risk of dementia. Eur J Epidemiol. 2000;16:357–363. doi: 10.1023/a:1007614613771. [DOI] [PubMed] [Google Scholar]
- D'Angelo B, Santucci S, Benedetti E, Di Loreto S, Phani RA, Falone S, Amicarelli F, Cerù MP, Cimini A. Cerium oxide nanoparticles trigger neuronal survival in a human alzheimer disease model by modulating BDNF pathway current nanoscience. Curr Nanosci. 2009;5:167–176. [Google Scholar]
- Dajas F, Rivera F, Blasina F, Arredondo F, Echeverry C, Lafon L, Morquio A, Heizen H. Cell culture protection and in vivo neuroprotective capacity of flavonoids. Neurotox Res. 2003;5:425–432. doi: 10.1007/BF03033172. [DOI] [PubMed] [Google Scholar]
- de Rijk MC, Breteler MM, den Breeijen JH, Launer LJ, Grobbee DE, van der Meché FG, Hofman A. Dietary antioxidants and Parkinson disease. The Rotterdam Study. Arch Neurol. 1997;54:762–765. doi: 10.1001/archneur.1997.00550180070015. [DOI] [PubMed] [Google Scholar]
- Deschamps V, Barberger-Gateau P, Peuchant E, Orgogozo JM. Nutritional factors in cerebral aging and dementia: Epidemiological arguments for a role of oxidative stress. Neuroepidemiology. 2001;220:7–15. doi: 10.1159/000054752. [DOI] [PubMed] [Google Scholar]
- Desideri G, Kwik-Uribe C, Grassi D, Necozione S, Ghiadoni L, Mastroiacovo D, Raffaele A, Ferri L, Bocale R, Lechiara MC, Marini C, Ferri C. Benefits in cognitive function, blood pressure, and insulin resistance through cocoa flavanol consumption in elderly subjects with mild cognitive impairment: The Cocoa, Cognition, and Aging (CoCoA) study. Hypertension. 2012;60:794–801. doi: 10.1161/HYPERTENSIONAHA.112.193060. [DOI] [PubMed] [Google Scholar]
- Engelhart MJ, Geerlings MI, Ruitenberg A, van Swieten JC, Hofman A, Witteman JC, Breteler MM. Dietary intake of antioxidants and risk of Alzheimer disease. JAMA. 2002;287:3223–3229. doi: 10.1001/jama.287.24.3223. [DOI] [PubMed] [Google Scholar]
- Fu Y, Koo MW. EGCG protects HT-22 cells against glutamate-induced oxidative stress. Neurotox Res. 2006;10:23–30. doi: 10.1007/BF03033331. [DOI] [PubMed] [Google Scholar]
- Guedj F, Sébrié C, Rivals I, Ledru A, Paly E, Bizot JC, Smith D, Rubin E, Gillet B, Arbones M, Delabar JM. Green tea polyphenols rescue of brain defects induced by overexpression of DYRK1A. PLoS ONE. 2009;4:e4606. doi: 10.1371/journal.pone.0004606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammerstone JF, Lazarus SA, Schmitz HH. Procyanidin content and variation in some commonly consumed foods. J Nutr. 2000;130:2086S–2092S. doi: 10.1093/jn/130.8.2086S. [DOI] [PubMed] [Google Scholar]
- Haque AM, Hashimoto M, Katakura M, Hara Y, Shido O. Green tea catechins prevent cognitive deficits caused by Abeta 1–40 in rats. J Nutr Biochem. 2008;19:619–626. doi: 10.1016/j.jnutbio.2007.08.008. [DOI] [PubMed] [Google Scholar]
- Heo HJ, Lee CY. Epicatechin and catechin in cocoa inhibit amyloid β protein induced apoptosis. J Agric Food Chem. 2005;53:1445–1448. doi: 10.1021/jf048989m. [DOI] [PubMed] [Google Scholar]
- Inestrosa NC, Alvarez A, Perez CA, Moreno RD, Vicente M, Linker C, Casanueva OI, Soto C, Garrido J. Acetylcholinesterase accelerates assembly of amyloid-beta-peptides into Alzheimer's fibrils: Possible role of the peripheral site of the enzyme. Neuron. 1996;16:881–891. doi: 10.1016/s0896-6273(00)80108-7. [DOI] [PubMed] [Google Scholar]
- Jenner P. Oxidative stress in Parkinson's disease and other neurodegenerative disorders. Pathol Biol. 1996;44:57–64. [PubMed] [Google Scholar]
- Joseph JA, Shukitt-Hale B, Casadesus G. Reversing the deleterious effects of aging on neuronal communication and behavior: Beneficial properties of fruit polyphenolic compounds. Am J Clin Nutr. 2005;81:313S–316S. doi: 10.1093/ajcn/81.1.313S. [DOI] [PubMed] [Google Scholar]
