

Abstract
Background
Both inhaled steroids (ICS) and long‐acting beta2‐agonists (LABA) are used in the management of chronic obstructive pulmonary disease (COPD). This updated review compared compound LABA plus ICS therapy (LABA/ICS) with the LABA component drug given alone.
Objectives
To assess the efficacy of ICS and LABA in a single inhaler with mono‐component LABA alone in adults with COPD.
Search methods
We searched the Cochrane Airways Group Specialised Register of trials. The date of the most recent search was November 2011.
Selection criteria
We included randomised, double‐blind controlled trials. We included trials comparing compound ICS and LABA preparations with their component LABA preparations in people with COPD.
Data collection and analysis
Two authors independently assessed study risk of bias and extracted data. The primary outcomes were exacerbations, mortality and pneumonia, while secondary outcomes were health‐related quality of life (measured by validated scales), lung function, withdrawals due to lack of efficacy, withdrawals due to adverse events and side‐effects. Dichotomous data were analysed as random‐effects model odds ratios or rate ratios with 95% confidence intervals (CIs), and continuous data as mean differences and 95% CIs. We rated the quality of evidence for exacerbations, mortality and pneumonia according to recommendations made by the GRADE working group.
Main results
Fourteen studies met the inclusion criteria, randomising 11,794 people with severe COPD. We looked at any LABA plus ICS inhaler (LABA/ICS) versus the same LABA component alone, and then we looked at the 10 studies which assessed fluticasone plus salmeterol (FPS) and the four studies assessing budesonide plus formoterol (BDF) separately. The studies were well‐designed with low risk of bias for randomisation and blinding but they had high rates of attrition, which reduced our confidence in the results for outcomes other than mortality.
Primary outcomes There was low quality evidence that exacerbation rates in people using LABA/ICS inhalers were lower in comparison to those with LABA alone, from nine studies which randomised 9921 participants (rate ratio 0.76; 95% CI 0.68 to 0.84). This corresponds to one exacerbation per person per year on LABA and 0.76 exacerbations per person per year on ICS/LABA. Our confidence in this effect was limited by statistical heterogeneity between the results of the studies (I2 = 68%) and a risk of bias from the high withdrawal rates across the studies. When analysed as the number of people experiencing one or more exacerbations over the course of the study, FPS lowered the odds of an exacerbation with an odds ratio (OR) of 0.83 (95% CI 0.70 to 0.98, 6 studies, 3357 participants). With a risk of an exacerbation of 47% in the LABA group over one year, 42% of people treated with LABA/ICS would be expected to experience an exacerbation. Concerns over the effect of reporting biases led us to downgrade the quality of evidence for this effect from high to moderate.
There was no significant difference in the rate of hospitalisations (rate ratio 0.79; 95% CI 0.55 to 1.13, very low quality evidence due to risk of bias, statistical imprecision and inconsistency). There was no significant difference in mortality between people on combined inhalers and those on LABA, from 10 studies on 10,680 participants (OR 0.92; 95% CI 0.76 to 1.11, downgraded to moderate quality evidence due to statistical imprecision). Pneumonia occurred more commonly in people randomised to combined inhalers, from 12 studies with 11,076 participants (OR 1.55; 95% CI 1.20 to 2.01, moderate quality evidence due to risk of bias in relation to attrition) with an annual risk of around 3% on LABA alone compared to 4% on combination treatment. There were no significant differences between the results for either exacerbations or pneumonia from trials adding different doses or types of inhaled corticosteroid.
Secondary outcomes ICS/LABA was more effective than LABA alone in improving health‐related quality of life measured by the St George's Respiratory Questionnaire (1.58 units lower with FPS; 2.69 units lower with BDF), dyspnoea (0.09 units lower with FPS), symptoms (0.07 units lower with BDF), rescue medication (0.38 puffs per day fewer with FPS, 0.33 puffs per day fewer with BDF), and forced expiratory volume in one second (FEV1) (70 mL higher with FPS, 50 mL higher with BDF). Candidiasis (OR 3.75) and upper respiratory infection (OR 1.32) occurred more frequently with FPS than SAL. We did not combine adverse event data relating to candidiasis for BDF studies as the results were very inconsistent.
Authors' conclusions
Concerns over the analysis and availability of data from the studies bring into question the superiority of ICS/LABA over LABA alone in preventing exacerbations. The effects on hospitalisations were inconsistent and require further exploration. There was moderate quality evidence of an increased risk of pneumonia with ICS/LABA. There was moderate quality evidence that treatments had similar effects on mortality. Quality of life, symptoms score, rescue medication use and FEV1 improved more on ICS/LABA than on LABA, but the average differences were probably not clinically significant for these outcomes. To an individual patient the increased risk of pneumonia needs to be balanced against the possible reduction in exacerbations.
More information would be useful on the relative benefits and adverse event rates with combination inhalers using different doses of inhaled corticosteroids. Evidence from head‐to‐head comparisons is needed to assess the comparative risks and benefits of the different combination inhalers.
Keywords: Adult; Humans; Adrenal Cortex Hormones; Adrenal Cortex Hormones/administration & dosage; Adrenal Cortex Hormones/adverse effects; Adrenergic beta‐Agonists; Adrenergic beta‐Agonists/administration & dosage; Adrenergic beta‐Agonists/adverse effects; Albuterol; Albuterol/administration & dosage; Albuterol/adverse effects; Albuterol/analogs & derivatives; Androstadienes; Androstadienes/administration & dosage; Androstadienes/adverse effects; Bronchodilator Agents; Bronchodilator Agents/administration & dosage; Bronchodilator Agents/adverse effects; Budesonide; Budesonide/administration & dosage; Budesonide/adverse effects; Drug Combinations; Drug Therapy, Combination; Drug Therapy, Combination/methods; Ethanolamines; Ethanolamines/administration & dosage; Ethanolamines/adverse effects; Fluticasone; Formoterol Fumarate; Nebulizers and Vaporizers; Pneumonia; Pneumonia/chemically induced; Pulmonary Disease, Chronic Obstructive; Pulmonary Disease, Chronic Obstructive/drug therapy; Pulmonary Disease, Chronic Obstructive/mortality; Quality of Life; Randomized Controlled Trials as Topic; Salmeterol Xinafoate
Plain language summary
Do combined inhalers (steroid plus bronchodilator) offer additional benefits or harms in people with COPD compared with the bronchodilator alone?
Chronic obstructive pulmonary disease (COPD) includes chronic bronchitis and emphysema. People with COPD have damaged (inflamed) or narrowed airways (tubes in the lungs), which makes breathing difficult. The symptoms are breathlessness, coughing and phlegm and can vary from mild to severe (where day‐to‐day activities become limited). The most common cause of COPD is smoking, however COPD may also be caused by occupational exposure to dust.
This review is about regular treatment with the combined delivery of inhaled steroids and long‐acting beta agonists in one inhaler (a combination inhaler). Combination inhalers contain two medications normally given in separate inhalers, a long‐acting beta2‐agonist (LABA), which is a bronchodilator that widens the tubes in the lungs, and a steroid which helps control the underlying inflammation in the lungs. This combination inhaler may make it easier to take the medication than using separate inhalers. Two combination inhalers containing two different medications are currently licensed for COPD, budesonide and formoterol (marketed as Symbicort) and fluticasone and salmeterol (marketed as Advair, Viani or Seretide).
We searched for randomised controlled trials (RCTs) that compared any combination inhaler versus the same LABA component inhaler used by people with COPD. The studies were well‐designed with low risk of bias for randomisation and blinding but there were high numbers of people who dropped out of the trials, which affected our confidence in the results for the outcomes.
Overall, we found 14 trials involving 11,794 people with COPD.
The results of the studies showed that combined inhalers reduced the frequency of exacerbations compared with their LABA component alone, from, for example, an average of one exacerbation per year on a long‐acting beta2‐agonist to an average of 0.76 exacerbations per year on a combined inhaler. The risk of mortality was similar between the treatments, although the overall result was not precise enough to rule out an effect in favour of either treatment. There was evidence of an overall increased risk of pneumonia with combined inhalers, from around three per 100 people per year on LABA to four per 100 per year on combined inhalers.
There was no significant difference between treatments in terms of hospitalisations although the results of the three studies were inconsistent so we cannot be certain what this means. Combined treatment was more effective than LABA in improving health‐related quality of life, symptoms such as breathlessness and cough, some measures of lung function, and also reduced rescue medication use, but it is difficult to tell whether these differences would be meaningful for individual people with COPD. Fluticasone/salmeterol led to more candidiasis and chest infections compared with salmeterol.
Future research is required to show whether combined therapy reduces hospitalisations, and to better estimate the increased risks of pneumonia. This will need more trials with different doses of inhaled corticosteroids and including direct comparisons of different combination inhalers. The conclusions of the review are current to November 2011.
Summary of findings
Summary of findings for the main comparison. Combined inhalers compared to LABAs for COPD.
| Combined inhalers compared to LABAs for COPD | ||||||
|
Patient or population: COPD
Intervention: Combined inhalers
Comparison: LABA inhalers Setting: community | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| LABA inhalers | Combined inhalers | |||||
| Annual Exacerbation Rates Follow‐up: median 1 year | 1 per person per year |
0.76 per person per year (0.32 exacerbations fewer to 0.16 exacerbations fewer) |
Rate ratio 0.76 (0.68 to 0.84) |
99211 (9 studies) | ⊕⊕⊝⊝ low2,3 | The control arm exacerbation rates ranged from 0.9 to 1.5 per year in the studies of at least one year duration that included participants with severe COPD who had at least one exacerbation in the previous year.4 |
|
Number of people experiencing one or more exacerbations Follow up: median 1 year |
47 per 100 |
42 per 100 (38 to 46) |
OR 0.83 (0.70 to 0.98) | 3357 (6 studies) |
⊕⊕⊕⊝ moderate5 | The evidence summarised for this outcome comes only from studies evaluating fluticasone/salmeterol. As such evidence for this outcome does not apply to budesonide/formoterol. |
|
Annual hospitalisation rates Follow‐up: 1 to 3 years |
0.16 per person per year6 |
0.15 per person per year (0.1 to 0.21) |
Rate ratio 0.79 (0.55 to 1.13) |
48791 (3 studies) |
⊕⊕⊝⊝ very low2,3,7 |
|
| Mortality Follow‐up: median 1 year | 8 per 1000 | 7 per 1000 (5 to 9) | OR 0.92 (0.76 to 1.11) | 10681 (10 studies) | ⊕⊕⊕⊝ moderate7 | The majority of data on mortality was derived from TORCH (vital status was ascertained in the patients who withdrew from treatment in this study). Control arm event rates varied from 0.4% to 5% in the one year studies. |
| Pneumonia Follow‐up: median 1 year | 27 per 1000 | 41 per 1000 (32 to 54)8 | OR 1.55 (1.2 to 2.01) | 11076 (12 studies) | ⊕⊕⊕⊝ moderate2 | See Table 3 for control arm event rates in all studies |
| *The basis for the assumed risk (was the median control group risk across studies of one year duration). The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 This is the number of participants randomised in the studies. The analysis took account of the amount of time the participants were enrolled for prior to withdrawal.
2 Risk of bias (‐1): High withdrawal rates in all the longer trials. 3 Inconsistency (‐1): Significant heterogeneity between trial results. 4 Exacerbations were moderate or severe requiring oral corticosteroids, antibiotics or leading to hospital admission.
5 Publication bias (‐1): Data from a key study of fluticasone and salmeterol (TORCH) and from studies evaluating budesonide/formoterol were not available as binary data and did not contribute to this measurement of exacerbations. Whilst the analysis of the data as rates in these studies is likely to reflect the individual trial protocols, we cannot be sure that their absence from this outcome measure has little or no impact on the pooled odds ratio.
6 Annualised rates of hospitalisation have been estimated from the three year study TORCH.
7 Imprecision (‐1): Confidence interval includes possible harm and benefit.
8 See Table 3 for a range of NNT(H) results for risk of pneumonia across the trials of different durations.
2. Rates and NNT(H) for pneumonia.
| Study ID | Study duration | Rate on LABA alone % | NNT (calculated from pooled OR using Visual Rx) |
| O'Donnell 2006 | 8 weeks | 0 | NA |
| Hanania 2003 | 24 weeks | 0.6 | 291 (192 to 525) |
| Mahler 2002 | 24 weeks | 0 | NA |
| SCO100470 | 24 weeks | 0.8 | 219 (145 to 395) |
| Tashkin 2008 | 26 weeks | 1.76 | 107 (59 to 291) |
| Kardos 2007 | 48 weeks | 1.4 | 126 (84 to 228) |
| TRISTAN | 52 weeks | 2.4 | 75 (50 to 135) |
| Calverley 2003 | 52 weeks | 2.7 | 67 (45 to 120) |
| Anzueto 2009 | 52 weeks | 2.5 | 76 (42 to 207) |
| Rennard 2009 | 52 weeks | 3.43 | 56 (31 to 152) |
| Ferguson 2008 | 52 weeks | 3.9 | 50 (28 to 135) |
| TORCH | 156 weeks | 13.3 | 17 (12 to 29) |
Background
Many people with asthma and chronic obstructive pulmonary disease (COPD) are treated with both an inhaled long‐acting beta2‐agonist (LABA) and an inhaled corticosteroid (ICS), and combinations of a LABA and ICS at fixed doses are available using a single inhaler device. Two commercially available preparations are fluticasone and salmeterol (FPS) (marketed as Advair, Seretide and Viani) and budesonide and formoterol (BDF) (marketed as Symbicort and Vannair).
Both component treatments are considered to confer some benefit in COPD. LABAs are recommended by NICE 2010 and the Global Initiative for Chronic Obstructive Lung Disease (GOLD) (GOLD 2010) as regular treatment because they are more effective and convenient than treatment with short‐acting bronchodilators (Mahler 1999; Dahl 2001).
Inhaled steroids reduce the frequency and severity of exacerbations (Yang 2007), although prolonged treatment with inhaled glucocorticosteroids does not slow the decline in forced expiratory volume in one second (FEV1) (Burge 2000; Sutherland 2003; Yang 2007; Celli 2008). However, the clinical impact of the effect size remains unclear and in the absence of randomised head‐to‐head comparisons the dosage at which steroids are effective in COPD remains a source of contention. NICE 2010 and GOLD 2010 recommend the addition of inhaled corticosteroids to long‐acting bronchodilators in symptomatic COPD patients with an FEV1 < 50% predicted and two or more exacerbations in the previous 12 months, or three exacerbations in the last three years (GOLD 2010). Recent Cochrane reviews (Spencer 2011; Welsh 2011) have compared the effects of treatment with a LABA with ICS treatment, and combination LABA and ICS (LABA/ICS) therapy with tiotropium.
In practice, at stages II and III (FEV1% predicted post‐bronchodilator > 30% and < 80%) or grades III and IV in the 2003 updated stages of severity (GOLD 2010), patients may seek medical attention because of dyspnoea or exacerbations of their disease that have an impact on their quality of life. Therefore, clinicians and patients are faced with the choice between starting treatment with either a LABA alone or its co‐administration with an ICS. The availability of new studies has prompted us to split the previous version of this review (Nannini 2004) in order to consider separate comparisons of combination LABA/ICS therapy with placebo (Nannini 2007a), inhaled steroids (Nannini 2010) and LABAs.
LABAs and ICS may also be used with tiotropium. As this is the subject of other Cochrane reviews (Karner 2011; Karner 2011a; Karner 2011b), treatment in addition to regular tiotropium has not been considered in this review.
Objectives
To assess the efficacy of combined inhaled corticosteroids (ICS) and long‐acting beta2‐agonist (LABA) preparations with LABAs alone in adults with COPD.
Methods
Criteria for considering studies for this review
Types of studies
We included randomised, double‐blind trials comparing combined ICS and LABA inhalers (LABA/ICS) with the same LABA alone.
Types of participants
Eligible studies had to recruit adult patients (age over 40 years) with known, stable COPD fulfilling American Thoracic Society (ATS), European Respiratory Society (ERS) and Global Initiative for Chronic Obstructive Lung Disease (GOLD) diagnostic criteria and who had not had an exacerbation for one month prior to study entry. We excluded studies that enrolled patients with significant diseases other than COPD; or a diagnosis of asthma, cystic fibrosis, bronchiectasis, thoracic surgery or other lung diseases. We included studies with people that had partial reversibility on pulmonary function testing.
Types of interventions
Fluticasone and salmeterol (FPS) versus salmeterol
Budesonide and formoterol (BDF) versus formoterol
Concomitant therapy was permitted provided it was not part of the randomised treatment.
Types of outcome measures
Primary outcomes
Exacerbations, urgent visits and hospitalisations
Mortality
Pneumonia
Secondary outcomes
Change in forced expiratory volume in one second (FEV1) and change in forced ventilatory capacity (FVC): trough, peak and average; and other measures of pulmonary function
Exercise performance, six‐minute walk test and other measures
Health‐related quality of life, measured on a validated questionnaire e.g. St George's Respiratory Questionnaire (SGRQ), Chronic Respiratory Disease Questionnaire (CRQ)
Symptoms
Rescue medication use
Withdrawal due to lack of efficacy
Withdrawal due to adverse events
Adverse events ‐ palpitations, tremor, hoarseness or dysphonia, oral candidiasis, cataracts, skin bruising, bone fracture, bone density, plasma cortisol level
Search methods for identification of studies
Electronic searches
We identified trials using the Cochrane Airways Group Specialised Register of trials, which is derived from systematic searches of bibliographic databases including the Cochrane Central Register of Controlled Trials (CENTRAL), MEDLINE, EMBASE, CINAHL, AMED and PsycINFO and handsearching of respiratory journals and meeting abstracts (see Appendix 1). All records in the Specialised Register coded as 'COPD' were searched using the following terms:(((beta* and agonist*) and long*) or ((beta* and adrenergic*) and long*) and (*steroid or steroid* OR corticosteroid*)) or (fluticasone and salmeterol) or Seretide or Advair or (formoterol and budesonide) or Symbicort or (mometasone and formoterol) or Dulera
The most recent search on the Register was run in November 2011. In addition, we performed a search of LILACS (all years to March 2011) and CENTRAL (The Cochrane Library 2011, Issue 1).
