

Summary
von Willebrand’s disease (VWD) is probably the most common bleeding disorder, with some studies indicating that up to 1% of the population may have the condition. Over recent years interest in VWD has fallen compared to that of haemophilia, partly the result of focus on blood-borne diseases such as HIV and hepatitis. Now the time has come to revisit VWD, and in view of this some 60 international physicians with clinical and scientific interest in VWD met over 4 days in 2010 in the Åland islands to discuss state-of-the-art issues in the disease. The Åland islands are where Erik von Willebrand had first observed a bleeding disorder in a number of members of a family from Föglö, and 2010 was also the 140th anniversary of his birth. This report summarizes the main papers presented at the symposium; topics ranged from genetics and biochemistry through to classification of VWD, pharmacokinetics and laboratory assays used in the diagnosis of the disease, inhibitors, treatment guidelines in different age groups including the elderly who often have comorbid conditions that present challenges, and prophylaxis. Other topics included managing surgeries in patients with VWD and the role of FVIII in VWF replacement, a controversial subject.
Keywords: factor VIII, prophylaxis, treatment, von Wille-brand factor, von Willebrand’s disease
Introduction (Erik Berntorp)
In September 2010, a group of around 60 physicians from all over the world with clinical and scientific interest in von Willebrand’s disease (VWD) met to give an update on current research and treatment in VWD. It was also the 140th anniversary of the birth of Erik von Willebrand (1870–1949) who in 1926 published his first article on a bleeding disorder that he had observed in a number of members of a family from Föglö in the Åland islands (1; see Fig. 1). His first case was a 5-year-old girl, Hjördis S., who had bleeding symptoms, as did most of her 11 siblings [2]. During her fourth menstrual period at the age of 13, Hjö rdis bled to death, as had four of her sisters before her from bleeding from the nose, wounds and/or the intestinal canal (Fig. 2). von Willebrand published several articles describing the disease [3,4], and his descriptions are still relevant today.
Fig. 1.
The Åland islands.
Fig. 2.

Hjördis’ grave.
Progress in the understanding of VWD over the last 50 years (Ian Peake)
von Willebrand’s disease is a common inherited bleeding disorder, characterized by a deficiency of plasma (and platelet) von Willebrand factor (VWF) and factor VIII (FVIII) which result in mucocutaneous bleeding. Classification and diagnosis of VWD is important to identify the prognosis and correct treatment for the individual patient. There have been many articles published on the classification of VWD over the years. The earliest was an article by Soulier and Larrieu in 1954 [5]; later articles include a study by Rodeghiero et al. [6]. Landmark technologies in VWD classification and diagnosis include VWF antigen (VWF:Ag) and ristocetin cofactor activity (VWF:RCo) in the 1970s, VWF multimer analysis in the 1980s, and VWF DNA and RNA analysis in the 1990s/2000s. In the late 1970s, Arthur Bloom and I suggested seven diagnostic criteria for VWD:
A prolonged bleeding time.
Autosomal inheritance.
Reduced FVIII.
Reduced VWF:Ag.
A secondary rise in FVIII following transfusion.
Impaired ristocetin-induced platelet aggregation.
Reduced platelet adhesiveness.
In 1981 Ruggeri and Zimmerman published an article classifying variant VWD subtypes by analysing functional characteristics and multimeric composition of VWF [7]. Later in 1987, they extended their research and published an article specifying 11 subtypes of type 1 VWD and 13 for type 2 [8]. However, they also suggested a possible general classification as follows:
Patients with quantitative abnormalities and no evidence of intrinsic functional abnormality of VWF.
Patients whose VWF has low VWF:RCo.
Patients with enhanced responsiveness to ristocetin.
Patients with type 3 (severe) VWD.
Later, in 1994, the modern classification of the disease was published for the VWF Scientific and Standardization Committee (SSC) Subcommittee of the ISTH [9]. It was proposed that all VWD is caused by mutations at the VWF locus and divided quantitative defects into partial deficiency (type 1) and severe deficiency (type 3). Qualitative defects were divided into four subcategories: type 2A, 2B, 2M and 2N. In 2006, there was an update on the classification [10] when it was stated that VWD is not restricted to VWF gene mutations. Types 2A, 2B, 2M and 2N remain the same, and type 1 VWD includes partial quantitative deficiency of VWF. Plasma VWF may contain mutant subunits, but has normal functional activity relative to antigen level. The proportion of large multimers is not decreased significantly. Treatment was proposed, based on the underlying type of VWD summarized as in Table 1.
Table 1.
Recommended treatment of VWD.
| Type of VWD | Treatment | Alternative |
|---|---|---|
| 1 | DDAVP | VWF/FVIII concentrate |
| 2A | VWF/FVIII concentrate | DDAVP |
| 2B | VWF/FVIII concentrate | None |
| 2M | VWF/FVIII concentrate | DDAVP |
| 2N | VWF/FVIII concentrate | DDAVP |
| 3 | VWF/FVIII concentrate | Platelet concentrate |
VWD, von Willebrand’s disease; DDAVP, desmopressin; FVIII, factor VIII.
With regard to how to test for VWD, various tests have been devised including VWF:RCo, platelet function analyser (PFA–100, Siemens Healthcare Diagnostics, Tarrytown, NY, USA), ristocetin-induced platelet aggregation (RIPA) and VWF collagen binding assay (VWF:CB). These could in the future be replaced or complemented by VWF gene analysis, but there has been limited success to date. When differentiating between type 2 VWD variants, a VWF:RCo/VWF:Ag ratio <0.6 is characteristic of types 2A and 2M, RIPA can be used for type 2B, a low FVIII:C/VWF:Ag ratio for type 2N and loss of high-molecular weight (HMW) multimers for types 2A and 2B. In type 3, there is virtually undetectable VWF in the plasma. For type 1 VWD with a VWF:Ag level of <30 IU dL−1, phenotypic and genetic assays can be used, as well as response to desmopressin (DDAVP) test. For type 1 VWD with a VWF:Ag level of >30 IU dL−1, phenotypic assays are of limited value, but genetic assays can be used to study penetrance. For types 2 and 3 VWD, both phenotypic and genetic assays can be used, and in the case of type 3 patients, genetic analysis can be used for family studies and to aid in prenatal diagnosis.
What developments can we expect to happen over the next 50 years? These should include full VWF locus sequencing and analysis including haplotypes, glycosylation patterns etc.; a ‘better’ VWF:RCo assay involving flow; a standardized VWF:CB, an understanding of the role of non-VWF genes/loci in the regulation of VWF levels, designer treatments and gene therapy.
Multimers (Ulrich Budde)
Circulating VWF is the product of synthesis, storage, secretion, circulation (shear stress), interaction with its receptors and ADAMTS13 and other proteases. It is therefore important to analyse the structure of VWF from the patient’s plasma. Decreased synthesis of VWF occurs in inherited type 1 VWD, and can also occur as acquired VWD as a result of hypothyroidism, or ingestion of valproate. Increased clearance of VWF occurs in type 1, 2M, Vicenza and 2A VWD, and in the acquired syndrome in monoclonal gammopathy of undetermined significance (MGUS) type IgG and myeloma, MGUS type IgM and in Waldenströ m’s macroglobinaemia. Increased proteolysis occurs in type 2A VWD, and in the acquired syndrome (AVWS) as a result of receptor increase, thrombocythaemia, inherited cardiac defects, aortic stenosis, cardiac assist devices or as a result of treatment with ciprofloxacin. Abnormal folding occurs in a subset of type 1 VWD (20–25% of type 1 patients) with a peculiar smeary pattern and in many cases an increased velocity of the protein in the gel. Increased binding occurs in type 2B VWD (platelet type VWD) and there have been sporadic case reports in acquired VWD. Dimerization and multimerization defects occur in type 2A (IIC, IIC Miami and IID).