- Kim J, Lee HJ, Lee KW. Naturally occurring phytochemicals for the prevention of Alzheimer's Disease. J Neurochem. 2010;112:1415–1430. doi: 10.1111/j.1471-4159.2009.06562.x. [DOI] [PubMed] [Google Scholar]
- Lamuela-Raventós RM, Andrés-Lacueva C, Permanyer J, Izquierdo-Pulido M. More antioxidants in cocoa. J Nutr. 2001;131:834. doi: 10.1093/jn/131.3.834. [DOI] [PubMed] [Google Scholar]
- Lee KW, Kim YJ, Lee HJ, Lee CY. Cocoa has more phenoloc phytochemicals and a higher antioxidant capacity than teas and red wine. J Agric Food Chem. 2003;51:7292–7295. doi: 10.1021/jf0344385. [DOI] [PubMed] [Google Scholar]
- Lovell MA, Ehmann WD, Butler SM, Markesbery WR. Elevated thiobarbituric acid-reactive substances and antioxidant enzyme activity in the brain in Alzheimer's disease. Neurology. 1995;45:1594–1601. doi: 10.1212/wnl.45.8.1594. [DOI] [PubMed] [Google Scholar]
- Manach C, Donovan JL. Pharmacokinetics and metabolism of dietary flavonoids in humans. Free Radic Res. 2004;38:771–785. doi: 10.1080/10715760410001727858. [DOI] [PubMed] [Google Scholar]
- Mandel S, Youdim MB. Catechin polyphenols: Neurodegeneration and neuroprotection in neurodegenerative diseases. Free Radic Biol Med. 2004;37:304–317. doi: 10.1016/j.freeradbiomed.2004.04.012. [DOI] [PubMed] [Google Scholar]
- Marcus DL, Thomas C, Rodriguez C, Simberkoff K, Tsai JS, Strafaci JA, Freedman ML. Increased peroxidation and reduced antioxidant enzyme activity in Alzheimer's disease. Exp Neurol. 1998;150:40–44. doi: 10.1006/exnr.1997.6750. [DOI] [PubMed] [Google Scholar]
- Markesbery WR. Oxidative stress hypothesis in Alzheimer's disease. Free Radic Biol Med. 1997;23:134–147. doi: 10.1016/s0891-5849(96)00629-6. [DOI] [PubMed] [Google Scholar]
- Munõz FJ, Inestrosa NC. Neurotoxicity of acetylcholinesterase amyloid beta-peptide aggregates is dependent on the type of Abeta peptide and the AChE concentration present in the complexes. FEBS Lett. 1999;450:205–209. doi: 10.1016/s0014-5793(99)00468-8. [DOI] [PubMed] [Google Scholar]
- Naiki H, Higuchi K, Nakakuki K, Takeda T. Kinetic analysis of amyloid fibril polymerization. Lab Invest. 1991;65:104–110. [PubMed] [Google Scholar]
- Noé V, Penuelas S, Lamuela-Raventós RS, Permanyer J, Ciudad CJ, Izquierdo-Pulido M. Epicatechin and a cocoa polyphenolic extract modulate gene expression in human Caco-2. Cells J Nutr. 2004;134:2509–2516. doi: 10.1093/jn/134.10.2509. [DOI] [PubMed] [Google Scholar]
- Nunomura A, Perry G, Pappolla MA, Wade R, Hirai K, Chiba S, Smith MA. RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer's disease. J Neurosci. 1999;19:1959–1964. doi: 10.1523/JNEUROSCI.19-06-01959.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oken BS, Storzbach DM, Kaye JA. The efficacy of Ginkgo biloba on cognitive function in Alzheimer disease. Arch Neurol. 1998;55:1409–1415. doi: 10.1001/archneur.55.11.1409. [DOI] [PubMed] [Google Scholar]
- Rahman K. Studies on free radicals, antioxidants, and co-factors. Clin Interv Aging. 2007;22:219–236. [PMC free article] [PubMed] [Google Scholar]
- Ramassamy C. Emerging role of polyphenolic compounds in the treatment of neurodegenerative diseases: A review of their intracellular targets. Eur J Pharmacol. 2006;545:51–64. doi: 10.1016/j.ejphar.2006.06.025. [DOI] [PubMed] [Google Scholar]
- Rice-Evans C. Flavonoid antioxidants. Curr Med Chem. 2001;8:797–807. doi: 10.2174/0929867013373011. [DOI] [PubMed] [Google Scholar]
- Ross JA, Kasum CM. Dietary flavonoids: Bioavailability, metabolic effects, and safety. Annu Rev Nutr. 2002;22:19–34. doi: 10.1146/annurev.nutr.22.111401.144957. [DOI] [PubMed] [Google Scholar]
- Santos MJ, Quintanilla RA, Toro A, Grandy R, Dinamarca MC, Godoy JA, Inestrosa NC. Peroxisomal proliferation protects from β-Amyloid neurodegeneration. J Biol Chem. 2005;80:41057–41068. doi: 10.1074/jbc.M505160200. [DOI] [PubMed] [Google Scholar]