Searching other resources
We reviewed reference lists of all primary studies and review articles for additional references. We contacted authors of identified randomised trials about other published and unpublished studies. In addition, we consulted the online trial registries of GSK and AstraZeneca, manufacturers of FPS and BDF, respectively (www.ctr.gsk.co.uk; www.astrazenecaclinicaltrials.com). Since December 2007, we reviewed the ClinicalTrials.gov protocol registration system.
Data collection and analysis
Selection of studies
Two review authors independently identified abstracts of trials which appeared potentially relevant. Using the full text of each study, two review authors independently selected trials for inclusion in the review. Agreement was by simple agreement; third party adjudication was used to resolve differences.
Data extraction and management
Two review authors independently extracted data from included trials and entered results into the Cochrane Collaboration's software program (RevMan 5). In some cases, we estimated information regarding outcomes from graphs. This was performed independently by two review authors. Data extraction included the following items.
Population: age, gender, smoking status, study setting (country, practice setting), inclusion and exclusion criteria.
Intervention: dose, delivery device, duration.
Control: concurrent treatments (ipratropium, beta2‐agonist, inhaled and systemic corticosteroids).
Outcomes: exacerbations, hospitalisations, mortality, pulmonary function measures (baseline and follow‐up FEV1 and FVC), timing of pulmonary function measures, six‐minute walk test, urgent visits, admissions, self‐rated symptom score or symptoms, quality of life, withdrawal due to lack of efficacy and due to adverse events, adverse effects (pneumonia, palpitations, dry mouth, blurred vision, urinary obstruction, oral candidiasis, and constipation), assessors, adjudicator of clinical endpoints. Mortality outcome data were collected from studies of greater than one year's duration, where available.
Design: method of randomisation, presence and type of run‐in period, study design (parallel, cross‐over).
Assessment of risk of bias in included studies
We assessed the risk of bias for each study according to the extent to which the design of the trial had protected the findings from bias. Our assessments were made on the basis of allocation, allocation concealment, blinding and incomplete outcome data. We assessed the risk of bias for the following sources of bias (domains) in each study in accordance with recommendations made in Chapter 8 of the Cochrane Handbook of Systematic Reviews of Interventions (Higgins 2008).
Random sequence generation (selection bias).
Allocation concealment (selection bias).
Blinding (performance bias and detection bias).
Incomplete outcome data (attrition bias): mortality.
Incomplete outcome data (attrition bias): all other outcomes.
Selective reporting: primary outcomes.
Dealing with missing data
We used reported confidence intervals or P values to calculate standard deviations or standard errors for results that were not reported and could not be obtained from the authors of the papers.
Assessment of heterogeneity
For pooled effects, we tested heterogeneity using the I2 statistic,which indicates the degree of variation between the studies not attributable to the play of chance (Higgins 2008).
Data synthesis
For continuous variables, we used a random‐effects model weighted mean difference (MD) for outcomes measured on the same metric. The results of a fixed‐effect model were also considered as part of a sensitivity analysis.
For dichotomous variables, we calculated a random‐effects model odds ratio (OR) with 95% confidence interval (95% CI) for individual studies. We pooled similar studies using the random‐effects model ORs and 95% CIs. Where mean treatment differences were reported (rather than the changes reported separately in each group) we entered data as generic inverse variance (GIV) provided we could calculate a standard error for the difference. We added GIV methodology in the 2012 update; this method was not available at the time the protocol was written.
We calculated numbers needed to treat to prevent one event (NNT) or numbers needed to treat to cause one harm (NNT(H)) for individual studies from the pooled odds ratio, by using Visual Rx and using the control group event rate for each study as the baseline risk.
Subgroup analysis and investigation of heterogeneity
Whilst we performed separate analyses (as separate comparisons) according to the type of combined inhaler, we pooled studies with differing dosages of the same drug. We planned other a priori subgroup analyses as follows.
Disease severity (related to baseline FEV1 and placebo group exacerbation rate) according to the GOLD staging: II (moderate COPD, characterised by deteriorating lung function (FEV1 ≤ 80% predicted; > 50% predicted) and progression of symptoms); III (severe COPD, characterised by severe airflow limitation (FEV1 < 50% predicted) and presence of respiratory failure or clinical signs of right heart failure): and stage IV (very severe, FEV1 < 30%) (GOLD 2010).
Prior ICS plus LABA use (dichotomised as yes or no).
Concurrent therapy with routine beta2‐agonist use (short or long‐acting), corticosteroid (systemic or inhaled) or theophylline (dichotomised as yes or no).
Reversibility of airflow obstruction with beta2‐agonist therapy (dichotomised as partial or none). Definition: > 12% and > 200 mL from baseline FEV1 or > 12% as a per cent of the predicted normal value following metered dose inhaler (MDI) salbutamol 200 to 400 µg.
Dose, duration and delivery method of therapy.
Sensitivity analysis
A fixed‐effect model was compared to the random‐effects model to determine the impact of heterogeneity on the overall pooled effects. In addition, the robustness of the results was tested using a sensitivity analysis based on the risk of bias of the trials, where possible.
Results
Description of studies
Results of the search
For details of the search history see Table 2. For a flow diagram illustrating the separation of comparisons from the original review (Nannini 2004), see Figure 1. For a study flow diagram of the 2007 to 2011 literature search update see Figure 2.
1. Search history.
| Version | Detail |
| 1st published version ‐ Issue 4, 2003 (All years to April 2002) | References identified: 34 References retrieved: 7 Studies excluded 3 (Cazzola 2000; Chapman 2002; Soriano 2002) Studies identified from supplementary searching: 4 (Dal Negro 2003; Hanania 2003 ‐ both included; Cazzola 2002a; Cazzola 2004 ‐ both excluded). Studies included: 4 |
| Update: Issue 3, 2004 (April 2003‐April 2004) | References identified: 12 References retrieved: 3 (2 papers full publication of a previously included or cited studies study (Dal Negro 2003; Hanania 2003). Hand searching identified two further references to the COSMIC 2003 study. Studies identified from supplementary searching: 1 (TRISTAN 2003) New studies included: 2 Total studies included: 6 |
| Update: Issue 3, 2005 (April 2004‐April 2005) | References identified: 52 References retrieved: 46 (references to studies already included/excluded/ongoing: 24) New unique studies identified: 10 (ongoing studies: 2) New studies included: 0 Total studies included: 6 |
| Update: April 2005 ‐ April 2007 | References identified: 66 References retrieved: 27 (references to studies already included/excluded/ongoing: ) New unique studies identified: 8 (ongoing studies: 0) New studies included: 4 Total studies included: 10 |
| Update: April 2007‐ November 2011 | References identified: 207 References retrieved: 4 (references to studies already included/excluded/ongoing: ) New unique studies identified: 4 (ongoing studies: 0) New studies included: 4 Total studies included: 14 |
1.
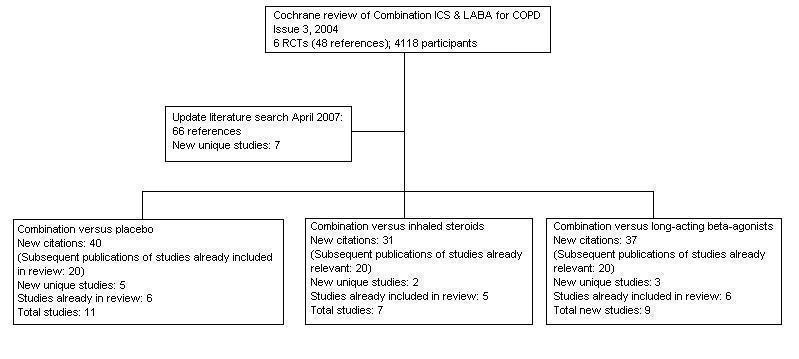
Flow chart to illustrate separation of review between three comparisons. Six RCTs met the original entry criteria of the review. All of these had a placebo and long‐acting beta2‐agonist arm, and five assessed combination against steroids. Seven new studies with one or more control comparisons were identified: five had a placebo arm, three had a long‐acting beta2agonist arm, and two had an inhaled steroid treatment arm.
2.
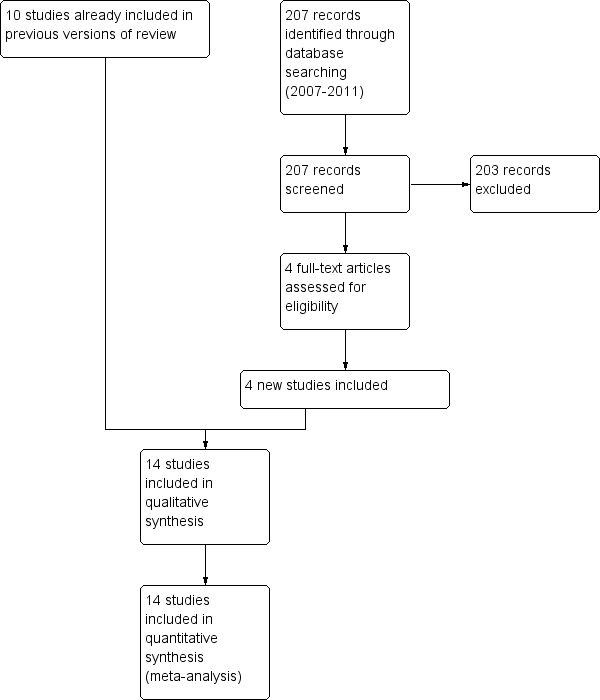
Study flow diagram for 2007‐2011 literature searches.
Included studies
Fourteen studies recruiting 11,794 people with COPD were included in this review, including four new studies added for the 2012 update (Ferguson 2008; Tashkin 2008; Anzueto 2009; Rennard 2009). For a full description of baseline characteristics, methods used and inclusion and exclusion entry criteria see Characteristics of included studies.
Design
All trials had a randomised, double‐blind parallel group design.
Participants
Participants suffered from COPD, with the definition of COPD and threshold response to short‐acting beta2‐agonist varying between the studies. In all studies, COPD was defined by national or international criteria: American Thoracic Society (ATS) (Mahler 2002; Hanania 2003; O'Donnell 2006; Ferguson 2008); European Respiratory Society (ERS) (TORCH; TRISTAN); or GOLD (SCO100470; Calverley 2003; Dal Negro 2003; Szafranski 2003; Kardos 2007). Patient populations in the studies suffered from moderate and severe COPD (GOLD 2010). Six trials enrolled participants with both partially reversible and non‐reversible COPD (Mahler 2002; Hanania 2003; Ferguson 2008; Tashkin 2008; Anzueto 2009; Rennard 2009). The baseline reversibility in O'Donnell 2006 indicated that the study population had a significant bronchodilator response of around 18%.
Interventions
Concomitant therapy permitted was as needed short‐acting beta2‐agonist (salbutamol) or oral steroids or antibiotics, or both, in the case of exacerbations. In three studies, theophylline was used in some patients in both arms. Eleven per cent of participants in Hanania 2003 and all 18 participants in Dal Negro 2003 received theophylline in addition to the study drugs. The exact proportion of patients in TRISTAN who were taking theophylline was not reported.
In four studies, the combination inhaled LABA/ICS was fluticasone and salmeterol (FPS) 250 µg/50 µg twice daily (Dal Negro 2003; Hanania 2003; Ferguson 2008; Anzueto 2009) versus 50 µg salmeterol. In the remainder of the FPS studies the dose was 500 µg/50 µg twice daily versus 50 µg salmeterol. In two studies the combination inhaled LABA/ICSwas budesonide and formoterol (BDF) (320 µg/9 µg twice daily) (Calverley 2003; Szafranski 2003) versus formoterol (9 µg twice daily). Two further studies included separate BDF arms with 320 µg/9 µg daily and 160 µg/9 µg daily (Tashkin 2008; Rennard 2009) also versus formoterol (9 µg twice daily). The dosage of the combined preparation and the separate medications remained stable throughout the studies.
Duration
8 weeks: O'Donnell 2006 24 weeks: Mahler 2002; SCO100470; Hanania 2003; Tashkin 2008 52 weeks: Calverley 2003; Dal Negro 2003; TRISTAN; Szafranski 2003; Kardos 2007; Ferguson 2008; Anzueto 2009; Rennard 2009 156 weeks: TORCH
Outcomes
Exacerbations were defined and stratified either by the medication given: oral steroids, antibiotics, or both in nine trials (Calverley 2003; Dal Negro 2003; TRISTAN; Szafranski 2003; Kardos 2007; TORCH; Ferguson 2008; Anzueto 2009; Rennard 2009); or hospitalisation in seven trials (Dal Negro 2003; TRISTAN; Kardos 2007; TORCH; Ferguson 2008; Anzueto 2009; Rennard 2009). Two trials withdrew participants who experienced an exacerbation (Mahler 2002; Hanania 2003). Lung function was measured as FEV1 or peak expiratory flow (PEF) in all the studies. Quality of life assessment by the SGRQ or CRQ were available for 12 studies (Mahler 2002; SCO100470; Calverley 2003; TRISTAN; Hanania 2003; Szafranski 2003; Kardos 2007; TORCH; Ferguson 2008; Tashkin 2008; Anzueto 2009; Rennard 2009). Only Tashkin 2008 used the validated Breathlessness Diary, a single‐item dyspnoea measure derived from the breathlessness, cough, and sputum scale (BCSS) (Leidy 2003). All cause mortality was available for 10 studies including the largest (TORCH).
Pneumonia was confirmed by chest X‐ray (as pre‐specified in the trial protocol) in one trial (Anzueto 2009). Pneumonia events were reported by physicians based on the Medical Dictionary for Regulatory Activities (version 10.0) pneumonia‐related preferred terms in two trials (Ferguson 2008; Rennard 2009). Pneumonia was not defined in Tashkin 2008. There was no prospective definition of pneumonia in the study protocol (for example confirmation on chest radiography) for TORCH, and pneumonia as an adverse event was not consistently reported in this study.
Excluded studies
Risk of bias in included studies
An overview of our assessment of four domains (allocation generation, allocation concealment, blinding and incomplete data) is given in Figure 3. Effective means of blinding study participants to treatment were undertaken in all the studies. Information describing the processes for randomising participants to treatment groups were not commonly available.
3.
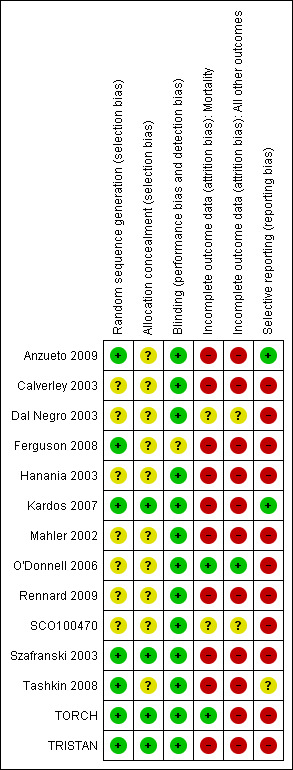
Methodological quality summary: review authors' judgements about each methodological quality item for each included study.
Methods of randomisation were described in eight studies (Mahler 2002; TRISTAN; Szafranski 2003; Kardos 2007; TORCH; Ferguson 2008; Tashkin 2008; Anzueto 2009). The method of blinding was not fully described in all studies. Following correspondence from GSK, trial methodology was confirmed for TRISTAN, and AstraZeneca confirmed the methodology for Szafranski 2003. Study characteristics were sufficiently described in one study without full text journal citation to justify inclusion in the review (SCO100470).
Drop‐out rates were uniformly high in the long‐term studies (Figure 3) and most studies were judged to be at high risk of attrition bias, but TORCH ascertained the mortality results for those participants that withdrew from the study and so was judged at low risk of attrition bias for mortality. For studies which reported dichotomised data on exacerbations (that is the number of participants experiencing one or more exacerbations), we did not consider attrition to have affected the direction or magnitude of effect across the studies which contributed to this outcome.
Taken together (high withdrawal rates and high risk of attrition bias) most studies had some potential for bias. The analysis of data for the primary outcome of exacerbations as dichotomous data was at risk of reporting bias.
Effects of interventions
See: Table 1
Primary outcome: exacerbations
Ratio of exacerbation rates
All combination inhalers versus LABA
The exacerbation rates with combined inhalers were reduced in comparison to LABA alone (rate ratio 0.76; 95% CI 0.68 to 0.84) (Figure 4). These results are based on data from nine trials. The number of participants randomised to the studies was 9921. However, the total number of years of follow‐up used to calculate the rates across the studies was not available. Assessment of the withdrawal rates suggests that this effect is at risk of attrition bias (see Figure 3). There was significant heterogeneity between the study results for this outcome (I2 = 68%). The range of average exacerbation rates on LABA alone (the control arm) in the trials lasting at least 12 months was between 0.9 and 1.5 exacerbations per person per year. The quality of evidence for this effect was graded as low due to the high rates of attrition and inconsistency between the results of different studies (see Table 1).
4.