VWF: glycosylation and function (Mike Laffan)
Protein glycosylation has several important roles:
Structural (cell walls, extracellular matrix).
Protein properties (solubility, stability, susceptibility to proteolysis).
Trafficking (intra/extracellular).
Adhesion (cell–cell, cell matrix).
Mediating and modulating signalling (intra/extra-cellular).
With regard to VWF, ~ 20% is glycan by weight and ~ 13% of N-linked glycans carry ABO blood sugars [11]. Occurrence of these asparagine-linked oligo-saccharides with blood group A and B structures suggest that the repeated use of factor VWF/FVIII pooled concentrate for the treatment of haemophiliacs could result in the production of antibodies against VWF with a different blood group from that of the patient, and that this development may be pathogenic. Out of the whole VWF protein comprised of 2050 amino acid residues, 12 N-linked and 10 O-linked glycan sites have been identified by cloning peptide sequencing of mature VWF [12]. There is one unused site in C2 and four more potential sites in propeptide from cDNA [12]. N-linked glycan sites are highly conserved. Caln-exin and calreticulin are related proteins that comprise an endoplasmic reticulum (ER) chaperone system that ensures the proper folding and quality control of newly synthesized glycoproteins. There is clear evidence that ERp57, an enzyme that catalyses disulfide bond formation, reduction and isomerization, participates in glycoprotein biogenesis either alone or in tandem with calnexin and calreticulin (13; Fig. 3).
Fig. 3.

N-linked glycosylation and endoplasmic reticulum quality control. Adapted with permission from The Company of Biologists Ltd [13].
N-linked glycosylation is required for VWF expression. Out of 17 N-linked glycan sites, four mutations affect secretion. These are N99Q (propeptide), N857Q (D′ domain), N2400Q (C1 domain) and N2790Q (cys-teine knot). Creatine kinase glycosylation at N2790 is required for dimerization. Functional effects of glycans include ADAMTS13 proteolysis. In a study of the influence of ABO blood group on the rate of proteolysis of VWF by ADAMTS13, it was found that the loss of collagen-binding activity was greater for VWF of group O compared with non-O VWF, in the rank order O ≥ B > A ≥ AB [14]. Specific N-linked glycans involved in ADAMTS13 cleavage are N1515 and N1574, which both occur in the A2 domain of VWF. Of the two, VWF–N1574Q is more susceptible to ADAMTS13 cleavage. VWF multimeric composition is regulated in plasma by ADAMTS13. A study by McGrath et al. [15] in 2010 showed that removal of sialic acid impairs ADAMTS13 cleavage and abolishes the ABO effect. Progressive shortening of N-linked glycan sugars (A–H–Bombay-Swainsonine–PNGase) results in more rapid cleavage of VWF. Conversely, removal of sialic acid makes VWF more resistant to ADAMTS13 cleavage. A loss of sugars is associated with increased affinity for ADAMTS13. There is a major effect from a single glycan at N1574.
With regard to VWF clearance, the ABO effect is probably mediated by an effect on clearance. A study by Sweeney et al. in 1990 showed that in the RIIIS/J mouse, glycans were able to modulate VWF clearance [16].
O-linked glycans attach to serine or threonine residues after protein folding. Long O-linked glycans are found in mucins. Short O-linked glycans occur at the hinge regions. O-linked glycans provide stiffness and protect against proteolysis. Thus, glycans regulate processing, ADAMTS13 cleavage, clearance and possibly other functional interactions.
Classification of VWD (Bob Montgomery)
According to the mechanistic classification of VWD, decreased synthesis of VWF is found in types 1 and 3 VWD, abnormal interaction with platelets occurs in type 2B (increased interaction) and type 2M (decreased in some cases; there are multiple causes of this variant), abnormal interaction with collagen is found in type 2M, also types 2A and 2B, abnormal clearance occurs in type 1C, also types 2A and 2B, and abnormal interaction with FVIII occurs in type 2N (which may be misdiagnosed as haemophilia). In type 1C VWD the half-life of VWF is reduced, but unlike type 1 VWD, FVIII and VWF are both reduced. Blood group is also an important consideration, as VWF is 25% lower in people with blood group O, and FVIII is similarly reduced in plasma secondary to this clearance. A common polymorphism present in >60% of African Americans renders the VWF:RCo assay unreliable for assessing VWF function (Fig. 4; 17). Assays developed by the Hamburg Group and the Milwaukee Group to circumvent this problem have been developed. Collagen-binding defects as a cause of type 2M VWD are underdiagnosed because VWF:CB assays are not commonly included in VWD screening.
Fig. 4.

VWF:RCo assay compared with GPIb complex-binding assay. The first two columns show VWF:RCo/VWF:Ag ratio (●) for subjects with and without the D1472H polymorphism. The second two columns show the GPIb complex-binding assay/VWF:Ag ratio (○) for subjects with and without the D1472H polymorphism. The mean value for each group is listed at the top of the graph. Error bars represent ± 1SD. Reproduced from [17] with permission from the American Society of Hematology (ASH).
Pharmacokinetics and laboratory assays when treating VWD (Jerzy Windyga)
The aim of therapy in VWD is to control or prevent bleeding through the specific correction of the dual defect of haemostasis, i.e. abnormal platelet adhesion-aggregation due to defect or deficiency of VWF and abnormal intrinsic coagulation due to low FVIII levels. Therapy aims to increase plasma levels of VWF. Treatment strategies are as follows: non-replacement therapy to increase VWF and FVIII with DDAVP, replacement therapy (pdVWF/FVIII concentrates, pdVWF concentrates), adjuvant treatments to promote haemo-stasis and wound healing (fibrinolysis inhibitors, platelet concentrates, oestrogen-progesterone preparations and topical agents) and combination therapy. If DDAVP is to be used, a DDAVP test should be performed to establish the patient’s individual response. The dose is 0.3 μg kg−1. Response to DDAVP should be assessed at 1 h after infusion. FVIII:C and VWF:RCo plasma levels should be assessed at 4 h post infusion to determine the pattern of clearance. Factors to consider when deciding on treatment are as follows:
The FVIII and VWF levels and VWD subtype.
The FVIII and VWF response to DDAVP if previously given.
The patient’s previous bleeding history and response to treatment.
The nature of the bleeding episode.
Presence of an inhibitor to VWF.
Potential risks of treatment (e.g. factor VIII accumulation after multiple injections [18]).
Patients who need to be monitored are those treated repeatedly with DDAVP, those with accelerated plasma clearance of VWF/FVIII and those in whom treatment is required for more than 3 days. Other indications for monitoring of therapy include major surgery, life-threatening bleeds, minor surgeries in severe VWD, and if there is a risk of delayed haemorrhage. For those patients in whom DDAVP is ineffective or prolonged treatments are required, or if DDAVP is contraindicated, treatment with VWF/FVIII or VWF concentrates is indicated. Treatments are Haemate-P, Wilate, Alphanate, Fandhi and Wilfactin. For treatment of spontaneous bleeding episodes, daily doses of 20–60 IU kg−1 of VWF/FVIII are required to maintain FVIII:C levels >30 U dL−1 until bleeding stops (usually 2–4 days). For prophylaxis for delivery and the puerperium, daily doses of 50 IU kg−1 are required to maintain FVIII:C level >50 U dL−1 for 3–4 days. For prophylaxis for dental extractions or invasive procedures, a single dose of 30 IU kg−1 of VWF/FVIII is required to maintain the FVIII:C level at > 50 U dL−1 for 12 h [19].