- Sayre LM, Zagorski MG, Surewicz WK, Krafft GA, Perry G. Mechanisms of neurotoxicity associated with amyloid beta deposition and the role of free radicals in the pathogenesis of Alzheimer's disease: A critical appraisal. Chem Res Toxicol. 1997;10:518–526. doi: 10.1021/tx970009n. [DOI] [PubMed] [Google Scholar]
- Schroeter H, Bahia P, Spencer JP, Sheppard O, Rattray M, Cadenas E, Rice-Evans C, Williams RJ. (_)Epicatechin stimulates ERK-dependent cyclic AMP response element activity and up-regulates GluR2 in cortical neurons. J Neurochem. 2007;101:1596–1606. doi: 10.1111/j.1471-4159.2006.04434.x. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Cell biology of protein misfolding: The examples of Alzheimer's and Parkinson's diseases. Nat Cell Biol. 2004;6:1054–1061. doi: 10.1038/ncb1104-1054. [DOI] [PubMed] [Google Scholar]
- Simonyi A, Wang Q, Miller RL, Yusof M, Shelat PB, Sun AY, Sun GY. Polyphenols in cerebral ischemia: Novel targets for neuroprotection. Mol Neurobiol. 2005;31:135–147. doi: 10.1385/MN:31:1-3:135. [DOI] [PubMed] [Google Scholar]
- Singh M, Arseneault M, Sanderson T, Murthy V, Ramassamy C. Challenges for research on polyphenols from foods in Alzheimer's disease: Bioavailability, metabolism, and cellular and molecular mechanisms. J Agric Food Chem. 2008;56:4855–4873. doi: 10.1021/jf0735073. [DOI] [PubMed] [Google Scholar]
- Smith MA, Tabaton M, Perry G. Early contribution of oxidative glycation in Alzheimer disease. Neurosci Lett. 1996;217:210–211. [PubMed] [Google Scholar]
- Steinberg FM, Bearden MM, Keen CL. Cocoa and chocolate flavonoids: Implications for cardiovascular health. J Am Diet Assoc. 2003;103:215–223. doi: 10.1053/jada.2003.50028. [DOI] [PubMed] [Google Scholar]
- Teng HK, Teng KK, Lee R, Wright S, Tevar S, Almeida RD, Kermani P, Torkin R, Chen ZY, Lee FS, Kraemer RT, Nykjaer A, Hempstead BL. ProBDNF induces neuronal apoptosis via activation of a receptor complex of p75NTR and sortilin. J Neurosci. 2005;25:5455–5463. doi: 10.1523/JNEUROSCI.5123-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadarajan S, Yatin S, Aksenova M, Butterfield DA. Alzheimer's amyloid beta-peptide-associated free radical oxidative stress and neurotoxicity. J Struct Biol. 2000;130:184–208. doi: 10.1006/jsbi.2000.4274. [DOI] [PubMed] [Google Scholar]
- Varadarajan S, Kanski J, Aksenova M. Different mechanisms of oxidative stress and neurotoxicity for Alzheimer's Aβ(1–42) and Aβ (25–35) J Am Chem Soc. 2001;123:5625–5631. doi: 10.1021/ja010452r. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ. A beta oligomers—A decade of discovery. J Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- Waterhouse AL, Shirley JR, Donovan JL. Antioxidants in chocolate. Lancet. 1996;348:834. doi: 10.1016/S0140-6736(05)65262-2. [DOI] [PubMed] [Google Scholar]
- White JA, Manelli AM, Holmberg KH, Van Eldik LJ, Ladu MJ. Differential effects of oligomeric and fibrillar amyloid-beta 1–42 on astrocyte-mediated inflammation. Neurobiol Dis. 2005;18:459–465. doi: 10.1016/j.nbd.2004.12.013. [DOI] [PubMed] [Google Scholar]
- Williams RJ, Spencer JP. Flavonoids, cognition, and dementia: Actions, mechanisms, and potential therapeutic utility for Alzheimer disease. Free Radic Biol Med. 2012;52:35–45. doi: 10.1016/j.freeradbiomed.2011.09.010. [DOI] [PubMed] [Google Scholar]
- Zhao BL, Li XJ, He RG, Cheng SJ, Xin WJ. Scavenging effect of extracts of green tea and natural antioxidants on active oxygen radicals. Cell Biophys. 1989;14:175–185. doi: 10.1007/BF02797132. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. 1 is the total gel staining for Western blotting presented in Figs. 1 and 5.
Supplementary Fig. 2 is the total gel staining for Western blotting presented in Figs. 6 and 7.