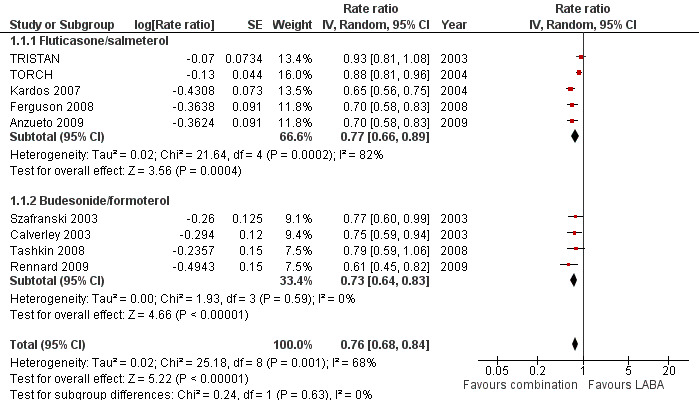
Forest plot of comparison: 1 Combined inhalers versus long‐acting beta2‐agonists (primary outcomes), outcome: 1.1 Exacerbation rates (combined treatment versus beta2‐agonist).
Subgroup analysis based on medication type
Combination FPS had a greater protective effect against exacerbations than salmeterol alone (rate ratio 0.77; 95% CI 0.66 to 0.89, 5 trials, 6391 participants) (Figure 4). There was a high level of statistical variation however (I2 = 82%), with the effect size from three studies (Kardos 2007; Ferguson 2008; Anzueto 2009) considerably greater than for either of the earlier studies (TORCH; TRISTAN). This disparity across the studies may reflect subgroups of COPD patients who are at greater risk of exacerbating. TORCH was the only study that did not require study participants to have had a history of exacerbations in the 12 months before enrolment. This does not explain the disparity between TRISTAN and the newer studies (since the inclusion criteria of TRISTAN specified at least one episode of acute COPD per year in the previous three years). When pooled data were re‐analysed using a fixed‐effect model, the result was very similar (rate ratio 0.80; 95% CI 0.76 to 0.85). The two trials using lower‐dose FPS (250 µg/50 µg) that contributed data to this outcome showed a significant reduction in exacerbations (rate ratio 0.70; 95% CI 0.61 to 0.79) (Ferguson 2008; Anzueto 2009).
The combination BDF was also significantly more effective than formoterol alone in preventing exacerbations (rate ratio 0.73; 95% CI 0.64 to 0.83). These results were based on 2622 participants from four trials. The results were unchanged with a fixed‐effect model (rate ratio 0.73; 95% CI 0.64 to 0.83) and heterogeneity among these results was low (I2 = 0%).
No significant difference in the effect on exacerbation rates was found between the results from studies using FPS or BDF (test for subgroup differences: Chi2 = 0.24, df = 1 (P = 0.63)).
Number of people experiencing one or more exacerbations
FPS versus salmeterol
Six studies on 3357 participants taking FPS versus salmeterol reported the number of people experiencing one or more exacerbations. There was a significant reduction in the odds of exacerbations between FPS and salmeterol (OR 0.83; 95% CI 0.70 to 0.98; Analysis 1.2). The TORCH and TRISTAN studies did not report exacerbations as dichotomous data and were not included in this analysis. There was some heterogeneity in this outcome (I2 = 17%) although analysis with a fixed‐effect model gave a similar result (OR 0.82; 95% CI 0.71 to 0.95).
1.2. Analysis.
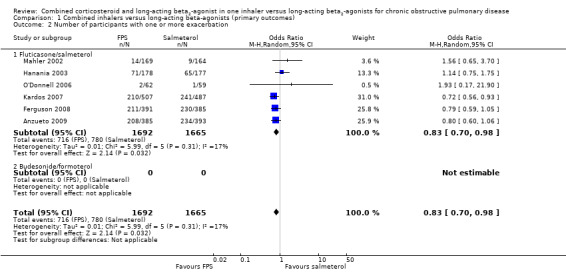
Comparison 1 Combined inhalers versus long‐acting beta‐agonists (primary outcomes), Outcome 2 Number of participants with one or more exacerbation.
BDF versus formoterol
Data on participants experiencing one or more exacerbations were not presented in the reports of trials of this comparison.
Exacerbation leading to hospital admission
FPS versus salmeterol
Compared with salmeterol, there was no significant difference in hospitalisations in people treated with FPS (rate ratio 0.79; 95% CI 0.55 to 1.13; Analysis 1.3), but there was substantial heterogeneity (I2 = 70%) between TORCH and the newer studies (Kardos 2007; Anzueto 2009). None of the studies discriminated between hospital admission and treatment in an intensive care unit (ICU). Ferguson 2008 did not distinguish between moderate and severe exacerbations, and Kardos 2007 did not report hospitalisations and emergency department (ED) visits separately, but the authors provided data on the number of participants admitted to hospital from each treatment arm and this was converted into a risk ratio and combined with the rate ratios from the other studies.
1.3. Analysis.
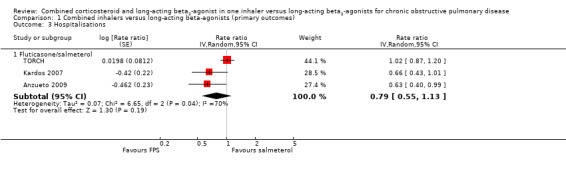
Comparison 1 Combined inhalers versus long‐acting beta‐agonists (primary outcomes), Outcome 3 Hospitalisations.
BDF versus formoterol
Data on severe exacerbations leading to hospitalisations were not available in any of the four studies.
Primary outcome: mortality
Pooled results for all combination inhalers versus LABA
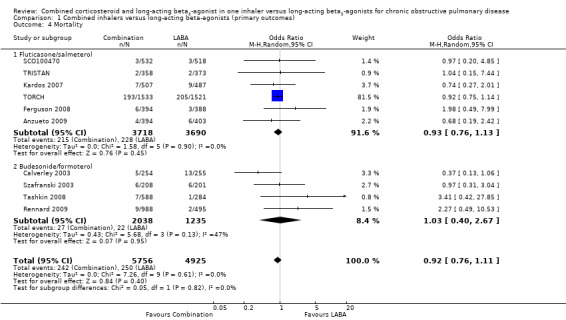
There was no significant difference in mortality between combination therapy and LABA (OR 0.92; 95% CI 0.76 to 1.11, N = 10681, 10 studies) (Figure 5). It should be noted that at the end of the large trial, TORCH investigators ascertained the vital status of participants who withdrew. Since TORCH contributed most of the data for this outcome, we felt that the overall risk of attrition bias for this outcome was lower than for the other outcomes. There was statistical imprecision in this outcome, with the wide CIs including the possibility of clinically important differences in mortality in both directions.
5.
Forest plot of comparison: 1 Combined inhalers versus Long‐acting beta2‐agonists (Primary Outcomes), outcome: 1.2 Mortality.
Subgroup analysis based on medication type
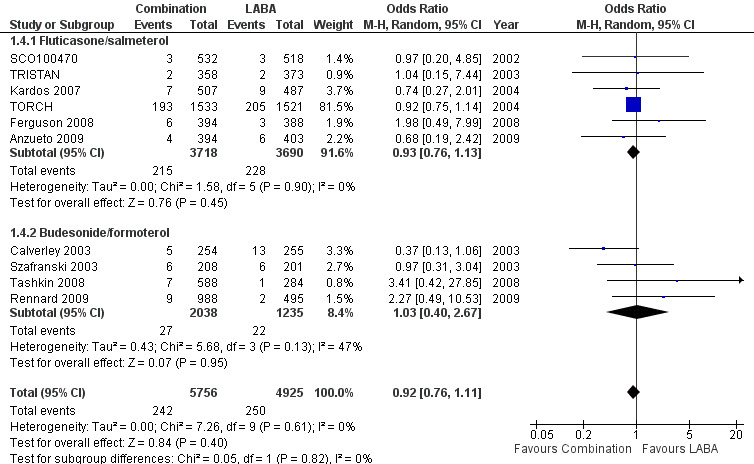
There was no significant difference between FPS and salmeterol in the odds of death from end of study data in six trials (OR 0.93; 95% CI 0.76 to 1.13, N = 7408; Analysis 1.4).
1.4. Analysis.
Comparison 1 Combined inhalers versus long‐acting beta‐agonists (primary outcomes), Outcome 4 Mortality.
There was no significant difference between BDF and formoterol for mortality (OR 1.03; 95% CI 0.40 to 2.67, N = 3273).
There was no significant difference between these subgroups (test for subgroup differences: Chi² = 0.05, df = 1 (P = 0.82)).
Primary outcome: pneumonia
Pooled results for all combination inhalers versus LABA
There was a significant increase in the odds of pneumonia on combination therapy compared with LABA as monotherapy (OR 1.55; 95% CI 1.20 to 2.01, 12 trials, N = 11,076) (Figure 6). A fixed‐effect model gave a similar overall result (OR 1.59; 95% CI 1.35 to 1.86).
6.

Forest plot of comparison: 1 Combined inhalers versus long‐acting beta2‐agonists (primary outcomes), outcome: 1.3 Pneumonia.
The NNT(H) for one additional participant to suffer pneumonia on combination treatment was calculated for each study with baseline risks drawn from the LABA only treatment groups (see Table 3). As might be anticipated, studies of a year or more were associated with lower NNT(H)s than those conducted over a shorter period of time.
For illustrative purposes, the NNT(H) over three years using the 20% baseline risk in TORCH is 17, whilst over one year in TRISTAN the baseline risk of 2% converts to an NNT(H) of 75. After TORCH raised the issue of risks of pneumonia, all subsequent studies added chest X‐ray for diagnosis of pneumonia.
Subgroup analyses based on medication type and dose
Pneumonia was more frequently reported in people on FPS treatment than by those on salmeterol in the studies using FPS 500/50 µg twice daily (OR 1.55; 95% CI 0.92 to 2.61). Three studies used a lower dose of ICS, FPS 250/50 µg twice daily (Hanania 2003; Ferguson 2008; Anzueto 2009) instead of 500/50 µg twice daily. Restricting the results to these studies still showed a significant increase in the risk of pneumonia even on the lower dose of fluticasone (OR 2.19; 95% CI 1.35 to 3.53) as shown in Analysis 1.6.
1.6. Analysis.

Comparison 1 Combined inhalers versus long‐acting beta‐agonists (primary outcomes), Outcome 6 Pneumonia subgrouped by dose.
Pneumonia was reported for Calverley 2003, Tashkin 2008 and Rennard 2009, and we have separated the results into participants treated with different doses of budesonide. The trials using higher dose budesonide, BDF 320/9 µg twice daily, showed an increase in the odds of pneumonia that was not statistically significant (OR 1.08; 95% CI 0.60 to 1.97), and this was similar to the results from trials using lower dose budesonide, BDF 160/9 µg twice daily (OR 1.10; 95% CI 0.53 to 2.26), see Analysis 1.6. Fewer participants were included in the BDF trials and the BDF CIs overlap those from the FPS studies. We have not demonstrated a significant difference in the risk of pneumonia between FPS and BDF, or any indication of a dose‐response effect (test for subgroup differences: Chi² = 4.22, df = 3 (P = 0.24), I² = 28.9%).
Such indirect comparisons must be interpreted with caution as they are subject to confounding by differences between the trials in the ascertainment of pneumonia, co‐interventions and patient characteristics. Moreover, the average duration of the FPS studies was longer than the BDF studies, as shown in Table 3, which may also make comparison between the subgroups unreliable.
Secondary outcome: quality of life
FPS versus salmeterol
Combination therapy led to a significant improvement in health‐related quality of life as measured on the SGRQ compared with salmeterol (‐1.58 SGRQ units; 95% CI ‐2.15 to ‐1.01, 6 studies, N = 10,681; Analysis 2.3). The data from TORCH were not available for the intention‐to‐treat (ITT) population. Removing this study from the outcome data gave a difference of ‐1.31 (95% CI ‐2.01 to ‐0.61). The minimal clinically important difference (MCID) for the SGRQ is around four units (Jones 2005). Furthermore, the trials in this analysis did not provide data on the number of participants that showed both a decrease and an increase in the threshold of four units over control in each treatment arm, so it was not possible to assess whether or not there was a difference in the proportion of participants with clinically important changes in quality of life on each treatment.
2.3. Analysis.
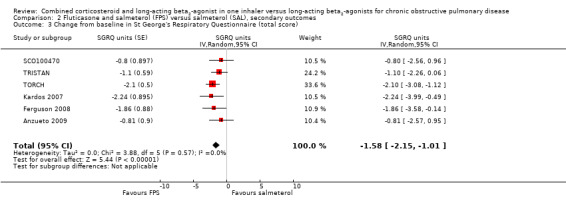
Comparison 2 Fluticasone and salmeterol (FPS) versus salmeterol (SAL), secondary outcomes, Outcome 3 Change from baseline in St George's Respiratory Questionnaire (total score).
The SGRQ domain data were available for four studies. Significant differences on three domains of the SGRQ were evident: symptoms ‐2.78 (95% CI ‐3.88 to ‐1.68); activity ‐1.31 (95% CI ‐2.38 to ‐0.24); and impact ‐1.41 (95% CI ‐2.33 to ‐0.50).
Health‐related quality of life was also measured using the CRQ. Pooled data from two trials on 680 participants showed a significant improvement in health‐related quality of life in people on FPS in comparison with those on salmeterol (2.83 units; 95% CI 0.25 to 5.41), but this difference is small in comparison with the minimal clinically important difference of 10 units on this scale (Hanania 2003).
BDF versus formoterol
There was a significant difference in the change from baseline in SGRQ scores favouring BDF (SGRQ ‐2.69; 95% CI ‐3.82 to ‐1.55; Analysis 3.1) from the three studies in 3442 participants that could be combined. However, comparisons with formoterol were not reported in detail in Szafranski 2003, which reported mean effects of 3.09 in the BDF group and 3.6 units in those treated with formoterol.
3.1. Analysis.
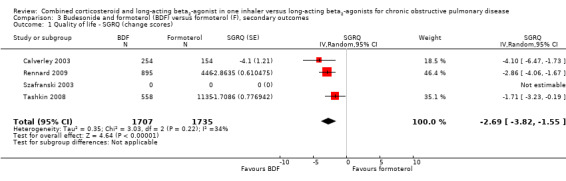
Comparison 3 Budesonide and formoterol (BDF) versus formoterol (F), secondary outcomes, Outcome 1 Quality of life ‐ SGRQ (change scores).
Secondary outcome: symptoms and nighttime awakenings
FPS versus salmeterol
Two studies on 677 participants reported changes in Transitional Dyspnoea index. There were conflicting findings between two studies, with a significant difference of 1.2 units in favour of people on FPS reported by Mahler 2002 and a non‐significant difference of 0.1 units from Hanania 2003. Change from baseline in the dyspnoea score was significantly in favour of FPS in two studies on 1554 participants (mean difference (MD) ‐0.09; 95% CI ‐0.13 to ‐0.05; Analysis 2.14). Mean change in nighttime awakenings was also significantly better on FPS in the same two studies (MD ‐1.34; 95% CI ‐2.07 to ‐0.61; Analysis 2.15).
2.14. Analysis.

Comparison 2 Fluticasone and salmeterol (FPS) versus salmeterol (SAL), secondary outcomes, Outcome 14 Change from baseline in dyspnoea score.
2.15. Analysis.
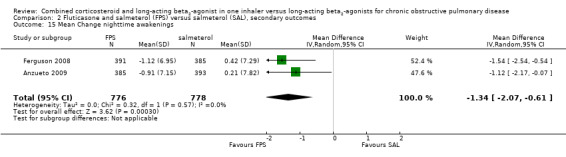
Comparison 2 Fluticasone and salmeterol (FPS) versus salmeterol (SAL), secondary outcomes, Outcome 15 Mean Change nighttime awakenings.
TRISTAN reported improvements in symptoms after treatment in favour of FPS versus salmeterol on breathlessness scores (FPS mean: 1.47; salmeterol mean: 1.58 (P = 0.010)). Improvement in nighttime awakenings was also reported (FPS mean number of nights per week: 2.31; salmeterol mean: 2.94 (P = 0.011)). Cough scores were not significantly different.
BDF versus formoterol
In the two studies on 2308 participants reporting on breathlessness, the score was significant lower in BDF in comparison with formoterol (MD ‐0.07; 95% CI ‐0.12 to ‐0.01; Analysis 3.8).
3.8. Analysis.
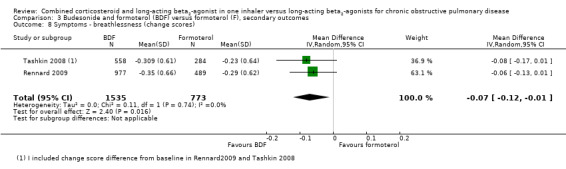
Comparison 3 Budesonide and formoterol (BDF) versus formoterol (F), secondary outcomes, Outcome 8 Symptoms ‐ breathlessness (change scores).
Secondary outcome: lung function
FPS versus salmeterol
Pooled analysis of data was conducted without findings from the Dal Negro 2003 study (which had only 12 participants but very low standard deviations). The mean change in pre‐dose FEV1 was significantly higher for people on FPS (0.07 L; 95% CI 0.05 to 0.10; Analysis 2.16) although the 70 mL mean difference in FEV1 would not be regarded as clinically significant (Cazzola 2008). Post‐dose FEV1 showed a similar small advantage for FPS (0.05 L; 95% CI 0.03 to 0.06; Analysis 2.17).
2.16. Analysis.

Comparison 2 Fluticasone and salmeterol (FPS) versus salmeterol (SAL), secondary outcomes, Outcome 16 Change from baseline in predose FEV1.