Genetics of VWD (Anne Goodeve)
This presentation examined where genetic testing in VWD can help clarify disease type and suggested appropriate management. In type 3 VWD, large deletions and other null mutations (nonsense, splice, small deletions and insertions) result in lack of VWF protein and there are also a small proportion of missense mutations. VWF dosage analysis using multiplex ligation-dependent probe amplification (MLPA) can be used to detect homozygous and heterozygous deletions and duplications of ≥1 exon. Sequencing of exons 2–52 is performed to identify point mutations. In type 3 VWD, the phenotype is usually clear. The cut-off between severe type 1 and type 3 may be equivocal, in which case mutation analysis may help to clarify the disease type.
In type 1 VWD, analysis of the entire VWF gene is required for complete point mutation analysis as mutations are found throughout the gene. Mutations are detected in up to 70% of patients.
Type 2 VWD largely results from missense mutations in specific VWF domains that affect a specific protein function. In type 2B VWD, enhanced ristocetin-induced platelet aggregation (RIPA) may be the only identifying feature. There is a loss of HMW multimers due to clearance of VWF from plasma along with platelets and enhanced ADAMTS13 sensitivity. Type 2B mutations are missense mutations in a discrete region of the A1 domain encoded by exon 28 (Fig. 5 [20]).
Fig. 5.
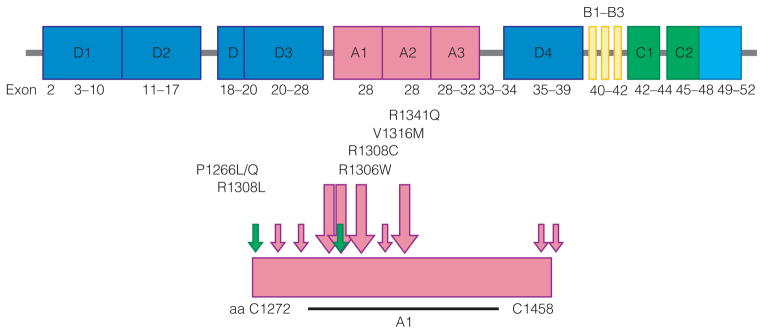
Type 2B VWD mutations. Missense mutations in discrete region of A1 domain, exon 28. Adapted from [20] with permission from Elsevier.
In type 2M VWD, mutations affect the ability of VWF to bind platelet GpIbα or to subendothelium. They are largely located in the A1 and A3 domains. There is no loss of HMW multimers.
Patients with type 2A VWD have a loss of HMW multimers that can result from different mutation types and locations that correlate with subtle differences in multimer profile. Dimerization, multimerization, extent of intracellular retention, clearance from the circulation and sensitivity to ADAMTS13 cleavage can all be affected. Most type 2A VWD is dominantly inherited.
In type 2N VWD, missense mutations are detected in the D′ and D3 domains. The R854Q mutation is particularly common in European populations. Mutations are recessively inherited. Genetic analysis is important to clarify or exclude VWD diagnosis and can indicate phenotype-genotype correlations, which can be helpful in establishing the response to DDAVP.
Treating the paediatric patient (Pia Petrini)
Should children with VWD be assessed differently to adults? According to the ISTH [10], the criteria for diagnosis are a significant bleeding history, a family history or relevant VWD mutation and laboratory determination, but only 4.5% of children fulfil these criteria [21]. This study concluded that ISTH criteria failed to identify the majority of children and adolescents who presented with significant mucocutaneous bleeding. The diagnosis is difficult in children, who are not yet challenged with operations, tooth extractions or menorrhagia, and 25% of the normal population may have bleeding symptoms such as epistaxis and bruising. Accordingly taking a mucocutaneous bleeding history is extremely important. With regard to neonates with a family history, they should be tested if type 3 VWD is a possibility or the child has bleeds, but not if the child is doing well after a normal delivery. VWF and FVIII can be raised in newborns, which complicates the diagnosis as type 1 subtypes may be missed. Treatment involves cyclokapron or DDAVP. Intranasal application of DDAVP is of special interest for children as it does not involve the use of needles. However, it is not used before school age in our centre because of the risk of hyponatraemia. Long-term prophylaxis, which has become a state-of-the-art approach in haemophilia, is not very common in VWD. However, more recent data suggest that many VWD patients could benefit from prophylactic treatment with VWF-containing concentrates. In a study of 35 Swedish VWD patients who required prophylaxis (mainly because of nose/mouth bleeds and joint bleeds, Fig. 6), there was a substantial overall reduction in bleeding episodes and there were no signs of arthropathy in children who began prophylaxis before the age of 5 years [22].
Fig. 6.
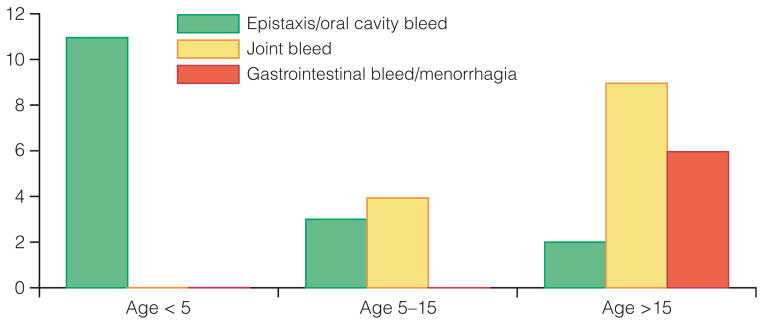
Clinical indication for prophylaxis by age at commencement of therapy (reproduced from ref. 22 with permission from Blackwell Publishing Ltd).
In a study of bleeding risk in 41 children with type 1 VWD undergoing adenotonsillar surgery [23], the children were treated according to the Children’s Hospital of Philadelphia protocol involving intravenous DDAVP, oral aminocaproic acid and overnight observation. Seven patients developed delayed (>24 h) postoperative haemorrhage requiring intervention, of whom five were treated with cautery and the remaining two responded to DDAVP and aminocaproic acid. The authors concluded that despite treatment to decrease the risk of postoperative haemorrhage, the incidence of haemorrhage was higher in pretreated patients with type 1 VWD than in children without bleeding disorders.
It is also important to consider the diagnosis of VWD in children presenting with head injury. In a study of an 11-month girl who presented to hospital with a massive subdural haematoma and bilateral haemorrhages following an allegedly minor fall, the parents were initially accused of child abuse [24]. Retinal haemorrhages and subdural haematoma are typical signs in the shaken baby syndrome. In this case, there was no bruising and no prior bleeding tendency was reported. Although initial analyses were normal, repeated testing led to a diagnosis of mild type 1 VWD. Police charges against the parents were then dropped. The case highlights the importance of considering VWD as a possible cause of head injury in young children.
Prophylaxis in VWD (Erik Berntorp)
Prophylaxis is well established in haemophilia A, but not in VWD. The Swedish experience of VWD prophylaxis is the largest cohort followed for the longest period of time. A total of 37 patients have been studied: three with type 1 VWD, three with type 2A, three with type 2B and 28 with type 3 [22,25]. Age at the start of prophylaxis was a median of 13 years (1–61 years). Indications for prophylaxis were mostly nose, mouth and joint bleeds, which were graded as serious to life-threatening. Treatment was with Haemate/Humate-P one to three times a week. Prophylaxis resulted in a substantial reduction of bleeding episodes. Patients who began prophylaxis prior to 5 years of age have had no joint bleeds and no clinical signs of arthropathy. The results show that long-term prophylaxis is warranted in most patients with type 3 VWD and in other subtypes with severe bleeding tendencies. Other small cohorts have been published with favourable results [26–29]. An international effort has been initiated [the von Willebrand’s Disease Prophylaxis Network (VWD PN)] and is ongoing as several issues remain such as dosing, indication and choice of concentrates. The aim of the Network is to investigate the role of prophylaxis in clinically severe VWD that is non-responsive to other treat-ment(s), involving a consensus of 30 centres in Europe and 44 in North America. From the collection of data from a large number of centres it will be possible to establish the number of people with VWD under care, the distribution of types of VWD and the frequency and indications for use of prophylaxis. Results to date show that there are a greater number of patients with type 3 VWD in Europe, 7% compared with 4% in North America. Use of prophylaxis in type 3 VWD varied significantly (P = 0.0004) by region: 28.7% in Europe vs. 12.2% in North America. Use of prophylaxis in types 1 and 2 VWD was rare in both regions. Indication for prophylaxis was mainly joint bleeds (40%), followed by epistaxis/oral (23%) and did not differ greatly by geographic region. When comparing prophylaxis in VWD to that in haemophilia, the dosing interval can be longer in VWD than in haemophilia.
The VWD International Prophylaxis (VIP) Study is a combination of prospective and retrospective studies. The primary aims are to identify subjects with VWD who may benefit from prophylaxis by determining patterns of bleeding prior to enrolment; study the effect of prophylaxis on bleeding frequency; and establish optimal treatment regimens for joint bleeding, gastrointestinal (GI) bleeding, epistaxis and menorrhagia. Secondary objectives include the determination of how quality of life at baseline is related to bleeding history and whether or not changes in bleeding frequency are associated with changes in quality of life. The prospective study is a non-randomized, dose-escalation study enrolling 40–50 patients for each bleeding indication: joint, epistaxis, gastrointestinal and menorrhagia. Follow-up is for 1 year, with visits every 3 months. Use of VWF/FVIII products licensed for the treatment of VWD is permitted. The retrospective study includes patients who have been on prophylaxis for at least 6 months, with data collected from study centres and diaries; the aim is to compare bleeding frequency pre and post prophylaxis. A third area is a retrospective study of the natural history of GI bleeding, looking at patients with a history of GI bleeding due to angiodysplasia or unexplained by other factors. As at 14 July 2010, a total of 69 centres with 83 patients were participating in the VIP study. The primary area for prophylaxis was epistaxis in the prospective study (66.7%) and joint bleeding in the retrospective study (25.7%; Table 2; [30]). It was concluded that the early data provide strong support for the use of prophylaxis for people most severely affected by VWD. They also support prospective efforts to identify optimal prophylactic treatment regimens for those at greatest risk for bleeding.
Table 2.
Primary indication for prophylaxis*.
| Prospective | Retrospective | Total | |
|---|---|---|---|
| Site | N (%) | N (%) | N (%) |
| Epistaxis | 4 (66.7) | 8 (22.9) | 12 (29.3) |
| GI bleeding | 1 (16.7) | 7 (20.0) | 8 (19.5) |
| Joint bleeding | 1 (16.7) | 9 (25.7) | 10 (24.4) |
| Menorrhagia | 0 (0.0) | 4 (11.4) | 4 (9.8) |
| Other | 0 (0.0) | 4 (11.4) | 4 (9.8) |
| Mixed | 0 (0.0) | 3 (8.6) | 3 (7.3) |
| Total | 6 (100.0) | 35 (100.0) | 41 (100.0) |
Defined as the most severe symptom, or the source of the most significant morbidity.
Managing surgeries and interventions (Mario von Depka)
In prospective clinical trials, 32 VWD patients undergoing 57 surgical procedures were treated with a VWF/FVIII concentrate (Wilate® Octapharma GmbH, Langenfeld, Germany). More than 68% of the procedures were performed in patients with severe VWD type 3. Ten of the 57 procedures were in paediatric patients. Thirty procedures were minor and the remaining 27 were major surgery. The mean dose per infusion was 34 IU kg−1 in total (36 IU kg−1 in minor, 31 IU kg−1 in major surgeries) and 31.9 IU kg−1 in paediatric patients. Dosing and monitoring was performed according to FVIII:C or VWF:RCo or both parameters. Wilate’s overall haemostatic efficacy was rated as excellent/good in 96% (51/53 of rated cases). There was no development of inhibitors to VWF or FVIII or any thrombotic events. After multiple dosing, no accumulation of FVIII was observed. It was concluded that Wilate provides effective cover in patients with VWD undergoing surgical procedures.
When VWD comes into age – VWD in geriatrics (Wolfgang Miesbach)
Between 1962 and 2002, average life expectancy in Western Germany increased from 67.1 to 75.6 years in men and from 72.7 to 81.3 years in women. The largest contribution to the increase in life expectancy came from the age group 65 years and older [31]. In the Swedish population, there were 948 patients with haemophilia for the period 1831–1980. For them life expectancy has increased fivefold from 11 years (1831–1920) to 56.8 years (1961–1980; 32]. Patients with VWD are also living longer, and the elderly VWD patient is also likely to have other co-morbidities such as hypertension, diabetes mellitus, osteoporosis, atherosclerotic and thromboembolic diseases and malignancies (Fig. 7). In the elderly, more patients have the severe form of VWD and more patients have acquired VWD as a result of medication, malignancies (clonal monoclonal gammopathy, or lymphoproliferative or myeloproliferative disorders; [33]) or operations. In younger patients the primary bleeding symptoms are oral cavity bleeding, epistaxis, cutaneous bleeding, menorrhagia, bleeding from minor wounds, tooth extraction and postpartum haemorrhage, whereas in the elderly the primary symptoms are GI bleeding, bleeding following surgery/tooth extraction and haemarthrosis [34,35]. Treatment with DDAVP is contraindicated in the elderly as it can cause vasoconstriction and hyponatraemia. An online survey is proposed to investigate elderly patients with VWD. This will improve our knowledge of the disease in the elderly.
Fig. 7.
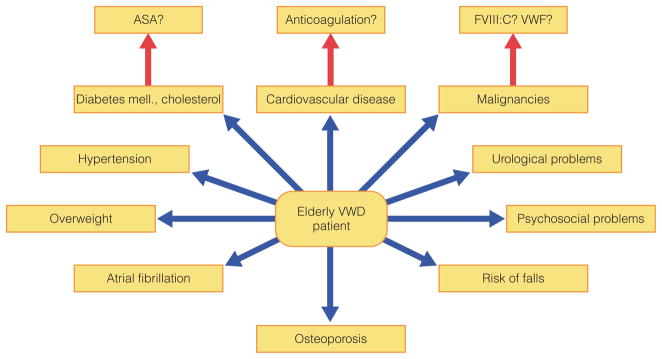
Co-morbidities in the elderly VWD patient.