2.17. Analysis.
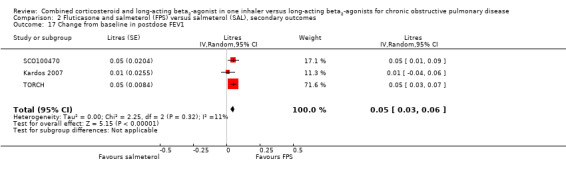
Comparison 2 Fluticasone and salmeterol (FPS) versus salmeterol (SAL), secondary outcomes, Outcome 17 Change from baseline in postdose FEV1.
One trial on only 12 participants reported change in % predicted FEV1, so we were unable to pool this outcome (Analysis 2.19). Our concerns over small study bias are supported by the very low variance in Dal Negro 2003. We therefore thought that it was not appropriate to combine the results on a standardised mean difference (SMD) scale. Two trials on 1780 participants reported data on pre‐dose FEV1 in mL, there was no significant difference (MD 0.01; 95% CI ‐0.02 to 0.03; Analysis 2.18).
2.19. Analysis.
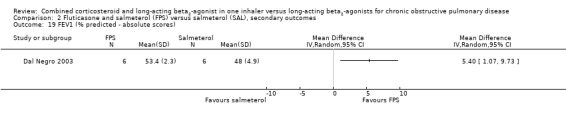
Comparison 2 Fluticasone and salmeterol (FPS) versus salmeterol (SAL), secondary outcomes, Outcome 19 FEV1 (% predicted ‐ absolute scores).
2.18. Analysis.
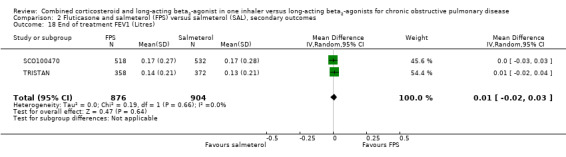
Comparison 2 Fluticasone and salmeterol (FPS) versus salmeterol (SAL), secondary outcomes, Outcome 18 End of treatment FEV1 (Litres).
BDF versus formoterol
There was a similar significant, small advantage for BDF in comparison to formoterol in change from baseline of pre‐dose FEV1 (MD 0.05 L; 95% CI 0.00 to 0.09).
Secondary outcome: rescue medication
FPS versus salmeterol
Pooled data from four studies on 2435 participants indicated that there was significantly less use of rescue medication on FPS compared to salmeterol (MD ‐0.38 puffs/day; 95% CI ‐0.61 to ‐0.16; Analysis 2.21).
2.21. Analysis.
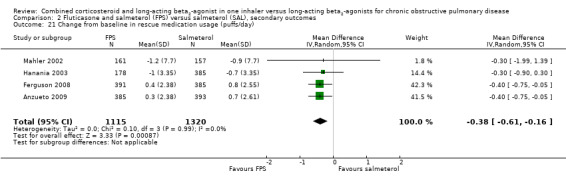
Comparison 2 Fluticasone and salmeterol (FPS) versus salmeterol (SAL), secondary outcomes, Outcome 21 Change from baseline in rescue medication usage (puffs/day).
TRISTAN reported a significant difference in median % of days without use of relief medication in favour of FPS of around 11% (P = 0.004).
BDF versus formoterol
On pooling data from two studies on 2310 participants, the change from baseline in rescue medication usage (puffs/day) was significantly lower in people on BDF (MD ‐0.33 puffs/day; 95% CI ‐0.57 to ‐0.09; Analysis 3.10). Calverley 2003 reported a significant difference versus formoterol of ‐0.3 puffs per day (P < 0.05), but we were unable to pool this data.
3.10. Analysis.
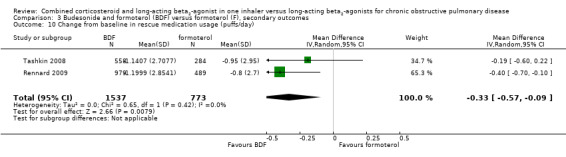
Comparison 3 Budesonide and formoterol (BDF) versus formoterol (F), secondary outcomes, Outcome 10 Change from baseline in rescue medication usage (puffs/day).
Secondary outcome: adverse events
FPS versus salmeterol
Candidiasis and upper respiratory tract infections occurred more frequently among people on FPS than in those on salmeterol only (OR 3.75; 95% CI 2.33 to 6.04, 6 studies, 3118 participants; Analysis 2.24; (OR 1.32; 95% CI 1.12 to 1.55, 7 studies, 6198 participants; Analysis 2.27), respectively. Headache did not occur more frequently with either therapy (OR 1.06; 95% CI 0.90 to 1.26; Analysis 2.26).
2.24. Analysis.
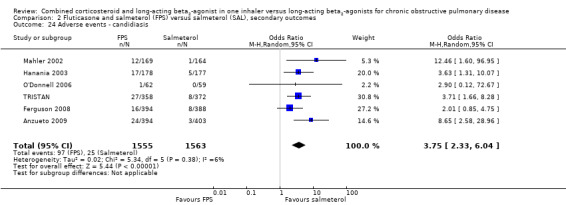
Comparison 2 Fluticasone and salmeterol (FPS) versus salmeterol (SAL), secondary outcomes, Outcome 24 Adverse events ‐ candidiasis.
2.27. Analysis.
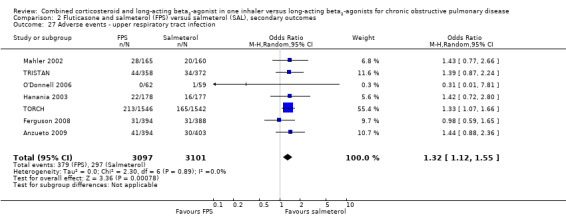
Comparison 2 Fluticasone and salmeterol (FPS) versus salmeterol (SAL), secondary outcomes, Outcome 27 Adverse events ‐ upper respiratory tract infection.
2.26. Analysis.
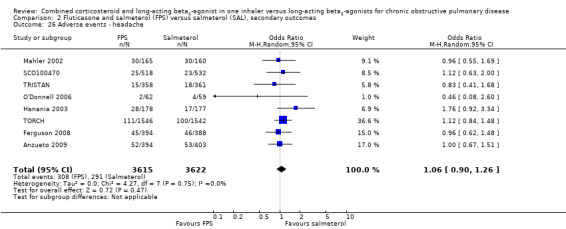
Comparison 2 Fluticasone and salmeterol (FPS) versus salmeterol (SAL), secondary outcomes, Outcome 26 Adverse events ‐ headache.
BDF versus formoterol
There was high and unexplained heterogeneity in the incidence of candidiasis among 2325 participant in two studies and therefore we did not pool these data (Analysis 3.12). There was no significant difference in serious adverse advents (OR 0.88; 95% CI 0.73 to 1.07) in four trials on 546 participants (Analysis 3.11).
3.12. Analysis.
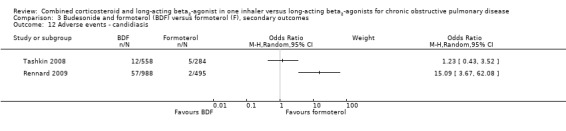
Comparison 3 Budesonide and formoterol (BDF) versus formoterol (F), secondary outcomes, Outcome 12 Adverse events ‐ candidiasis.
3.11. Analysis.
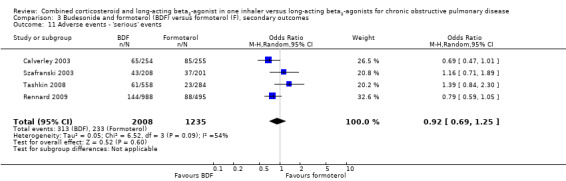
Comparison 3 Budesonide and formoterol (BDF) versus formoterol (F), secondary outcomes, Outcome 11 Adverse events ‐ 'serious' events.
Secondary outcome: withdrawals
FPS versus salmeterol
We were able to pool withdrawal data from nine trials on 8226 participants. Pooled analyses indicated that, overall, participant withdrawal occurred less frequently in people on FPS compared with those on salmeterol (OR 0.85; 95% CI 0.77 to 0.94; Analysis 2.28). Withdrawal due to lack of efficacy was less frequent in people on FPS than in those on salmeterol (OR 0.53; 95% CI 0.39 to 0.72, 6 studies, N = 6739; Analysis 2.29). The likelihood of withdrawal due to adverse events was not significantly different between FPS and salmeterol (OR 0.90; 95% CI 0.79 to 1.02, 8 studies, N = 7895 Analysis 2.30).
2.28. Analysis.

Comparison 2 Fluticasone and salmeterol (FPS) versus salmeterol (SAL), secondary outcomes, Outcome 28 Withdrawals.
2.29. Analysis.
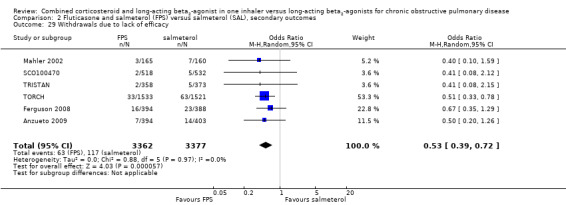
Comparison 2 Fluticasone and salmeterol (FPS) versus salmeterol (SAL), secondary outcomes, Outcome 29 Withdrawals due to lack of efficacy.
2.30. Analysis.
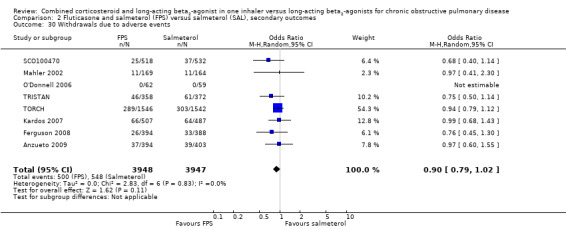
Comparison 2 Fluticasone and salmeterol (FPS) versus salmeterol (SAL), secondary outcomes, Outcome 30 Withdrawals due to adverse events.
BDF versus formoterol
Data for withdrawals due to adverse events were pooled from four studies on 3243 participants. There was no significant difference in the likelihood of withdrawal due to adverse events between people on BDF and those on formoterol (OR 0.88; 95% CI 0.65 to 1.19; Analysis 3.13) other than withdrawal due to COPD deterioration (OR 0.49; 95% CI 0.32 to 0.74; Analysis 3.14) in which treatment with BDF led to fewer withdrawals due to worsening of COPD symptoms compared with formoterol alone.
3.13. Analysis.

Comparison 3 Budesonide and formoterol (BDF) versus formoterol (F), secondary outcomes, Outcome 13 Withdrawals due to adverse events.
3.14. Analysis.
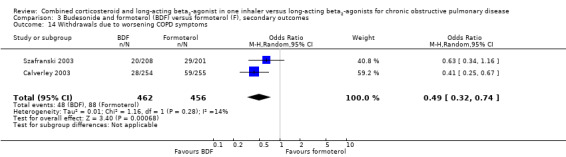
Comparison 3 Budesonide and formoterol (BDF) versus formoterol (F), secondary outcomes, Outcome 14 Withdrawals due to worsening COPD symptoms.
Discussion
Summary of main results
We have reviewed the data from 14 randomised trials (11,794 participants) assessing the effectiveness of combined inhaled corticosteroid and long‐acting beta2‐agonist (LABA/ICS) in the treatment of chronic obstructive pulmonary disease (COPD). The majority of trials contributing data to this review were in people with severe COPD, defined by a low FEV1 (< 50% predicted) and a history of exacerbations in the previous 12 months.
Primary outcomes
The exacerbation rates for participants on combined inhalers were reduced in comparison with the rates on LABAs alone, who had mean rates of around one to one and a half exacerbations per person per year. However, the size of the effect estimates varied across the studies. The odds of an exacerbation were lower with LABA/ICS, and all cause mortality was similar between treatment groups (Figure 5). There was no significant difference between LABA/ICS and LABA in hospitalisations, although there was considerable heterogeneity among the studies contributing data to this outcome (I2 = 74%). Differences in the eligibility criteria between the studies provide one possible explanation for this variation in average response to treatment. Anzueto 2009; Ferguson 2008; Kardos 2007; and TRISTAN stipulated a recent exacerbation as an inclusion criterion and may have recruited participants who were more at risk of future exacerbations than TORCH which did not recruit on the basis of exacerbation history.
Pneumonia occurred more often in people randomised to combined inhalers (OR 1.55; 95% CI 1.20 to 2.01) (Figure 6) with an annual risk in the one‐year studies of around 3% on LABAs alone compared to 4% on combination treatment. No significant differences were found between results of studies comparing FPS and salmeterol with those comparing BDF with formoterol for any of the primary outcomes.
Secondary outcomes
Combined treatment was more effective than LABAs alone in improving quality of life measured by the SGRQ and the CRQ, symptom score, pre‐dose and post‐dose FEV1 and rescue medication use. However, the mean differences in SGRQ and FEV1 between combination therapy and the LABAs were less than the minimal clinically significant difference for these outcomes. Withdrawals due to lack of efficacy and due to adverse events were significantly less with combined inhalers.
Overall completeness and applicability of evidence
The effect of exacerbations on health status measurements, the impact on daily activities and their value in predicting long‐term morbidity suggest that preventing or reducing exacerbations leads to improvements in quality of life (Osman 1997; Miravitlles 2004; Miravitlles 2007). The evidence summarized by this review indicates that there is an effect of combination therapy on exacerbations over that of long‐acting beta‐agonists, but the size of the effect is likely to be a source of ongoing debate.
There is some uncertainty as to what constitutes a minimal important difference for relative measures of exacerbations such as odds or rates. The minimal important difference for exacerbation rate has been suggested to be 22% (Cazzola 2008) and combined inhalers showed a 24% reduction. Cazzola 2008 also suggests that a reduction of one exacerbation per year could be considered a minimal important difference, greater than the difference we found with combined inhalers in this review. Taken together with evidence of the relationship between exacerbations and quality of life, there is a possible explanation for why the statistically significant difference we found for health status measurements with LABA/ICS did not exceed thresholds for minimal important differences on the questionnaires used.
Neither the mean effect of around 2.7 units (BDF) or 1.6 units (FPS) reached the designated 4 unit threshold of clinical importance. We are unable to fully interpret this without a comparison of the number of participants who achieved a clinically significant difference in SGRQ in each treatment group, as given in Karner 2012.
The most concerning side‐effect observed in this review was the increase in risk of pneumonia with combination therapy. TORCH represented 37% of the total weight in this analysis but did not use radiological findings to confirm the diagnosis of pneumonia. All the trials after TORCH included chest X‐ray as one diagnostic criterion for pneumonia. We did not find any significant differences in the risk of pneumonia between high and medium doses of FPS (500 µg fluticasone or 250 µg fluticasone) nor between FPS and BDF trials. The additional evidence from newer trials in this 2012 update, which used combined inhalers containing a lower dose of 250 µg fluticasone twice daily (Ferguson 2008; Anzueto 2009), still indicates a significant reduction in exacerbation rates and a (not significant) increase in the risk of pneumonia. We have, therefore, found no evidence of a dose‐related benefit or harm in the trials of FPS.
The data available for BDF comes from Calverley 2003; Tashkin 2008 and Rennard 2009, which did not find significant differences in the risk of pneumonia between the combined inhaler and formoterol. However, there was also no significant difference in the risk of pneumonia between the trials using FPS versus salmeterol and those comparing BDF to formoterol (test for subgroup differences: Chi² = 2.62, df = 1 (P = 0.11), I² = 61.8%) (Figure 6). Moreover, comparing the results from FPS and BDF trials may give misleading results as we cannot be sure that the ascertainment of pneumonia, severity of COPD, duration of treatment and other factors are comparable between the trials. Likewise, the data available for serious adverse events is limited by the absence of studies evaluating fluticasone and salmeterol, including the TORCH trial which dominated the results for pneumonia and mortality. The most reliable comparative evidence will have to come from trials randomising FPS and BDF within the same study. Others have also noted that evidence for any intra‐class differences in the risk of pneumonia between currently available formulations is inconclusive due to the absence of head‐to‐head trials (Singh 2010).
This review suggests that there may be a risk of pneumonia associated with the administration of inhaled steroid at the high doses that are licensed in combined inhalers for treating COPD. More work is needed on different doses of inhaled corticosteroids to assess the relative risks and benefits at lower doses. Also, the systematic, standardised collection of confirmed pneumonia cases in future studies will help to ascertain whether or not this risk is associated with additional morbidity leading to hospitalisation.
Quality of the evidence
The outcomes we included in the Table 1 relate to the exacerbation rate (low quality), number of people experiencing one or more exacerbations (moderate quality), hospitalisations (very low quality), pneumonia (moderate quality) and mortality (moderate quality). We downgraded the quality of evidence because of the risk of bias due to attrition in exacerbation rates and pneumonia, inconsistency in exacerbations rates and in hospitalisation, imprecision in mortality and hospitalisation, and for reporting bias in the number of people experiencing one or more exacerbations.
Although we rated the effect of LABA/ICS on exacerbations as low quality, the impact of inconsistency could be debated: it is the magnitude of the effect that varies across the studies and not the direction. Furthermore the effect was more consistent across the subgroup of studies comparing budesonide/formoterol with formoterol. The data on pneumonia are also subject to attrition, although evidence does point to a possible increase in the risk of pneumonia with inhaled steroids in people with COPD (Singh 2009). As noted above we remain uncertain as to whether this is a compound, class or dose effect in the absence of head‐to‐head comparisons. High or unbalanced withdrawal rates in long‐term trials in COPD introduce a possible threat to the validity of the trial findings. For this reason we downgraded the quality of evidence for the exacerbation and pneumonia findings of the review. For mortality most of the data were derived from TORCH and we are confident that ascertainment of vital status extended to patients who withdrew from this study as well as for those who completed it (see Table 1).