Gene therapy for VWD: one step at a time (David Lillicrap)
Type 3 VWD causes serious problems such as haemarthroses and GI tract bleeding. Since 1975, different virus vectors have been developed in order to carry functional genes for gene transfer. However, no successful clinical trials have been reported to date. Recently, a new method for altering a single basepair of target DNA was reported using chimeric RNA/DNA oligonucleotides. In patients with haemophilia A or B, the mutations (coagulation factors VIII and IX) are well characterized and the mutation-repair method using chimeric RNA/DNA oligo-nucleotides could provide an alternative for the treatment of haemophilia [36]. In VWD there are challenges to gene transfer, with VWF cDNA being irreducible at 8.4 kb. The advantages of a mouse model are genetic homogeneity, consistent VWF cDNA (no influence of polymorphisms), good study population numbers and good in vivo thrombosis models. The challenges of using the VWF KO mice are species-specific VWF processing and ligand interaction. Three common recurrent type 2B VWD mutations introduced into mouse VWF cDNA are V1316M, R1306W and R1341Q. Wild type residues are conserved between human and mouse. Early evidence suggests that reconstitution of the plasma compartment alone, with VWF expressed from the liver, recapitulates the phenotypes of both qualitative and quantitative forms of VWD.
Role of FVIII in VWF replacement (Erik Berntorp)
FVIII is needed in addition to VWF because of low levels in type 3 VWD and other subtypes. In patients with type 3 VWD (and those with type 2N VWD), the lack of FVIII–VWF binding results in a secondary defi-ciency of FVIII and the FVIII level is usually <10% of normal in patients with type 3 VWD [27,37]. Joint bleeds are common, especially in type 3 VWD. A common concern is that FVIII will accumulate if combined VWF/FVIII concentrates are given. Another concern is that there is an increased risk of venous thromboembolism (VTE) with combined concentrates. The overall incidence of VTE in VWD has been reported to be seven cases in 12 640 yearly treatments over 10 years [38], i.e. one case per 1806 treatment years. Only a few VTE events have been reported for Humate-P. In all cases, there was a connection with risk factors and levels of FVIII:C/VWF:RCo were above 100–200%. The efficacy record is outstanding for VWF/FVIII concentrates.
With regard to purified concentrates, the risk is that FVIII levels can be too low. In 1992, Lethagen showed that there was a lag time before achieving a good plasma FVIII level with Wilfactin, a pure VWF concentrate almost devoid of FVIII [39]. Use of purified VWF concentrates requires co-administration of FVIII, or an extra dose of concentrate. The addition of FVIII is probably superfluous in concentrates if used for continuous prophylaxis or used for scheduled surgery – but more studies are needed.
Inhibitors in VWD (Augusto B. Federici)
Compared to patients with moderate to severe haemophilia A, who may develop anti-factor VIII inhibitors in ~ 20–30% of cases, the antibodies against VWF (anti-VWF inhibitors) are a rare complication of replacement therapy in transfused patients with inherited type 3 VWD. These anti-VWF inhibitors are allo-antibodies and for many years have been associated only with deletions of the VWF gene causing VWD type 3: they have never been identified in patients with types 1 or 2 VWD.
The occurrence of an alloantibody against VWF in multitransfused patients with VWD type 3 was first reported in 1974 [40]. In the most recent retrospective study of the Italian Association of Hemophilia Centers (AICE), 96 VWD type 3 patients (5.8%) were identi-fied among the 1650 cases included in the registry [41] with a prevalence of 1.6 per million of population; in this registry anti-VWF inhibitors were identified in seven patients only, from three VWD type 3 families. In such cases, VWF concentrates are not only ineffective, but may even cause post infusion life-threatening anaphylaxis [42], due to the formation of immune complexes. Inhibitors can also interact with FVIII; if they occur, combined VWF/FVIII products should not be used, but pure VWF or rF7a should be used instead.
Unfortunately, no general consensus has been reached for testing these anti-VWF antibodies in VWD type 3. Assays are currently available in only a few specialized laboratories and they mimic the Bethesda assays for haemophilia inhibitors by performing VWF and FVIII activities in patient-normal pool plasma mixtures after 2 h’ incubation at 37°C. The titre of anti-VWF inhibitor is calculated by the current dilution of VWD plasma inhibiting 50% of normal plasma pool diluted 1:2 compared to control mixture. Due to the different functions of VWF, all its activities should always be measured, such as anti-VWF:antigen, anti-VWF:RCo, anti-VWF:CB and anti-FVIII. Negative results by mixing tests cannot exclude inhibitors completely, since they might affect non-functional regions of the VWF protein. Some authors have recently proposed an indirect assay to test these anti-VWF antibodies using a sandwich enzyme-linked immunosorbent assay (ELISA). The sensitivity of this assay is very promising, but its specificity is low (many false-positive results). All these techniques should be standardized prospectively by a multicentre study organized by the Members of the European Group on VWD3 on behalf of the ISTH-SSC Subcommittee on VWF.
Current treatment guidelines – experiences from the UK (Mike Laffan)
In the UK, there are 101 centres. Current treatment guidelines (2004) are being followed, but there are some concerns about practice. Guidelines on diagnosis were published in 2004 [43]. They were written by a working party, circulated to the UK Haemophilia Centre Doctors’ Organization (UKHCDO) and peer reviewed. Requirements for diagnosis of VWD are as follows:
A personal history of (mucosal) bleeding.
Decreased functional VWF levels.
A mutation in the VWF gene or family history of bleeding that segregates with (2).
There are problems with all of these criteria. A significant bleeding history includes epistaxis for more than 20 min, prolonged bleeding (>15 min) from trivial wounds and prolonged or excessive postoperative bleeding [44]. For family history, there should be a first-degree relative affected, or two-second-degree relatives affected. Preliminary tests include full blood count, coagulation screen, PFA–100, FVIII, VWF:Ag, VWF:RCo, VWF:CB and RIPA. Further tests may include FVIII binding assay and genetic analysis. The bleeding time does not have a role as a screening test for VWD, although it is of value in the composite assessment of haemostasis. The VWF:Ag should be measured using an assay whose limit of detection is <1 IU dL−1. A platelet-based assay for VWF:RCo should be used to assess VWF:RCo (not an ELISA assay). Both VWF:RCo and VWF:CB should be obtained to improve the ability to detect type 2 variants and to more clearly define type 1 VWD. The VWF multimer analysis has a role in sub-classification of VWD; it is inappropriate to perform this test until a diagnosis of VWD has been confirmed. To minimize the risk of misdiagnosis, VWF:Ag and function must be measured in samples obtained on at least two occasions, with consistent results. It is important to avoid circumstances that may be confounding (stress, pregnancy, etc.). The ISTH 1994 criteria are used to classify the type of VWD [9]. Rather than ask does this patient have VWD, it is important to assess whether they are at risk of bleeding, either spontaneously or following injury or invasive procedures, and then decide what treatment, if any, is required, and whether other family members are also at risk. Guidelines on management of VWD were published in 2004 by the UKHCDO [18]. Management of the patient with VWD depends on the following:
The nature of the bleeding episode.
The FVIII and VWF levels and the VWD type.
The patient’s previous bleeding history and response to treatment.
The FVIII and VWF response to DDAVP if given previously.
Presence of an inhibitor.
Potential risks of treatment.
Treatment options include tranexamic acid (useful alone or as an adjunct including as a mouth wash), DDAVP (preferred to blood products when effective), concentrates (need to avoid excessive rise in FVIII) and treatments for menorrhagia (e.g. tranexamic acid and Mirena IUCD). For pregnant patients, vaginal delivery is acceptable if VWF:RCo is >40 IU dL−1, and a Caesarean section is acceptable if VWF:RCo >50 IU dL−1. In type 1 VWD patients, it is important to watch out for rebleeding at 4–5 days post delivery. For patients with types 2 and 3 VWD, the patient should be advised against having an epidural. If the foetus is at risk of having types 2 or 3 VWD, foetal scalp monitoring, forceps and ventouse delivery should be avoided. Regardless of the mode of delivery, newborns at risk of types 2 and 3 VWD need to be tested for VWD using cord blood and assessed to exclude intracranial haemorrhage.
Current treatment guidelines – experiences from Scandinavia (Riitta Lassila)
In 1926, Erik von Willebrand published the first report of a patient with VWD [1]. Since then there have been key observations and commitment to research in VWD in Nordic countries. The Nordic hemophilia council (NHC) is a society of physicians and related experts from Nordic haemophilia centres in Denmark, Finland, Iceland, Norway and Sweden. The NHC holds a general annual meeting and forms a base for co-operation between the Nordic centres. The NHC is responsible for the management of the society and implementation of the Nordic guidelines for bleeding disorders. Joint projects include the evaluation of the prevalence of bleeding disorders, and the evaluation of the products used and their pricing in Nordic countries. A flow chart for the diagnosis of VWD is shown in Fig. 8 [45–47].
Fig. 8.

Flow chart of the diagnosis of von Willebrand’s disease. Adapted from [45] with permission from the Ferrata Storti Foundation.
Differential diagnosis of type 1 VWD includes platelet disorders and effect of concurrent medications such as selective serotonin reuptake inhibitors (SSRIs) and omega 3. Regular prophylaxis with a VWF concentrate should be considered in some patients with types 2 and 3 VWD. Prophylactic treatment should also be considered if nose bleeds, menorrhagia or gastrointestinal bleeds cause significant anaemia despite iron supplementation, have an impact on social life and/or all other treatment modalities have failed. As prophylaxis, a VWF concentrate in a dose of about 50 IU VWF:RCo kg−1 i.v. administered 2–3 times per week is used to prevent bleeds. Levels of VWF:RCo and FVIII:C should be monitored when repeated doses of the concentrate are given.
Current treatment guidelines – experience from the USA (Gil White)
In 2008, the National Heart, Lung and Blood Institute (NHLBI) in the USA published evidence-based guidelines on the diagnosis and management of VWD [48].
It was proposed that patients should be asked a series of questions:
Have you or a blood relative ever needed medical attention for a bleeding problem, or have you been told you had a bleeding disorder? During or after surgery? With dental work? With trauma?
Do you have or have you ever had liver or kidney disease? A blood or bone marrow disorder? High- or low platelet count?
Are you presently taking or have you taken anticoagulation? NSAIDs?
Do you have a blood relative with a bleeding disorder like VWD or haemophilia A?
Have you ever had prolonged bleeding from trivial wounds, lasting >15 min or recurring spontaneously within 7 days?
Have you ever had excessive bleeding after surgical procedures?
Have you ever had bruising, with minimal or no trauma?
Have you ever had a spontaneous nosebleed that lasted >10 min?
Have you ever had excessive bleeding after dental extractions?
Have you ever had unexplained blood in your stool?
Have you ever had anaemia that required a blood transfusion?
Have you ever had heavy menses, with clots >1 inch in diameter.
During the consultation, evidence for a bleeding disorder should be sought. Evidence of other causes of increased bleeding should also be looked for, such as jaundice, splenomegaly and arthropathy. If the diagnosis of VWD seems possible, the initial evaluation should include VWF:RCo, VWF:Ag and FVIII, as well as complete blood count, platelet count, prothrombin time, partial thromboplastin time and thrombin time. If after these tests the diagnosis of VWD is supported, other tests should be carried out including VWF:RCo/VWF:Ag ratio, VWF multimer analysis and VWF:CB. It is important to eliminate stress as much as possible when taking blood samples, as this may falsely elevate VWF and FVIII levels. The presence of an inflammatory illness may also elevate VWF and FVIII levels, as may pregnancy or administration of oral contraceptives.
Once the diagnosis is confirmed, treatment of patients with VWD is aimed at cessation of bleeding and prophylaxis for surgical procedures. Epistaxis and oropharyngeal, soft tissue, or minor bleeding should be treated with intravenous or intranasal DDAVP, if appropriate, based on trial testing. For prophylaxis for minor surgery, initial treatment should be expected to achieve VWF:RCo and FVIII activity levels of at least 30 IU dL−1 and preferably higher than 50 IU dL−1. For persons with mild to moderate VWD, antifibrinolytics combined with DDAVP are generally effective for oral surgery. VWF concentrates should be administered to those in whom DDAVP is contraindicated or who bleed on DDAVP. Whenever possible, all major surgical procedures and bleeding events should be treated in hospitals with 24-h laboratory capability and monitored by a team that includes a haematologist and surgeon skilled in the management of bleeding disorders. For severe bleeding or for prophylaxis during major surgery, initial target VWF:RCo and FVIII should be at least 100 IU dL−1. Subsequent dosing should maintain VWF:RCo and FVIII above a trough of 50 IU dL−1 for at least 7–10 days. In an adolescent or adult woman who does not desire pregnancy but may in the future, the first choice of therapy for menorrhagia should be combined oral contraceptives. For a woman who desires pregnancy, DDAVP, antifibrinolytics, or a VWF concentrate may be tried to control menorrhagia. For pregnant patients, those with type 1, type 2, or type 3 VWD, with FVIII or VWF:RCo levels < 50 IU dL−1 or a history of severe bleeding:
Should be referred to a centre that has high-risk obstetrics capabilities and expertise in haemostasis for prenatal care, delivery, termination of pregnancy, or miscarriage.
Should receive prophylaxis with DDAVP or VWF concentrate before invasive procedures.
Should achieve VWF:RCo and FVIII levels of at least 50 IU dL−1 before delivery and maintain those levels for 3–5 days.
Patients with AVWS and who require surgery should be considered for a trial of therapy with DDAVP and/or VWF concentrate, with monitoring of VWF:RCo and FVIII, to evaluate for accelerated clearance of VWF. For patients with AVWS and who bleed excessively despite DDAVP and VWF concentrate, treatment with high-dose IVIG should be considered.
Concluding remarks (Erik Berntorp)
The 2010 Åland meeting was as memorable as a previous meeting held in 1998 [49]. The historical atmosphere, the friendly and knowledgeable participants and the venue located among 5 000 beautiful and unspoiled islands will help us all to improve the situation for patients with VWD. Hopefully this was not the last Åland conference on VWD.
Acknowledgments
Writing support was provided by Ros Kenn, freelance medical editor/writer, and funded by Octapharma.
Footnotes
Disclosures
The authors have no interests to declare.
Declaration of funding interests: Full funding was provided by Octapharma.
Contributor Information
Prof. Dr. E. BERNTORP, Email: erik.berntorp@med.lu.se.