Potential biases in the review process
There is a well‐established debate over the definition and analysis of exacerbations which occur in clinical trials of therapies in COPD (Roisin 2000; Calverley 2005; Suissa 2006; Cazzola 2008). The statistical analysis of exacerbations as rate ratios and how exacerbations are defined at the study level bring into question some of the findings in this review. The large long‐term studies (that is those in excess of six months) which are adequately powered to detect statistically significant findings may overestimate the treatment effects of this therapy if they do not make allowances for duration of exposure to treatment, and within and between patient variability (Suissa 2006). The method of weighting counts of exacerbations as described by Suissa 2006 (using duration of person follow‐up time as a denominator in calculating the mean group rate of exacerbations rather than an unweighted approach) was undertaken in Calverley 2003; Szafranski 2003; and TORCH. The pooled analyses across the different interventions gave an effect size of 0.76 in the rate of exacerbations in favour of combination therapy. The relative reduction of 24% in the mean number of exacerbations may mean that for people who experience frequent loss of disease control, this will result in a potentially greater benefit than in patients who are relatively stable. It is noteworthy that in spite of the entry criteria stipulating a significant history of exacerbations, only 51% of participants from the salmeterol group in Kardos 2007 suffered one or more exacerbations after 48 weeks of treatment. Although the analysis of exacerbations as dichotomous data gave results which supported a protective effect of LABA/ICS, neither TORCH nor studies of budesonide and formoterol contribute to this outcome.
Agreements and disagreements with other studies or reviews
Both combination therapy and LABAs have been shown to reduce exacerbations compared with placebo, and therefore both represent effective treatments for people with COPD (Appleton 2006; Nannini 2007a). Whilst two related reviews have found a significant reduction in mortality when combined inhalers were compared to placebo or inhaled corticosteroids alone (Nannini 2007a; Nannini 2010), no significant difference was found in this review when combined inhalers were compared to LABAs alone.
Authors' conclusions
Implications for practice.
Our assessment of the quality of evidence for the primary outcomes has presented challenges to interpreting statistically significant results for exacerbations. Biases in the analysis and reporting of data bring the reliability of the observed reduction in exacerbations, measured as rates or as odds ratios, into question. The studies recruited people with moderate and severe COPD, many of whom had a history of exacerbations in the previous year. Combination therapy led to fewer exacerbations and withdrawals, and to better quality of life, lung function, symptom scores and less rescue medication use than with long‐acting beta2‐agonist treatment alone. Rates of side‐effects were similar between combination therapy and long‐acting beta2‐agonists with the exception of oral candidiasis, upper respiratory tract infection and, in particular, an increased risk of pneumonia seen with combination therapy. Clinicians and patients should weigh up the benefits of combination therapy in terms of reduction in COPD exacerbations against the risk it poses in terms of the possible increased risk of pneumonia.
Implications for research.
Reporting of exacerbations in future trials should be separated by type (that is requirement for medication and hospitalisation), allowing further exploration of how these drugs affect different severities of exacerbation and providing a clearer picture as to the resource implications of choosing either therapy. In the absence of a consensus regarding the choice of analysis of exacerbation rates, data reflecting both risk and rates should be available for evaluation.
Pneumonia requires careful monitoring in future trials. This may also help clarify to what extent the exacerbation rate is affected by an increase in the incidence of pneumonia. Further head‐to‐head trials making direct comparisons between the risks and benefits of FPS and BDF are needed, including at different doses.
Mortality should be ascertained in all randomised participants in future trials, not just for those who complete the trial.
Feedback
Analysis of exacerbations and quality of life, 23 March 2010
Summary
We read with interest the review by Nannini et al. (1), particularly because of its focus on desirable clinical endpoints in the management of COPD, which include exacerbations and quality of life. The authors of this review should be congratulated for their efforts in compiling the evidence on the efficacy and safety of the combination LABA and ICS as compared to LABA alone and for providing further information on the role of combination inhaled therapy in the management of COPD. Our concerns with this review lie in the analysis of exacerbations and the quality of life data from the included trials.
The TORCH trial (2) contributed the most weight to the exacerbation analysis. In this trial, 34 yo 44% of randomised participants withdrew from the study and only exacerbations for those who remained were counted. There was a differential rate of withdrawal between treatment arms in this trial and this has implications for how one might interpret any differences in exacerbations between treatments. It is clear from the reported data that people in each arm withdrew for different reasons. One cannot assume that the treatment arms were balanced when only participants that stayed in the trial were accounted for. As such, differences in rates of exacerbations between the groups cannot be attributed solely to differences in allocated treatment; rather the differences may be due to confounders.
In addition, the data used in the exacerbation meta‐analysis of this review does not allow readers to understand what the trials were measuring. The data used in Analysis 1.1 from each of the five trials appears to be the annualized rate per year of exacerbations. Further details on the types of exacerbations and incidence of exacerbations in studies are required to fully understand the impact of combination therapy on exacerbations. For example, it appears that data on the annual rate of all exacerbations in TRISTAN (3) was included in the analysis, whereas from TORCH, the annual rate of moderate/severe exacerbations was included.
More importantly, readers need to be alerted to the fact that annualized rates of exacerbations and subsequent reductions in these rates need to be interpreted with caution. In TORCH, the annual rate of exacerbations at baseline was approximately 1 per year for all patients. After treatment, LABA alone patients had a rate per year of 0.97 and LABA+ICS patients had a rate of 0.85. There are a number of concerns with this type of outcome measure. First, it is unclear what the clinical significance is of a statistical reduction of 0.12 exacerbations per year. Does this mean you would need to treat a patient for 8 years with LABA+ICS to prevent one additional exacerbation versus LABA alone? This review reports a 12% relative risk reduction in 'exacerbations' from TORCH. However, additional information is required to assess what the magnitude of the benefit is in absolute terms.
When one calculates a 'rate per year' of exacerbations, it is done by adding all the exacerbations that took place in a treatment arm and dividing by the number of years in the study. This method would count multiple exacerbations that occurred in a single patient. It is our opinion that the yearly rate of exacerbations cannot be interpreted in isolation. One needs to also know how many people had at least one exacerbation in each treatment arm at the end of the trial and then compare this measure between groups. In TORCH, which made up over 50% of the overall effect size, this information was not reported. The authors do report that annual rate of any exacerbation (in Table 4 of the TORCH trial) however these numbers are smaller than the reported annual rates of moderate to severe exacerbations. In our opinion, this obvious discrepancy in the reported numbers in TORCH needs to be clarified.
We are also concerned about the conclusion that there was a 'clinical benefit' with LABA+ICS versus LABA alone with respect to quality of life. Nannini et al report a St. George's Respiratory Questionnaire Score (SGRQ) improvement of ‐1.64 units; 95% CI ‐2.28 to ‐1, four studies, N = 4700. The minimum clinically important difference in SGRQ is thought to be a change of at least 4 points (4). In our opinion, readers of this review should be alerted to the fact that trials may have shown a statistical improvement in SGRQ with LABA+ICS but this difference may not be clinically perceptible.
In summary, we feel that although there may be statistical differences between LABA+ICS and LABA alone, there is insufficient evidence of a clinically important change in quality of life. As for exacerbations, more information needs to be provided within the review to adequately describe the data from included trials and only then can readers make sense of the information and form their own, well‐informed conclusions.
References
Nannini LJ, Cates CJ, Lasserson TJ, Poole P. Combined corticosteroid and long‐acting beta2‐agonist in one inhaler versus long‐acting beta agonist for chronic obstructive pulmonary disease. Cochrane Database of Systematic Reviews 2007;4:CD006829.
Calverley P, Anderson JA, Celli B, Ferguson GT, Jenkins C, Jones PW. Salmeterol and fluticasone propionate and survival in chronic obstructive pulmonary disease: TORCH. NEJM 2007;356:775‐89.
Calverley P, Pauwels R, Vestbo J, Jones P, Pride N, et al. Combined salmeterol and fluticasone in the treatment of chronic obstructive pulmonary disease: a randomised controlled trial. Lancet 2003;361:449‐56.
Jones PW. Health status measurement in chronic obstructive pulmonary disease. Thorax 2001;56:880?887.
Reply
We are grateful to Dr Tejani and Dr Bruchet for sending us their comments on the review. We have taken their feedback into account for this update of the review and have undertaken the following to enable readers to draw their own conclusions regarding the relative benefits, harms and quality of evidence for the studies that address our review question:
Included a Summary of Findings table to illustrate the risks of mortality and pneumonia with combination treatment and with long‐acting beta‐agonists.
Clarified that the analysis of exacerbation rates as ratios reflect an average per patient and provided additional estimates of baseline rates to indicate the size of effect with combination treatment in the Summary of Findings table.
Given further consideration to the size of effect of combination therapy on quality of life scores. We agree that the results obtained in our review do not reach the minimum clinically important difference in SGRQ scores, but note that small average differences may conceal important differences in the number in each treatment group that do achieve a minimally important improvement in quality of life.
Contributors
Aaron Tejani and Nicole Bruchet
What's new
| Date | Event | Description |
|---|---|---|
| 12 April 2013 | Amended | Funder acknowledgement added |
History
Protocol first published: Issue 3, 2002 Review first published: Issue 4, 2007
| Date | Event | Description |
|---|---|---|
| 24 April 2012 | New citation required and conclusions have changed | The addition of new evidence to this review has changed the results relating to pneumonia. |
| 11 November 2011 | New search has been performed | New literature search run. Four new included studies added. Two of these (Anzueto 2009; Ferguson 2008 total 1579 participants) added twice daily 250 µg fluticasone to salmeterol. The other two new studies (Rennard 2009; Tashkin 2008 total 2355 participants) added twice daily 160 µg or 320 µg budesonide to formoterol. |
| 23 March 2010 | Feedback has been incorporated | Comment from Aaron Tejani and Nicole Bruchet added |
| 22 July 2008 | Amended | Review converted to RevMan 5 |
| 22 January 2008 | Amended | Following the identification an error in the data analysis we have revised the odds ratio for pneumonia. The data were incorrectly entered in the formoterol arm from the study by Calverley 2003. This amendment changed the pooled OR for all studies from 1.62 (95% CI 1.35 to 1.94) to OR 1.58; (95% CI 1.32 to 1.88). This does not alter the conclusions of the review. |
| 22 August 2007 | New citation required and conclusions have changed | This review contains evidence from 5 studies previously included in a review of combination therapy in COPD (Nannini L, Cates CJ, Lasserson TJ, Poole P. Combined corticosteroid and long‐acting beta‐agonist in one inhaler for chronic obstructive pulmonary disease. Cochrane Database of Systematic Reviews 2004, Issue 3), with new data from four studies (Kardos 2007; O'Donnell 2006; SCO100470; TORCH). New findings There was no significant difference in the odds of death between combination therapy and LABA. Exacerbation rates are lower with combination therapy over LABA. Pneumonia was more frequent with combination therapy than with LABA. Additional work should focus on budesonide and formoterol, and the collection of confirmatory evidence on the frequency of pneumonia. |
Acknowledgements
The authors are indebted to the Hamamellis Trust who very generously funded the return travel to London for Dr Nannini to spend a week working on the development of the first review. Thanks to Liz Stovold, Susan Hansen, Karen Blackhall, Jo Picot and Sarah Tracy for technical and clerical support. We are grateful to Karla Soares‐Weiser who extracted data in duplicate for the 2011 update of this review and to Chris Cates for statistical advice and input with calculating SEMs for the included studies for use in GIV analyses.
We would also like to acknowledge the efforts of Inge Vestbo, Diane Grimley and Karen Richardson of GSK who helped us in our attempts to obtain unpublished information on TRISTAN, and of Goran Tornling, Moira Coughlan and Roger Metcalf of AstraZeneca in helping us obtain data for Szafranski 2003. We thank Dr Nick Hanania, Dr Peter Kardos and Prof Donald Mahler for corresponding with us in our attempts to obtain unpublished data from their studies, Dr Dal Negro (Dal Negro 2003) for confirming the SDs in his study and Dr Anzueto for confirming that Ferguson 2008 and Anzueto 2009 are replicate trials.
CRG Funding Acknowledgement: The National Institute for Health Research (NIHR) is the largest single funder of the Cochrane Airways Group.
Disclaimer: The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the NIHR, NHS or the Department of Health.
Appendices
Appendix 1. Sources and search methods for the Cochrane Airways Group Specialised Register (CAGR)
Electronic searches: core databases
| Database | Frequency of search |
| MEDLINE (Ovid) | Weekly |
| EMBASE (Ovid) | Weekly |
| CENTRAL (the Cochrane Library) | Quarterly |
| PsycINFO (Ovid) | Monthly |
| CINAHL (EBSCO) | Monthly |
| AMED (EBSCO) | Monthly |
Handsearches: core respiratory conference abstracts
| Conference | Years searched |
| American Academy of Allergy, Asthma and Immunology (AAAAI) | 2001 onwards |
| American Thoracic Society (ATS) | 2001 onwards |
| Asia Pacific Society of Respirology (APSR) | 2004 onwards |
| British Thoracic Society Winter Meeting (BTS) | 2000 onwards |
| Chest Meeting | 2003 onwards |
| European Respiratory Society (ERS) | 1992, 1994, 2000 onwards |
| International Primary Care Respiratory Group Congress (IPCRG) | 2002 onwards |
| Thoracic Society of Australia and New Zealand (TSANZ) | 1999 onwards |
MEDLINE search strategy used to identify trials for the CAGR
COPD search
1. Lung Diseases, Obstructive/
2. exp Pulmonary Disease, Chronic Obstructive/
3. emphysema$.mp.
4. (chronic$ adj3 bronchiti$).mp.
5. (obstruct$ adj3 (pulmonary or lung$ or airway$ or airflow$ or bronch$ or respirat$)).mp.
6. COPD.mp.
7. COAD.mp.
8. COBD.mp.
9. AECB.mp.
10. or/1‐9
Filter to identify RCTs
1. exp "clinical trial [publication type]"/
2. (randomised or randomised).ab,ti.
3. placebo.ab,ti.
4. dt.fs.
5. randomly.ab,ti.
6. trial.ab,ti.
7. groups.ab,ti.
8. or/1‐7
9. Animals/
10. Humans/
11. 9 not (9 and 10)
12. 8 not 11
The MEDLINE strategy and RCT filter are adapted to identify trials in other electronic databases
Appendix 2. Calculation of exacerbation rate ratios
For the 2012 update the rate ratios have been analysed using the Generic Inverse Variance method. Two new trials included three arms (Rennard 2009; Tashkin 2008) and the exacerbation rates from the two active arms were combined as a mean of the rate in each arm. The standard errors were calculated by using a P value of 0.001 in Rennard 2009 and a P value of 0.12 in Tashkin 2008. For the hospitalisation rates data were made available by Kardos 2007 on the number of patients with one or more exacerbations in each arm, and these were converted into a risk ratio which was combined with the rate ratios from the other studies.
Data and analyses
Comparison 1. Combined inhalers versus long‐acting beta‐agonists (primary outcomes).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Exacerbation rates (combined inhaler versus LABA alone) | 9 | Rate ratio (Random, 95% CI) | 0.76 [0.68, 0.84] | |
| 1.1 Fluticasone/salmeterol | 5 | Rate ratio (Random, 95% CI) | 0.77 [0.66, 0.89] | |
| 1.2 Budesonide/formoterol | 4 | Rate ratio (Random, 95% CI) | 0.73 [0.64, 0.83] | |
| 2 Number of participants with one or more exacerbation | 6 | 3357 | Odds Ratio (M‐H, Random, 95% CI) | 0.83 [0.70, 0.98] |
| 2.1 Fluticasone/salmeterol | 6 | 3357 | Odds Ratio (M‐H, Random, 95% CI) | 0.83 [0.70, 0.98] |
| 2.2 Budesonide/formoterol | 0 | 0 | Odds Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3 Hospitalisations | 3 | Rate ratio (Random, 95% CI) | Subtotals only | |
| 3.1 Fluticasone/salmeterol | 3 | Rate ratio (Random, 95% CI) | 0.79 [0.55, 1.13] | |
| 4 Mortality | 10 | 10681 | Odds Ratio (M‐H, Random, 95% CI) | 0.92 [0.76, 1.11] |
| 4.1 Fluticasone/salmeterol | 6 | 7408 | Odds Ratio (M‐H, Random, 95% CI) | 0.93 [0.76, 1.13] |
| 4.2 Budesonide/formoterol | 4 | 3273 | Odds Ratio (M‐H, Random, 95% CI) | 1.03 [0.40, 2.67] |
| 5 Pneumonia | 12 | 11076 | Odds Ratio (M‐H, Random, 95% CI) | 1.55 [1.20, 2.01] |
| 5.1 Fluticasone/salmeterol | 9 | 8242 | Odds Ratio (M‐H, Random, 95% CI) | 1.75 [1.25, 2.45] |
| 5.2 Budesonide/formoterol | 3 | 2834 | Odds Ratio (M‐H, Random, 95% CI) | 1.09 [0.69, 1.73] |
| 6 Pneumonia subgrouped by dose | 12 | 11076 | Odds Ratio (M‐H, Random, 95% CI) | 1.56 [1.26, 1.93] |
| 6.1 Lower dose FPS (250/50 bid) | 3 | 1934 | Odds Ratio (M‐H, Random, 95% CI) | 2.19 [1.35, 3.53] |
| 6.2 Higher dose FPS (500/50 bid) | 6 | 6308 | Odds Ratio (M‐H, Random, 95% CI) | 1.55 [0.92, 2.61] |
| 6.3 Lower dose BDF (160/9 bid) | 2 | 1164 | Odds Ratio (M‐H, Random, 95% CI) | 1.10 [0.53, 2.26] |
| 6.4 Higher dose BDF (320/9 bid) | 3 | 1670 | Odds Ratio (M‐H, Random, 95% CI) | 1.08 [0.60, 1.97] |
1.1. Analysis.
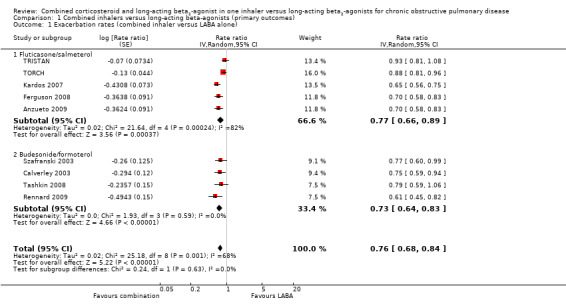
Comparison 1 Combined inhalers versus long‐acting beta‐agonists (primary outcomes), Outcome 1 Exacerbation rates (combined inhaler versus LABA alone).
1.5. Analysis.
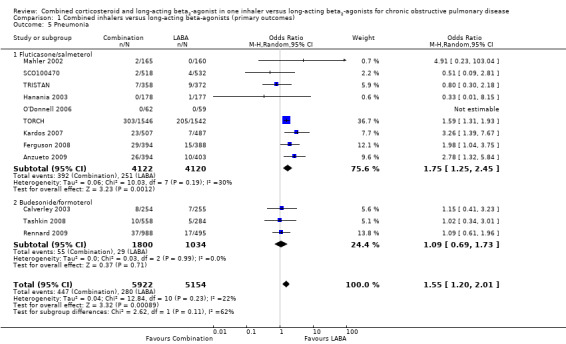
Comparison 1 Combined inhalers versus long‐acting beta‐agonists (primary outcomes), Outcome 5 Pneumonia.
Comparison 2. Fluticasone and salmeterol (FPS) versus salmeterol (SAL), secondary outcomes.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Exacerbations by type | 5 | Rate ratio (Random, 95% CI) | Subtotals only | |
| 1.1 Requirement for oral steroids | 4 | Rate ratio (Random, 95% CI) | 0.71 [0.62, 0.81] | |
| 1.2 Hospitalisation | 3 | Rate ratio (Random, 95% CI) | 0.79 [0.55, 1.13] | |
| 2 Mortality by duration | 6 | 7408 | Odds Ratio (M‐H, Random, 95% CI) | 0.93 [0.76, 1.13] |
| 2.1 Mortality: three year data | 1 | 3054 | Odds Ratio (M‐H, Random, 95% CI) | 0.92 [0.75, 1.14] |
| 2.2 Mortality: >one and <three year data | 0 | 0 | Odds Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 2.3 Mortality: one year data | 5 | 4354 | Odds Ratio (M‐H, Random, 95% CI) | 0.94 [0.51, 1.70] |
| 2.4 Mortality: 6 month data | 0 | 0 | Odds Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3 Change from baseline in St George's Respiratory Questionnaire (total score) | 6 | SGRQ units (Random, 95% CI) | ‐1.58 [‐2.15, ‐1.01] | |
| 4 Change from baseline in St George's Respiratory Questionnaire (domain ‐ symptoms) | 4 | St George's RQ score (Random, 95% CI) | ‐2.78 [‐3.88, ‐1.68] | |
| 5 Change from baseline in St George's Respiratory Questionnaire (domain ‐ activity) | 4 | SGRQ units (Random, 95% CI) | ‐1.31 [‐2.38, ‐0.24] | |
| 6 Change from baseline in St George's Respiratory Questionnaire (domain ‐ impact) | 4 | SGRQ units (Random, 95% CI) | ‐1.41 [‐2.33, ‐0.50] | |
| 7 End of treatment St George's Respiratory Questionnaire scores (total score) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 8 End of treatment St George's Respiratory Questionnaire scores (domain ‐ symptoms) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 9 Change from baseline in Chronic Respiratory Disease Questionnaire scores | 2 | 680 | Mean Difference (IV, Random, 95% CI) | 2.83 [0.25, 5.41] |
| 10 End of treatment Transitional dyspnea index (TDI) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 11 End of treatment symptom scores | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 12 Change from baseline in Transitional Dyspnoea Index (TDI) | 2 | 677 | Mean Difference (IV, Random, 95% CI) | 0.61 [‐0.47, 1.68] |
| 13 Change in MRC rated dyspnoea | 1 | symptoms (Random, 95% CI) | Totals not selected | |
| 14 Change from baseline in dyspnoea score | 2 | Mean Difference (Random, 95% CI) | ‐0.09 [‐0.13, ‐0.05] | |
| 15 Mean Change nighttime awakenings | 2 | 1554 | Mean Difference (IV, Random, 95% CI) | ‐1.34 [‐2.07, ‐0.61] |
| 16 Change from baseline in predose FEV1 | 5 | Litres (Random, 95% CI) | 0.07 [0.05, 0.10] | |
| 16.1 Reversible population | 3 | Litres (Random, 95% CI) | 0.07 [0.02, 0.12] | |
| 16.2 Partially reversible population (mixed population) | 2 | Litres (Random, 95% CI) | 0.08 [0.04, 0.12] | |
| 16.3 Poorly reversible population | 2 | Litres (Random, 95% CI) | 0.06 [0.01, 0.12] | |
| 17 Change from baseline in postdose FEV1 | 3 | Litres (Random, 95% CI) | 0.05 [0.03, 0.06] | |
| 18 End of treatment FEV1 (Litres) | 2 | 1780 | Mean Difference (IV, Random, 95% CI) | 0.01 [‐0.02, 0.03] |
| 19 FEV1 (% predicted ‐ absolute scores) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 20 Change from baseline in am PEF (L/min) | 2 | L/min (Random, 95% CI) | 11.61 [7.91, 15.30] | |
| 21 Change from baseline in rescue medication usage (puffs/day) | 4 | 2435 | Mean Difference (IV, Random, 95% CI) | ‐0.38 [‐0.61, ‐0.16] |
| 22 End of treatment rescue medication usage (puffs/day) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 23 Adverse events ‐ any event | 9 | 8250 | Odds Ratio (M‐H, Random, 95% CI) | 1.05 [0.93, 1.19] |
| 24 Adverse events ‐ candidiasis | 6 | 3118 | Odds Ratio (M‐H, Random, 95% CI) | 3.75 [2.33, 6.04] |
| 25 Adverse events ‐ pneumonia | 9 | 8242 | Odds Ratio (M‐H, Random, 95% CI) | 1.75 [1.25, 2.45] |
| 26 Adverse events ‐ headache | 8 | 7237 | Odds Ratio (M‐H, Random, 95% CI) | 1.06 [0.90, 1.26] |
| 27 Adverse events ‐ upper respiratory tract infection | 7 | 6198 | Odds Ratio (M‐H, Random, 95% CI) | 1.32 [1.12, 1.55] |
| 28 Withdrawals | 9 | 8226 | Odds Ratio (M‐H, Random, 95% CI) | 0.85 [0.77, 0.94] |
| 29 Withdrawals due to lack of efficacy | 6 | 6739 | Odds Ratio (M‐H, Random, 95% CI) | 0.53 [0.39, 0.72] |
| 30 Withdrawals due to adverse events | 8 | 7895 | Odds Ratio (M‐H, Random, 95% CI) | 0.90 [0.79, 1.02] |
2.1. Analysis.
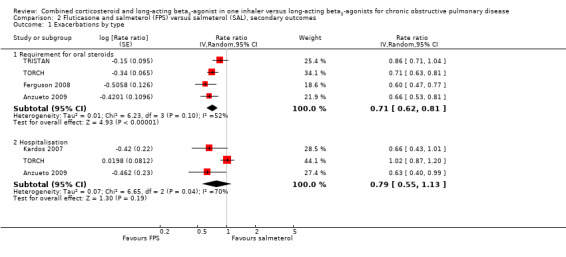
Comparison 2 Fluticasone and salmeterol (FPS) versus salmeterol (SAL), secondary outcomes, Outcome 1 Exacerbations by type.
2.2. Analysis.
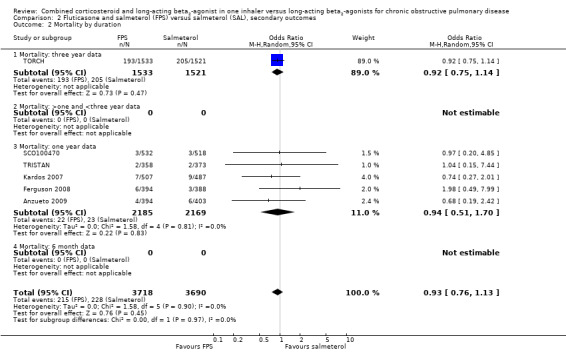
Comparison 2 Fluticasone and salmeterol (FPS) versus salmeterol (SAL), secondary outcomes, Outcome 2 Mortality by duration.
2.4. Analysis.
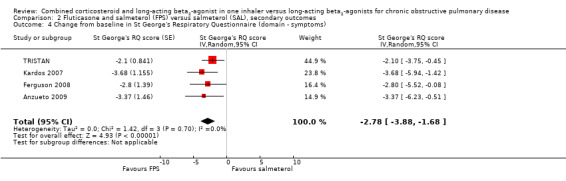
Comparison 2 Fluticasone and salmeterol (FPS) versus salmeterol (SAL), secondary outcomes, Outcome 4 Change from baseline in St George's Respiratory Questionnaire (domain ‐ symptoms).
2.5. Analysis.
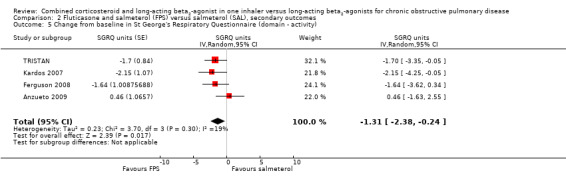
Comparison 2 Fluticasone and salmeterol (FPS) versus salmeterol (SAL), secondary outcomes, Outcome 5 Change from baseline in St George's Respiratory Questionnaire (domain ‐ activity).
2.6. Analysis.
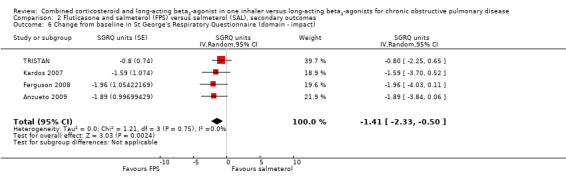
Comparison 2 Fluticasone and salmeterol (FPS) versus salmeterol (SAL), secondary outcomes, Outcome 6 Change from baseline in St George's Respiratory Questionnaire (domain ‐ impact).
2.7. Analysis.
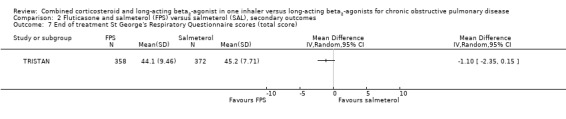
Comparison 2 Fluticasone and salmeterol (FPS) versus salmeterol (SAL), secondary outcomes, Outcome 7 End of treatment St George's Respiratory Questionnaire scores (total score).
2.8. Analysis.
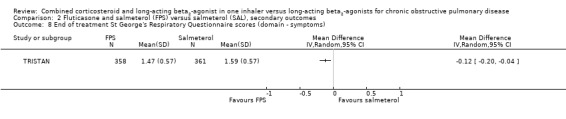
Comparison 2 Fluticasone and salmeterol (FPS) versus salmeterol (SAL), secondary outcomes, Outcome 8 End of treatment St George's Respiratory Questionnaire scores (domain ‐ symptoms).
2.9. Analysis.
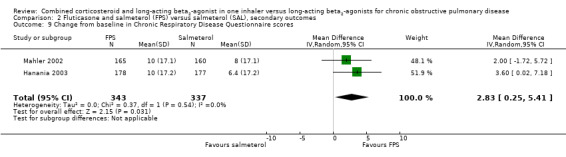
Comparison 2 Fluticasone and salmeterol (FPS) versus salmeterol (SAL), secondary outcomes, Outcome 9 Change from baseline in Chronic Respiratory Disease Questionnaire scores.
2.10. Analysis.
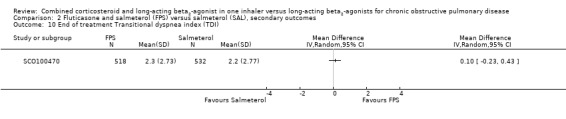
Comparison 2 Fluticasone and salmeterol (FPS) versus salmeterol (SAL), secondary outcomes, Outcome 10 End of treatment Transitional dyspnea index (TDI).
2.11. Analysis.
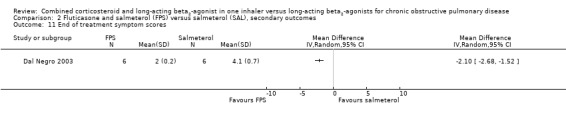
Comparison 2 Fluticasone and salmeterol (FPS) versus salmeterol (SAL), secondary outcomes, Outcome 11 End of treatment symptom scores.
2.12. Analysis.
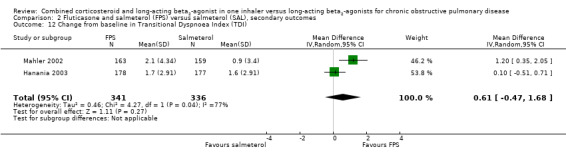
Comparison 2 Fluticasone and salmeterol (FPS) versus salmeterol (SAL), secondary outcomes, Outcome 12 Change from baseline in Transitional Dyspnoea Index (TDI).
2.13. Analysis.
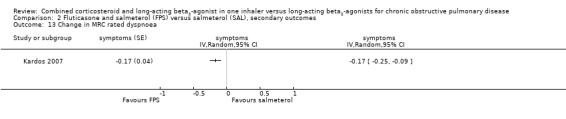
Comparison 2 Fluticasone and salmeterol (FPS) versus salmeterol (SAL), secondary outcomes, Outcome 13 Change in MRC rated dyspnoea.
2.20. Analysis.

Comparison 2 Fluticasone and salmeterol (FPS) versus salmeterol (SAL), secondary outcomes, Outcome 20 Change from baseline in am PEF (L/min).
2.22. Analysis.
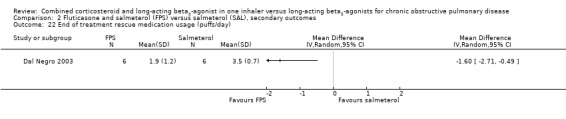
Comparison 2 Fluticasone and salmeterol (FPS) versus salmeterol (SAL), secondary outcomes, Outcome 22 End of treatment rescue medication usage (puffs/day).
2.23. Analysis.
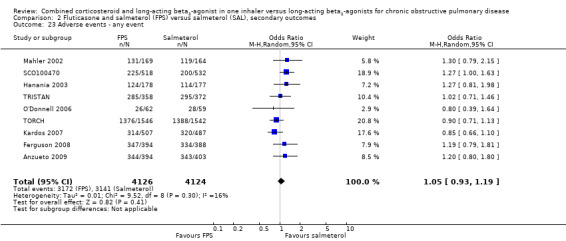
Comparison 2 Fluticasone and salmeterol (FPS) versus salmeterol (SAL), secondary outcomes, Outcome 23 Adverse events ‐ any event.
2.25. Analysis.

Comparison 2 Fluticasone and salmeterol (FPS) versus salmeterol (SAL), secondary outcomes, Outcome 25 Adverse events ‐ pneumonia.
Comparison 3. Budesonide and formoterol (BDF) versus formoterol (F), secondary outcomes.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Quality of life ‐ SGRQ (change scores) | 4 | 3442 | SGRQ (Random, 95% CI) | ‐2.69 [‐3.82, ‐1.55] |
| 2 Quality of life ‐ SGRQ (change scores) | 4 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 3 Change from baseline in St George's Respiratory Questionnaire (domain ‐ symptoms) | 2 | 2233 | Mean Difference (IV, Random, 95% CI) | ‐3.43 [‐5.13, ‐1.72] |
| 4 Change from baseline in St George's Respiratory Questionnaire (domain ‐ activity) | 2 | 2215 | Mean Difference (IV, Random, 95% CI) | ‐1.57 [‐2.87, ‐0.27] |
| 5 Change from baseline in St George's Respiratory Questionnaire (domain ‐ impact) | 2 | 2222 | Mean Difference (IV, Random, 95% CI) | ‐2.28 [‐3.60, ‐0.95] |
| 6 Mean FEV1 (% increase from baseline) | 1 | Std. Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 7 Mean change from baseline in pre dose FEV1 to the average over the randomised treatment period. | 2 | 1203 | Mean Difference (IV, Random, 95% CI) | 0.05 [0.00, 0.09] |
| 8 Symptoms ‐ breathlessness (change scores) | 2 | 2308 | Mean Difference (IV, Random, 95% CI) | ‐0.07 [‐0.12, ‐0.01] |
| 9 Change from baseline in cough score | 2 | 2308 | Mean Difference (IV, Random, 95% CI) | ‐0.06 [‐0.11, ‐0.00] |
| 10 Change from baseline in rescue medication usage (puffs/day) | 2 | 2310 | Mean Difference (IV, Random, 95% CI) | ‐0.33 [‐0.57, ‐0.09] |
| 11 Adverse events ‐ 'serious' events | 4 | 3243 | Odds Ratio (M‐H, Random, 95% CI) | 0.92 [0.69, 1.25] |
| 12 Adverse events ‐ candidiasis | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 13 Withdrawals due to adverse events | 4 | 3243 | Odds Ratio (M‐H, Random, 95% CI) | 0.88 [0.65, 1.19] |
| 14 Withdrawals due to worsening COPD symptoms | 2 | 918 | Odds Ratio (M‐H, Random, 95% CI) | 0.49 [0.32, 0.74] |
3.2. Analysis.

Comparison 3 Budesonide and formoterol (BDF) versus formoterol (F), secondary outcomes, Outcome 2 Quality of life ‐ SGRQ (change scores).