Prof. em Dr I. PEAKE, Email: i.r.peake@sheffi eld.ac.uk.
Prof. U. BUDDE, Email: u.budde@asklepios.com.
Prof. Dr M. LAFFAN, Email: m.laffan@imperial.ac.uk.
Prof.Dr R. MONTGOMERY, Email: bob.montgomery@bcw.edu.
Prof. Dr J. WINDYGA, Email: jwindyga@ihit.waw.pl.
Dr A. GOODEVE, Email: a.goodeve@sheffi eld.ac.uk.
Dr P. PETRINI, Email: pia.petrini@karolinska.se.
Dr M.VON DEPKA, Email: mvd@werlhof-institut.de.
Dr W. MIESBACH, Email: wolfgang.miesbach@kgu.de.
Prof. Dr D. LILLICRAP, Email: lillicrap@cliff.path.queensu.ca.
Prof. Dr A. B. FEDERICI, Email: augusto.federici@unimi.it.
Dr R. LASSILA, Email: riitta.lassila@hus.fi.
Dr G. WHITE, Email: gcwhite@bcw.edu.
References
- 1.Von Willebrand EA. Hereditär pseudohemofili. Finska Läkarsällskapets Handl. 1926;67:7–112. [Google Scholar]
- 2.Nilsson IM. The history of von Willebrand disease. Haemophilia. 1999;5(Suppl 2):7–11. doi: 10.1046/j.1365-2516.1999.0050s2007.x. [DOI] [PubMed] [Google Scholar]
- 3.Von Willebrand EA. Über hereditäre Pseudohämaphilie. Acta Med Scand. 1931;67 :521–50. [Google Scholar]
- 4.Von Willebrand EA. Über ein vererbbares Blutungsübel: Die konstitutionelle Thrombopathie. Dtsch Arch Klin Med. 1933;175:453–83. [Google Scholar]
- 5.Soulier JP, Larrieu MJ. Willebrand-Jürgens syndrome and thrombopathies, study of 66 cases; attempt at classification. Rev Hematol. 1954;9:77–122. [PubMed] [Google Scholar]
- 6.Rodeghiero F, Castaman G, Tosetto A. Optimizing treatment of von Willebrand disease by using phenotype and molecular data. Hematology Am Soc Hematol Educ Program. 2009 Jan 1;:113–23. doi: 10.1182/asheducation-2009.1.113. [DOI] [PubMed] [Google Scholar]
- 7.Ruggeri ZM, Zimmerman TS. Classification of variant von Willebrand’s disease subtypes by analysis of functional characteristics and multimeric composition of factor VIII/von Willebrand factor. Ann N Y Acad Sci. 1981;370:205–9. doi: 10.1111/j.1749-6632.1981.tb29733.x. [DOI] [PubMed] [Google Scholar]
- 8.Ruggeri ZM, Zimmerman TS. Von Wille-brand factor and von Willebrand disease. Blood. 1987;70:895–904. [PubMed] [Google Scholar]
- 9.Sadler JE. A revised classification of von Willebrand disease. For the Subcommittee on von Willebrand Factor of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis. Thromb Haemost. 1994;71:520–5. [PubMed] [Google Scholar]
- 10.Sadler JE, Budde U, Eikenboom JC, et al. Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand factor. J Thromb Haemost. 2006;4:2103–14. doi: 10.1111/j.1538-7836.2006.02146.x. [DOI] [PubMed] [Google Scholar]
- 11.Matsui T, Titani K, Mizuochi T. Structures of the asparagine-linked oligosaccharide chains of human von Willebrand factor. Occurrence of blood group A, B, and H(O) structures. J Biol Chem. 1992;267:8723–31. [PubMed] [Google Scholar]
- 12.Titani K, Kumar S, Takio K, et al. Amino acid sequence of human von Willebrand factor. Biochemistry. 1986;25:3171–84. doi: 10.1021/bi00359a015. [DOI] [PubMed] [Google Scholar]
- 13.Williams DB. Beyond lectins: the calnexin/calreticulin chaperone system of the endoplasmic reticulum. J Cell Sci. 2006;119:615–23. doi: 10.1242/jcs.02856. [DOI] [PubMed] [Google Scholar]
- 14.Bowen DJ. An influence of ABO blood group on the rate of proteolysis of von Willebrand factor by ADAMTS13. J Thromb Haemost. 2003;1:33–40. doi: 10.1046/j.1538-7836.2003.00007.x. [DOI] [PubMed] [Google Scholar]
- 15.McGrath RT, McKinnon TA, Byrne B, et al. Expression of terminal alpha2-6-linked sialic acid on von Willebrand factor specifically enhances proteolysis by ADAMTS13. Blood. 2010;115:2666–73. doi: 10.1182/blood-2009-09-241547. [DOI] [PubMed] [Google Scholar]
- 16.Sweeney JD, Novak EK, Reddington M, Takeuchi KH, Swank RT. The RIIIS/J inbred mouse strain as a model for von Willebrand disease. Blood. 1990;76:2258–65. [PubMed] [Google Scholar]
- 17.Flood VH, Gill JC, Morateck PA, et al. Common VWF exon 28 polymorphisms in African Americans affecting the VWF activity assay by ristocetin cofactor. Blood. 2010;116:280–6. doi: 10.1182/blood-2009-10-249102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pasi KJ, Collins PW, Keeling DM, et al. Management of von Willebrand disease: a guideline from the UK Haemophilia Centre Doctors’ Organization. Haemophilia. 2004;10 :218–31. doi: 10.1111/j.1365-2516.2004.00886.x. [DOI] [PubMed] [Google Scholar]
- 19.Mannucci PM, Franchini M, Castaman G, Federici AB Italian Association of Hemo-philia Centers. Evidence-based recommendations on the treatment of von Willebrand disease in Italy. Blood Transfus. 2009;7:117–26. doi: 10.2450/2008.0052-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goodeve AC. The genetic basis of von Willebrand disease. Blood Rev. 2010;24:123–34. doi: 10.1016/j.blre.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 21.Hyatt SA, Wang W, Kerlin BA, O’Brien SH. Applying diagnostic criteria for type 1 von Willebrand disease to a pediatric population. Pediatr Blood Cancer. 2009;52:102–7. doi: 10.1002/pbc.21755. [DOI] [PubMed] [Google Scholar]
- 22.Berntorp E. Prophylaxis in von Willebrand disease. Haemophilia. 2008;14(Suppl 5):47–53. doi: 10.1111/j.1365-2516.2008.01851.x. [DOI] [PubMed] [Google Scholar]
- 23.Witmer CM, Elden L, Butler RB, Manno CS, Raffini LJ. Incidence of bleeding complications in pediatric patients with type 1 von Willebrand disease undergoing adenotonsillar procedures. J Pediatr. 2009;155:68–72. doi: 10.1016/j.jpeds.2009.01.051. [DOI] [PubMed] [Google Scholar]
- 24.Stray-Pedersen A, Omland S, Nedregaard B, Klevberg S, Rognum TO. An infant with subdural haematoma and retinal haemorrhages: does von Willebrand disease explain the findings? Forensic Sci Med Pathol. 2011;7:37–41. doi: 10.1007/s12024-010-9176-7. [DOI] [PubMed] [Google Scholar]
- 25.Berntorp E, Petrini P. Long-term prophylaxis in von Willebrand disease. Blood Coagul Fibrinolysis. 2005;16(Suppl 1):S23–6. doi: 10.1097/01.mbc.0000167659.23262.18. [DOI] [PubMed] [Google Scholar]
- 26.Dunkley S, Baker RI, Pidcock M, et al. Clinical efficacy and safety of the factor VIII/von Willebrand factor concentrate BIOSTATE in patients with von Wille-brand’s disease: a prospective multi-centre study. Haemphilia. 2010;16:615–24. doi: 10.1111/j.1365-2516.2010.02206.x. [DOI] [PubMed] [Google Scholar]
- 27.Berntorp E, Windyga J European Wilate Study Group. Treatment and prevention of acute bleedings in von Willebrand disease – efficacy and safety of Wilate, a new generation von Willebrand factor/ factor VIII concentrate. Haemophilia. 2009;15:122–30. doi: 10.1111/j.1365-2516.2008.01901.x. [DOI] [PubMed] [Google Scholar]
- 28.Federici AB. Prophylaxis of bleeding episodes in patients with von Willebrand’s disease. Blood Transfus. 2008;6(Suppl 2):S26–32. doi: 10.2450/2008.0034-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Borel-Derlon A, Federici AB, Roussel-Robert V, et al. Treatment of severe von Willebrand disease with a high-purity von Willebrand factor concentrate (Wilfactin): a prospective study of 50 patients. J Thromb Haemost. 2007;5:1115–24. doi: 10.1111/j.1538-7836.2007.02562.x. [DOI] [PubMed] [Google Scholar]
- 30.Berntorp E. Personal communication.