3.3. Analysis.

Comparison 3 Budesonide and formoterol (BDF) versus formoterol (F), secondary outcomes, Outcome 3 Change from baseline in St George's Respiratory Questionnaire (domain ‐ symptoms).
3.4. Analysis.
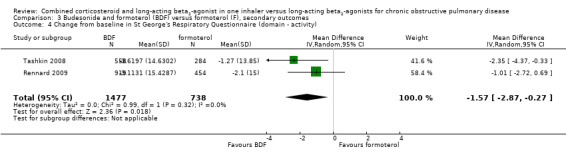
Comparison 3 Budesonide and formoterol (BDF) versus formoterol (F), secondary outcomes, Outcome 4 Change from baseline in St George's Respiratory Questionnaire (domain ‐ activity).
3.5. Analysis.
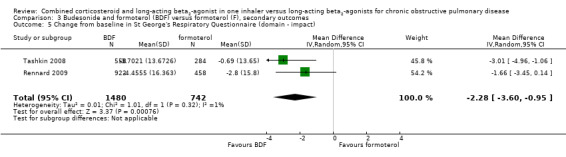
Comparison 3 Budesonide and formoterol (BDF) versus formoterol (F), secondary outcomes, Outcome 5 Change from baseline in St George's Respiratory Questionnaire (domain ‐ impact).
3.6. Analysis.
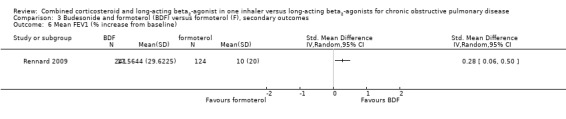
Comparison 3 Budesonide and formoterol (BDF) versus formoterol (F), secondary outcomes, Outcome 6 Mean FEV1 (% increase from baseline).
3.7. Analysis.
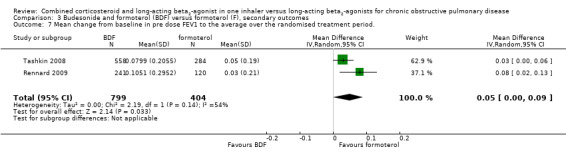
Comparison 3 Budesonide and formoterol (BDF) versus formoterol (F), secondary outcomes, Outcome 7 Mean change from baseline in pre dose FEV1 to the average over the randomised treatment period..
3.9. Analysis.
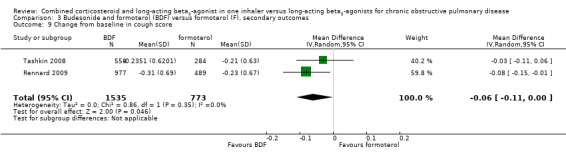
Comparison 3 Budesonide and formoterol (BDF) versus formoterol (F), secondary outcomes, Outcome 9 Change from baseline in cough score.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Anzueto 2009.
| Methods | Randomised, double‐blind parallel study. Duration: 52 weeks. | |
| Participants |
|
|
| Interventions | Run‐in 4 weeks with open‐label fluticasone/salmeterol 250/50 Diskus BID. Following run‐in, subjects were randomised to FPS 250/50 or salmeterol twice‐daily via DISKUS for 52 weeks. Clinic visits were conducted at screening, day 1 (randomisation), and after 4, 8, 12, 20, 28, 36, 44, and 52 weeks. Treatments were assigned in blocks using a centre‐based randomisation schedule. Assignment to blinded study medication was stratified based on subjects' FEV1 response to albuterol at screen visit. Oral corticosteroids and antibiotics were allowed for the acute treatment of a COPD exacerbation. | |
| Outcomes | The primary efficacy endpoint was the annual rate of moderate/severe exacerbations. Secondary endpoints were the time to first moderate/severe exacerbation, the annual rate of exacerbations requiring oral corticosteroids, and pre‐dose FEV1. Related endpoints included the annual rate of all exacerbations, time to onset of each moderate/severe exacerbation, and diary records of dyspnoea scores, nighttime awakenings due to COPD, and use of supplemental albuterol. | |
| Notes | The run‐in with combined therapy precluded exacerbations due to ICS withdrawal. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Assigned in block centre‐based randomisation schedule and stratified according to salbutamol response in FEV1 at baseline. |
| Allocation concealment (selection bias) | Unclear risk | Information not available. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Identical Diskus inhaler device. |
| Incomplete outcome data (attrition bias) Mortality | High risk | 39% discontinued on salmeterol and 32% on FPS. |
| Incomplete outcome data (attrition bias) All other outcomes | High risk | 39% discontinued on salmeterol and 32% on FPS. |
| Selective reporting (reporting bias) | Low risk | Contributes data to all primary outcomes. |
Calverley 2003.
| Methods | Parallel group study. Randomisation: unclear. Blinding: double‐blind (identical inhaler devices). Trial duration: 12 months with two week run‐in of treatment optimisation. Allocation concealment: unclear. Withdrawals: stated. Intention‐to‐treat analysis: stated. | |
| Participants |
|
|
| Interventions | Run‐in phase: All participants received 30 mg oral prednisolone BiD and 2 x 4.5 mcg formoterol BiD (2 weeks).
Additional treatment groups not covered in this review:
Inhaler device: Turbuhaler. |
|
| Outcomes | Time to first exacerbation; change in post‐medication FEV1; number of exacerbations; time to and number of OCS‐treated episodes; am and pm PEF, slow VC, HRQL, symptoms, use of reliever medication, AEs. | |
| Notes | Classified as 'poorly reversible population'. P values used to calculate pooled SEMs for the following outcomes: Health related quality of life; FEV1; rescue medication. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as randomised; no other information reported. |
| Allocation concealment (selection bias) | Unclear risk | Information not available. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Identical inhaler devices. |
| Incomplete outcome data (attrition bias) Mortality | High risk | 44% withdrew on formoterol and 29% on BDF. |
| Incomplete outcome data (attrition bias) All other outcomes | High risk | 44% withdrew on formoterol and 29% on BDF. |
| Selective reporting (reporting bias) | High risk | No data available for analysis of exacerbations as dichotomous data or hospitalisations. |
Dal Negro 2003.
| Methods | Parallel group study. Trial duration: 12 months. Baseline characteristics: comparable. Intention‐to‐treat analysis: Yes. | |
| Participants |
|
|
| Interventions | Run‐in: 2 weeks treatment with theophylline and prn SABA.
Additional treatment groups not covered in this review
Participants were on concomitant therapy: SABA prn and theophylline 400ug/day, for 12 months. Inhaler device: Diskus. |
|
| Outcomes | FEV1, Delta FEV1, PEF am, symptom scores, rescue medication use, exacerbations (event rate and mean number per year). | |
| Notes | Classified as 'poorly reversible population'. Mild exacerbation: requirement for increase in SABA prn by >2 occasions/24hrs on two or more consecutive days compared with baseline mean of last seven days of run‐in Moderate exacerbation: condition requiring treatment with antibiotics and/or oral corticosteroids Severe exacerbation: Condition requiring emergency hospital treatment and/or hospitalisation. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as randomised; no other information reported. |
| Allocation concealment (selection bias) | Unclear risk | Information not available. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Identical inhaler devices. |
| Incomplete outcome data (attrition bias) Mortality | Unclear risk | Only 12 participants. |
| Incomplete outcome data (attrition bias) All other outcomes | Unclear risk | Only 12 participants. |
| Selective reporting (reporting bias) | High risk | No data available for the primary outcomes. |
Ferguson 2008.
| Methods | Randomised, double‐blind, parallel group study (study code SCO40043). Treatments were assigned in blocks using a centre based randomisation schedule. Patients were stratified based on FEV1 response to albuterol at screening. Study duration: 52 weeks. | |
| Participants |
|
|
| Interventions | 4‐week run‐in period with open‐label FPS 250/50 via DISKUS. FPS 250/50 or salmeterol 50 mg twice daily via DISKUS for 12 months. As‐needed albuterol was provided for use throughout the study. Nine visits after screening. | |
| Outcomes | Annual rate of moderate to severe exacerbations. Time to first moderate to severe exacerbation, the annual rate of exacerbations requiring oral corticosteroids, and pre‐dose FEV1. Related endpoints were the annual rate of all exacerbations (mild and moderate to severe), duration of moderate to severe exacerbations, time to onset of each moderate to severe exacerbation. Exacerbation recovery time (determined by length of oral corticosteroid and antibiotic courses and hospital stays) and diary records of dyspnoea, nighttime awakenings due to COPD, and use of supplemental albuterol. | |
| Notes | In 782 patients with COPD (mean FEV1 Z0.94 0.36 L, 33% predicted normal), treatment with fluticasone propionate/salmeterol 250/50 significantly reduced (1) the annual rate of moderate to severe exacerbations by 30.5% compared with salmeterol (1.06 and 1.53 per subject per year, respectively, P < 0.001. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Treatments were assigned in blocks using a centre based randomisation schedule. Patients were stratified based on FEV1 response to albuterol at screening. |
| Allocation concealment (selection bias) | Unclear risk | No details in the paper. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Following run in, patients were randomised to FPS 250/50 or salmeterol 50 mg (Serevent; GlaxoSmithKline) twice daily via DISKUS. |
| Incomplete outcome data (attrition bias) Mortality | High risk | 38% discontinued on salmeterol and 30% on FPS. |
| Incomplete outcome data (attrition bias) All other outcomes | High risk | 38% discontinued on salmeterol and 30% on FPS. |
| Selective reporting (reporting bias) | High risk | Does not contribute data to the analysis of hospitalisations. |
Hanania 2003.
| Methods | Parallel group study. Trial duration: 24 weeks with 2‐week run‐in period. Baseline characteristics: comparable. Intention‐to‐treat analysis: not stated. | |
| Participants |
|
|
| Interventions | Run‐in: 2 weeks treatment with placebo inhaler and prn SABA.
Additional treatment groups not covered in this review
Inhaler device: Diskus. |
|
| Outcomes | Lung function: Change in FEV1 from baseline to end of study (M). PEF data not stratified by reversibility. Quality of life: CRDQ, CBSQ not stratified by reversibility. Dyspnoea and symptoms: Transitional Dyspnoea index, Baseline dyspnoea index not stratified by reversibility. Exacerbations. Rescue salbutamol use. | |
| Notes | FEV1 reversibility < 12% or 200 mL (of baseline FEV1) Reversibility stratified data. Mean % increase non‐reversible patients = 8.8. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as randomised; other information not available. |
| Allocation concealment (selection bias) | Unclear risk | Information not available. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Identical inhaler devices. |
| Incomplete outcome data (attrition bias) Mortality | High risk | 32% withdrew on salmeterol and 30% on FPS. |
| Incomplete outcome data (attrition bias) All other outcomes | High risk | 32% withdrew on salmeterol and 30% on FPS. |
| Selective reporting (reporting bias) | High risk | Does not contribute data to analysis of exacerbations as rate ratios, mortality or hospitalisation. |
Kardos 2007.
| Methods | Randomised controlled trial, parallel group design. Trial duration: 52 weeks (4 week run‐in). Baseline characteristics: comparable. Intention‐to‐treat analysis stated. | |
| Participants |
|
|
| Interventions | Run‐in: 4 weeks on stable medication.
Inhaler device: DPI. |
|
| Outcomes | Withdrawals; Exacerbations (defined according to Rodriguez Roisin Chest article at stages II and III); FEV1; Rescue medication; symptoms; SGRQ; adverse events. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer generated randomisation schedule. |
| Allocation concealment (selection bias) | Low risk | Centrally generated schedule. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Identicial inhaler devices. |
| Incomplete outcome data (attrition bias) Mortality | High risk | 21% discontinued on salmeterol and 20% on FPS. |
| Incomplete outcome data (attrition bias) All other outcomes | High risk | 21% discontinued on salmeterol and 20% on FPS. |
| Selective reporting (reporting bias) | Low risk | Contributes data to all primary outcomes. |
Mahler 2002.
| Methods | Randomised controlled parallel group study. Trial duration: 24 weeks. Baseline characteristics: comparable. Intention‐to‐treat analysis: stated. | |
| Participants |
|
|
| Interventions | Run‐in: 2 weeks treatment with placebo inhaler and prn SABA.
Additional treatment groups not covered in this review
Inhaler device: Diskus. |
|
| Outcomes | Lung function: Change in FEV1 from baseline to end of study (M). Quality of life: CRDQ, CBSQ not stratified by reversibility. Dyspnoea and symptoms: End of study dyspnoea (TDI). Exacerbations. Rescue salbutamol use. | |
| Notes | COPD subjects reversible and non‐reversible, < 15% (baseline) improvement in FEV1 to salbutamol. Reversibility stratified data. Mean FEV1 reversibility 11.0%. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as randomised; stratified by reversibility and investigative site. |
| Allocation concealment (selection bias) | Unclear risk | Information not available. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Identical inhaler devices. |
| Incomplete outcome data (attrition bias) Mortality | High risk | 28% withdrew on salmeterol and 32% on FPS. |
| Incomplete outcome data (attrition bias) All other outcomes | High risk | 28% withdrew on salmeterol and 32% on FPS. |
| Selective reporting (reporting bias) | High risk | Does not contribute data to analysis of exacerbations as rate ratios, mortality or hospitalisations. |
O'Donnell 2006.
| Methods | Randomised controlled parallel group design. Trial duration: 8 weeks. Baseline characteristics: comparable. Intention‐to‐treat analysis stated. | |
| Participants |
|
|
| Interventions | Run‐in: 2 weeks, single‐blind placebo.
Additional treatment groups not covered in this review
Inhaler device: DPI. |
|
| Outcomes | Withdrawals; exercise time; FEV1; adverse events. | |
| Notes | Study downloaded from ctr.gsk.co.uk | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as randomised; other information not available. |
| Allocation concealment (selection bias) | Unclear risk | Information not available. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Identical inhaler devices. |
| Incomplete outcome data (attrition bias) Mortality | Low risk | One patient withdrew on salmeterol and three on FPS. |
| Incomplete outcome data (attrition bias) All other outcomes | Low risk | One patient withdrew on salmeterol and three on FPS. |
| Selective reporting (reporting bias) | High risk | Does not contribute data to the analysis of exacerbations as rate ratios, mortality or hospitalisations. |
Rennard 2009.
| Methods | Randomised, double‐blind, double dummy, parallel group, active and placebo‐controlled, multi‐centre trial. | |
| Participants |
|
|
| Interventions | 4 weeks run‐in where ICS was permitted. Four groups: Budesonide/formoterol HFA pressurized metered‐dose inhaler 160/4.5 BID. Budesonide/formoterol HFA pressurized metered‐dose inhaler 80/4.5 BID. Formoterol dry powder inhaler 4.5 BID. Placebo. Duration: 12 months. | |
| Outcomes | 1) pre‐dose and 1‐hour post‐dose FEV1 2) FVC measured at all clinic visits, and 3) morning and evening peak expiratory flow(PEF) recorded daily. 4)exacerbations. 5) dyspnoea. 6) health status. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation in one of four treatments. |
| Allocation concealment (selection bias) | Unclear risk | Information not available. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Patients received both a pressurized metered‐dose inhaler (pMDI) and a dry powder inhaler (DPI) containing either active treatment or double‐dummy placebo as appropriate. |
| Incomplete outcome data (attrition bias) Mortality | High risk | 32% discontinued on formoterol and 28% on BDF. |
| Incomplete outcome data (attrition bias) All other outcomes | High risk | 32% discontinued on formoterol and 28% on BDF. |
| Selective reporting (reporting bias) | High risk | Does not contribute data to analysis of exacerbations as dichotomous data or hospitalisations. |
SCO100470.
| Methods | RCT, parallel group design. Trial duration: 24 weeks. Baseline characteristics: comparable. Intention‐to‐treat analysis: stated. | |
| Participants |
|
|
| Interventions |
Inhaler device: DPI. |
|
| Outcomes | Withdrawals; FEV1; morning PEF; quality of life (SQRG); symptoms (TDI) adverse events. | |
| Notes | Unpublished study downloaded from ctr.gsk.co.uk | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as randomised; other information not available. |
| Allocation concealment (selection bias) | Unclear risk | Information not available. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Identical inhaler devices. |
| Incomplete outcome data (attrition bias) Mortality | Unclear risk | 14% withdrew on salmeterol and 11% on FPS. |
| Incomplete outcome data (attrition bias) All other outcomes | Unclear risk | 14% withdrew on salmeterol and 11% on FPS. |
| Selective reporting (reporting bias) | High risk | Does not contribute data to analysis of exacerbations as rate ratio, dichotomous data or hospitalisations. |
Szafranski 2003.
| Methods | Parallel group study. Intention‐to‐treat analysis: stated. | |
| Participants |
|
|
| Interventions | Run‐in: 2 weeks. Treatment with prn SABA only.