- 31.Klenk J, Rapp K, Büchele G, Keil U, Weiland SK. Increasing life expectancy in Germany: quantitative contributions from changes in age- and disease-specific mortality. Eur J Public Health. 2007;17:587–92. doi: 10.1093/eurpub/ckm024. [DOI] [PubMed] [Google Scholar]
- 32.Larsson SA. Life expectancy of Swedish haemophiliacs, 1831–1980. Br J Haematol. 1985;59:593–602. doi: 10.1111/j.1365-2141.1985.tb07353.x. [DOI] [PubMed] [Google Scholar]
- 33.Franchini M, Lippi G, Favaloro EJ. Advances in hematology. Etiology and diagnosis of acquired von Willebrand syndrome. Clin Adv Hematol Oncol. 2010;8:20–4. [PubMed] [Google Scholar]
- 34.Tosetto A, Rodgehiero F, Castaman G, et al. A quantitative analysis of bleeding symptoms in type 1 von Willebrand disease: results from a multicenter European study (MCMDM–1 VWD) J Thromb Haemost. 2006;4:766–73. doi: 10.1111/j.1538-7836.2006.01847.x. [DOI] [PubMed] [Google Scholar]
- 35.Rodeghiero F, Tosetto A, Castaman G. How to estimate bleeding risk in mild bleeding disorders. J Thromb Haemost. 2007;5(Suppl 1):157–66. doi: 10.1111/j.1538-7836.2007.02520.x. [DOI] [PubMed] [Google Scholar]
- 36.Zhang Z, Eriksson M, Blombäck M, Anvret M. A new approach to gene therapy. Blood Coagul Fibrinolysis. 1997;8 (Suppl 2):S39–42. [PubMed] [Google Scholar]
- 37.Federici AB, Barillari G, Zanon E, et al. Efficacy and safety of highly purified, doubly virus-inactivated VWF/FVIII concentrates in inherited von Willebrand’s disease: results of an Italian cohort study on 120 patients characterized by bleeding severity score. Haemophilia. 2010;16:101–10. doi: 10.1111/j.1365-2516.2009.02088.x. [DOI] [PubMed] [Google Scholar]
- 38.Mannucci PM. Venous thromboembolism in von Willebrand disease. Thromb Haemost. 2002;88:378–9. [PubMed] [Google Scholar]
- 39.Lethagen S, Berntorp E, Nilsson IM. Pharmacokinetics and hemostatic effect of different factor VIII/von Willebrand factor concentrates in von Willebrand’s disease type III. Ann Hematol. 1992;65:253–9. doi: 10.1007/BF01836069. [DOI] [PubMed] [Google Scholar]
- 40.Sarji KE, Stratton RD, Wagner RH, Brinkhous KM. Nature of von Willebrand factor: a new assay and a specific inhibitor. Proc Natl Acad Sci USA. 1974;71:2937–41. doi: 10.1073/pnas.71.8.2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Iorio A, Oliovecchio E, Morfini M, Mannucci PM Association of Italian Hemophilia Centres Directors. Italian Registry of Haemophilia and Allied Disorders. Objectives, methodology and data analysis. Haemophilia. 2008;14:444–53. doi: 10.1111/j.1365-2516.2008.01679.x. [DOI] [PubMed] [Google Scholar]
- 42.Bergamaschini L, Mannucci PM, Federici AB, Coppola R, Guzzoni S, Agostoni A. Posttransfusion anaphylactic reactions in a patient with severe von Willebrand disease: role of complement and alloantibodies to von Willebrand factor. J Lab Clin Med. 1995;125:348–55. [PubMed] [Google Scholar]
- 43.Laffan M, Brown SA, Collins PW, et al. The diagnosis of von Willebrand disease: a guideline from the UK Haemophilia Centre Doctors’ Organization. Haemophilia. 2004;10:199–217. doi: 10.1111/j.1365-2516.2004.00894.x. [DOI] [PubMed] [Google Scholar]
- 44.Dean JA, Blanchette VS, Carcao MD, et al. von Willebrand disease in a pediatric-based population – comparison of type 1 diagnostic criteria and use of the PFA–100 and a von Wiillebrand factor/collagen binding assay. Thromb Haemost. 2000;84:401–9. [PubMed] [Google Scholar]
- 45.Berntorp E, Önundarson PT. Prevalence of von Willebrand disease in the Nordic region. Haematologica Rep. 2005;1:4–6. [Google Scholar]
- 46.Lassila R, Holme PA, Landorph A, Petrini P, Önundarson PT, Hillarp A. Nordic Haemophilia Council’s practical guidelines on diagnosis and management of von Wille-brand disease. Semin Thromb Haemost. 2011;37:495–502. doi: 10.1055/s-0031-1281034. [DOI] [PubMed] [Google Scholar]
- 47.Lethagen S, Ingerslev J, Holme PA, Petrini P, Lassila R, Önundarson PT. Nordic Guidelines for Diagnosis and Management of von Willebrand Disease. [Accessed July 26, 2012];Guidelines of the Nordic Hemophilia Council. Available at: http://nordhaemophilia.org/document/NordicGuidelinesVWD_SL23APR2008b.pdf.
- 48.Nichols WL, Hultin MB, James AH, et al. von Willebrand disease (VWD): evidence-based diagnosis and management guidelines, the National Heart, Lung, and Blood Institute (NHLBI) Expert Panel report (USA) Haemophilia. 2008;14:171–232. doi: 10.1111/j.1365-2516.2007.01643.x. [DOI] [PubMed] [Google Scholar]
- 49.Berntorp E, Ingerslev J, Schulman S. von Willebrand disease: an update in the Åland islands. Summary of a Nordic symposium on von Willebrand disease, 24–25 September 1998, Mariehamn, Åland. Haemophilia. 1999;6(Suppl 2):1–6. doi: 10.1046/j.1365-2516.1999.0050s2001.x. [DOI] [PubMed] [Google Scholar]