Additional treatment groups not covered in this review
Inhaler device: Turbuhaler. |
|
| Outcomes | Symptoms, adverse events, exacerbations, lung function. | |
| Notes | Classified as 'poorly reversible' subgroup. Exacerbation defined as requirement of oral steroids and/or antibiotics and/or hospitalisation for respiratory symptoms. Mild exacerbation defined as requirement of >/= 4 inhalations per day. P values used to calculate pooled SEMs for following outcomes: Symptoms; rescue medication usage. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated scheme. |
| Allocation concealment (selection bias) | Low risk | At each centre, eligible patients received an enrolment code and then after run‐in, participants were allocated the next consecutive patient number. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Identical inhaler devices. |
| Incomplete outcome data (attrition bias) Mortality | High risk | 32% withdrew on formoterol and 28% on BDF. |
| Incomplete outcome data (attrition bias) All other outcomes | High risk | 32% withdrew on formoterol and 28% on BDF. |
| Selective reporting (reporting bias) | High risk | Does not contribute data to analysis of exacerbations as dichotomous data, hospitalisations or pneumonia. |
Tashkin 2008.
| Methods | 6‐month, randomised, double‐blind, double‐dummy, placebo‐controlled, parallel group, multi‐centre study. | |
| Participants |
|
|
| Interventions | After 2 weeks of treatment based on previous therapy (ICSs and short‐acting bronchodilators allowed during the run‐in period), patients received one of the following treatments administered twice daily: budesonide/formoterol pressurised metered dose inhaler (pMDI) 160/4.5 μg BID; budesonide/formoterol pMDI 80/4.5 μg BID; budesonide pMDI 160 μg BID plus formoterol dry powder inhaler (DPI) 4.5 μg BID; budesonide pMDI 160 μg BID; formoterol DPI 4.5 μg BID; or placebo. | |
| Outcomes | Pre‐dose and 1 hour post‐dose FEV1. 12‐hour spirometry, pre‐dose and 1‐hour post‐dose Inspiratory Capacity. Pre‐dose morning and evening PEF. Dyspnoea, health‐related quality of life. COPD exacerbations. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Balanced blocks according to a computer‐generated randomisation scheme at each site. |
| Allocation concealment (selection bias) | Unclear risk | Information not available. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | To maintain blinding, patients received both a pressurized metered‐dose inhaler (pMDI) and a dry powder inhaler (DPI) containing either active treatment or placebo, or combinations of active treatment and placebo, as appropriate. (Double blind, double dummy design) |
| Incomplete outcome data (attrition bias) Mortality | High risk | 21% discontinued on LABA and 14% on combination therapy. |
| Incomplete outcome data (attrition bias) All other outcomes | High risk | 21% discontinued on LABA and 14% on combination therapy. |
| Selective reporting (reporting bias) | Unclear risk | Does not contribute data to analysis of exacerbations as dichotomous data or hospitalisations. |
TORCH.
| Methods | RCT, parallel group design. Trial duration: 156 weeks. Baseline characteristics: comparable. Intention‐to‐treat analysis: stated. | |
| Participants |
|
|
| Interventions | Run‐in: 2 weeks. All maintenance treatment with ICS and LABA ceased.
Additional treatment groups not covered in this review
Inhaler device: DPI. |
|
| Outcomes | All cause mortality; change in SGRQ; exacerbations (requiring antibiotics, steroids, hospitalisation or combination of these); lung function; withdrawals; adverse events. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated scheme. Permuted block randomisation with stratification for smoking status and country. |
| Allocation concealment (selection bias) | Low risk | Centralised randomisation scheme. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Identical inhaler devices. |
| Incomplete outcome data (attrition bias) Mortality | Low risk | Mortality was the primary outcome and vital status was checked in those who withdrew. |
| Incomplete outcome data (attrition bias) All other outcomes | High risk | 36.9% withdrew on salmeterol and 34.1% on FPS. |
| Selective reporting (reporting bias) | High risk | Does not contribute data to analysis of exacerbations as dichotomous data. |
TRISTAN.
| Methods | RCT, parallel group design. Trial duration: 2 week run‐in period, 52 weeks treatment, 2‐week follow‐up. Baseline characteristics: comparable. Intention‐to‐treat analysis: stated. | |
| Participants |
|
|
| Interventions | Run‐in: 2 weeks. All maintenance treatment with ICS and LABA ceased.
Additional treatment groups not covered in this review
Inhaler device: DPI. |
|
| Outcomes | FEV1; PEF; exercise tolerance; quality of life: SGRQ; dyspnoea and symptoms (symptom score for shortness of breath, cough and sputum production); exacerbations (defined as requirement for antibiotics, oral steroids or both); rescue salbutamol use. | |
| Notes | FEV1 reversibility (% predicted normal). Mean Reversibility (% predicted) = 3.8. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation scheme. |
| Allocation concealment (selection bias) | Low risk | Centralised, third party randomisation. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Identical inhaler devices. |
| Incomplete outcome data (attrition bias) Mortality | High risk | 32% withdrew on salmeterol and 25% on FPS. |
| Incomplete outcome data (attrition bias) All other outcomes | High risk | 32% withdrew on salmeterol and 25% on FPS. |
| Selective reporting (reporting bias) | High risk | Does not contribute data to analysis of exacerbations as dichotomous data or hospitalisations. |
DPI: Dry powder inhaler
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Aaron 2007 | All the arms had tiotropium |
| Bourbeau 2007 | Inflammatory outcomes for FPS |
| Cukier 2007 | Combination of oxygen and bronchodilators |
| Golabi 2006 | Combined with tiotropium and different outcomes |
| Haque 2006 | Bench research of glucocorticoids receptors |
| INSPIRE | ARM with LAMA (tiotropium) |
| Lindberg 2007 | Short term. Outcome was onset of action. No LABA alone arm |
| Schermer 2007 | Only one arm with FPS |
| Sethi 2006 | Outcome was bacterial colonization for FPS |
| Sutherland 2006 | Genetic study of FPS |
| Trofimenko 2006 | FPS with no comparison |
| Zheng 2006 | FPS with no comparison |
Differences between protocol and review
We updated the risk of bias assessment, adding an evaluation of incomplete outcome data which was stratified by outcome. In view of the clinical heterogeneity between the studies and the statistical heterogeneity, the 2012 update has reported the results of random‐effects models throughout (with a fixed‐effect model for sensitivity analysis).
We removed subgroups based on reversibility from the forest plots for the 2012 review.
The GIV method was not available when the protocol was developed.
Contributions of authors
LN and PP developed the protocol. Studies were assessed by LN and TJL. TJL and LN checked data and entered data into RevMan. TJL and LN conducted the analysis. TJL and LN developed the discussion with input from PP. PP assisted with the write‐up of the 2012 update.
Sources of support
Internal sources
NHS Cochrane Grant scheme, UK.
External sources
No sources of support supplied
Declarations of interest
The authors who have been involved in this review have done so without any known conflicts of interest. None of the authors are considered a paid consultant by any pharmaceutical company which produces agents discussed in this review.
Edited (no change to conclusions)
References
References to studies included in this review
Anzueto 2009 {published data only}
- Anzueto A, Ferguson GT, Feldman G, Chinsky K, Seibert A, Emmett A, et al. Effect of fluticasone propionate/salmeterol (250/50) on COPD exacerbations and impact on patient outcomes. Journal of Chronic Obstructive Pulmonary Disease 2009;6(5):320‐9. [DOI] [PubMed] [Google Scholar]
Calverley 2003 {published and unpublished data}
- Borgstrom L, Asking L, Olsson H, Peterson S. Lack of interaction between disease severity and therapeutic response with budesonide/formoterol in a single inhaler [Abstract]. American Thoracic Society 100th International Conference, May 21‐26. 2004:C22 [Poster 505].
- Calverley PM, Bonsawat W, Cseke Z, Zhong N, Peterson S, Olsson H. Maintenance therapy with budesonide and formoterol in chronic obstructive pulmonary disease. European Respiratory Journal 2003;22(6):912‐9. [DOI] [PubMed] [Google Scholar]
- Calverley PMA, Cseke Z, Peterson S. Budesonide/formoterol reduces the use of oral corticosteroids in the treatment of COPD [Abstract]. European Respiratory Journal 2003;22 Suppl 45:P436. [DOI] [PubMed] [Google Scholar]
- Calverley PMA, Kuna P, Olsson H. COPD exacerbations are reduced by budesonide/formoterol in a single inhaler [Abstract]. European Respiratory Journal 2003;22 Suppl 45:P1587. [Google Scholar]
- Calverley PMA, Olsson H, Symbicort International COPD Study Group. Budesonide/formoterol ina single inhaler sustains improvements in lung function over 12 months compared with monocomponents and placebo in patients with COPD [abstract]. American Thoracic Society 99th International Conference. 2003:B024 [Poster 418].
- Calverley PMA, Peterson S. Combining budesonide/formoterol in a single inhale reduces exacerbation frequency in COPD [abstract]. American Thoracic Society 99th International Conference. 2003:D092 [Poster 211].
- Calverley PMA, Ståhl E, Jones PW. Budesonide/formoterol improves the general health status of patients with COPD [Abstract]. American Thoracic Society 2005 International Conference; May 20‐25; San Diego, California. 2005:B93 [Poster 303].
- Calverley PMA, Szafranski W, Andersson A. Budesonide/formoterol is a well‐tolerated long term maintenance therapy for COPD. European Respiratory Journal 2005;26 Suppl 49:Poster 1917. [Google Scholar]
- Calverley PMA, Thompson NC, Olsson H. Budesonide/formoterol in a single inhaler sustains lung function improvements in COPD [Abstract]. European Respiratory Journal 2003;22 Suppl 45:P435. [Google Scholar]
- Halpin D, Stahl E, Lundback B, Anderson F, Peterson S. Treatment costs and number needed to treat (NNT) with budesonide/formoterol to avoid one exacerbation of COPD [Abstract]. American Thoracic Society 100th International Conference, May 21‐26. 2004:D22 [Poster 525].
- Halpin DMG, Larsson T, Calverley PMA. How many patients with COPD must be treated with budesonide/formoterol compared with formoterol alone to avoid 1 day of oral steroid use? [Abstract]. American Thoracic Society 2005 International Conference; May 20‐25; San Diego, California. 2005:B93 [Poster 314].
- Jones PW, Stahl E. Budesonide/formoterol in a single inhaler improves health status in patients with COPD [abstract]. American Thoracic Society 99th International Conference. 2003:B024 [Poster 419].
- Jones PW, Ståhl E. Budesonide /formoterol sustains clinically relevant improvements in health status in COPD [Abstract]. European Respiratory Journal 2005;26 Suppl 49:Abstract 1352. [Google Scholar]
- Jones PW, Ståhl E. Reducing exacerbations leads to a better health‐related quality of life in patients with COPD. 13th ERS Annual Congress, 27th September, 2003, Vienna. 2003:P1586.
- Lofdahl CG. Reducing the impact of COPD exacerbations: Clinical efficacy of budesonide/formoterol. European Respiratory Review 2004;13(88):14‐21. [Google Scholar]
- Lofdahl CG, Andreasson E, Svensson K, Ericsson A. Budesonide/formoterol in a single inhaler improves health status in patients with COPD without increasing healthcare costs [Abstract]. European Respiratory Journal 2003;22 Suppl 45:P433. [Google Scholar]
- Lofdahl CG, Ericsson A, Svensson K, Andreasson E. Cost effectiveness of budesonide/formoterol in a single inhaler for COPD compared with each monocomponent used alone. Pharmacoeconomics 2005;23(4):365‐75. [DOI] [PubMed] [Google Scholar]
Dal Negro 2003 {published data only}
- Dal Negro R, Micheletto C, Trevsian F, Tognella S. [A98] Salmeterol and fluticasone 50ug/250ug BiD versus salmeterol 50ug bid and versus placebo in the long term treatment of COPD. Proceedings of the 98th International American Thoracic Society Conference. 2002:http://www.abstracts‐on‐line.com/abstracts/ATS.
- Dal Negro RW, Pomari C, Tognella S, Micheletto C. Salmeterol & fluticasone 50 microg/250 microg bid in combination provides a better long‐term control than salmeterol 50 microg bid alone and placebo in COPD patients already treated with theophylline. Pulmonary Pharmacology and Therapeutics 2003;16(4):241‐6. [DOI] [PubMed] [Google Scholar]
Ferguson 2008 {published data only}
- Ferguson GT, Anzueto A, Fei R, Emmett A, Knobil K, Kalberg C. Effect of fluticasone propionate/salmeterol (250/50 microg) or salmeterol (50 microg) on COPD exacerbations. Respiratory Medicine 2008;102:1099‐108. [DOI] [PubMed] [Google Scholar]
- SCO40043. A randomized, double‐blind, parallel‐group, 52‐week study to compare the effect of fluticasone propionate/salmeterol DISKUS® 250/50mcg bid with salmeterol DISKUS® 50mcg bid on the annual rate of moderate/severe exacerbations in subjects with chronic obstructive pulmonary disease (COPD). http://ctr.gsk.co.uk (accessed 8th April 2008).
Hanania 2003 {published and unpublished data}
- Hanania NA, Darken P, Horstman D, Reisner C, Lee B, Davis S, et al. The efficacy and safety of fluticasone propionate (250 micro g)/salmeterol (50 micro g) combined in the diskus inhaler for the treatment of COPD. Chest 2003;124(3):834‐43. [DOI] [PubMed] [Google Scholar]
- Hanania NA, Ramsdell J, Payne K, Davis S, Horstman D, Lee B, et al. Improvements in airflow and dyspnea in COPD patients following 24 weeks treatment with salmeterol 50mcg and fluticasone propionate 250mcg alone or in combination via the diskus. American Journal of Respiratory and Critical Care Medicine 2001;163 Suppl(5):A279. [Google Scholar]
- Horstman D, Darken P, Davis S, Lee B. Improvements in FEV1 and symptoms in poorly reversible COPD patients following treatment with salmeterol 50mcg/fluticasone propionate 250mcg combination [Abstract]. European Respiratory Journal 2003;22 Suppl 45:P434. [Google Scholar]
- Mahler DA, Darken P, Brown CP, Knobil K. Predicting lung function responses to combination therapy in chronic obstructive pulmonary disease (COPD) [Abstract]. National COPD Conference; Arlington, Virginia. 2003:Abstract 1081.
- Mahler DA, Darken P, Brown CP, Knobil K. Predicting lung function responses to salmeterol/fluticasone propionate combination therapy in COPD [Abstract]. European Respiratory Journal 2003;22 Suppl 45:P429. [Google Scholar]
- SFCA3007. A randomized, double‐blind, placebo‐controlled, parallel‐group trial evaluating the safety and efficacy of the DISKUS formulations of Salmeterol (SAL) 50mcg BID and Fluticasone Propionate (FP) 250mcg BID individually and in combination as Salmeterol 50mcg/Fluticasone Propionate 250mcg BID (SFC 50/250) compared to placebo in COPD subjects. GlaxoSmithKline Clinical Trials Register (http:ctr.gsk.co.uk) 2005.
- Spencer M, Wire P, Lee B, Chang CN, Darken P, Horstman D. Patients with COPD using salmeterol/fluticasone propionate combination therapy experience improved quality of life. European Respiratory Journal 2003;22 Suppl 45:51s. [Google Scholar]
- Spencer MD, Karia N, Anderson J. The clinical significance of treatment benefits with the salmeterol/fluticasone propionate 50/500mcg combination in COPD. European Respiratory Journal 2004;24 Suppl 48:290s. [Google Scholar]
Kardos 2007 {published and unpublished data}
- Kardos P, Wencker M. Combination therapy with salmeterol and fluticasone propionate (SFC) is more effective than salmeterol (SAL) alone in reducing exacerbations of COPD. European Respiratory Journal 2005;26 Suppl 49:Abstract 1944. [Google Scholar]
- Kardos P, Wencker M, Glaab T, Vogelmeier C. Impact of salmeterol/fluticasone propionate versus salmeterol on exacerbations in severe chronic obstructive pulmonary disease. American Journal of Respiratory and Critical Care Medicine 2007;175(2):144‐9. [DOI] [PubMed] [Google Scholar]
- SCO30006. A randomised, double‐blind, parallel‐group study to investigate the protective effect of the combination of fluticasone and salmeterol (500/50µg bid via the DISKUS) compared with salmeterol (50µg bid via the DISKUS) on the incidence of moderate to severe exacerbations in patients with severe chronic obstructive pulmonary disease (COPD) (GOLD III/IV). GlaxoSmithKline Clinical Trials Register (http:ctr.gsk.co.uk) 2005.
- Vogelmeier C. Combination therapy with salmeterol and fluticasone propionate (SFC) improves quality of life (QoL) more than salmeterol (SAL) alone in COPD. 42nd Nordic Lung Conference Trondheim. 2005; Vol. 13 Suppl 22.
- Vogelmeier CF, Wencker M, Glaab TH, Kardos P. Number needed to treat (NNT) to reduce exacerbations in severe COPD comparing salmeterol/fluticasone propionate (SFC) with salmeterol (SAL) treatment. Proceedings of the American Thoracic Society. 2006:A110.
Mahler 2002 {published and unpublished data}
- Mahler DA, Darken P, Brown CP, Knobil K. Predicting lung function responses to combination therapy in chronic obstructive pulmonary disease (COPD). http://www.abstracts2view.com 2003.
- Mahler DA, Wire P, Horstman D, Chang CN, Yates J, Fischer T, et al. Effectiveness of fluticasone propionate and salmeterol combination delivered via the diskus device in the treatment of chronic obstructive pulmonary disease. American Journal of Respiratory and Critical Care Medicine 2002;166(8):1084‐91. [DOI] [PubMed] [Google Scholar]
- SFCA3006. A randomized, double‐blind, placebo‐controlled, parallel‐group trial evaluating the safety and efficacy of the DISKUS formulations of Salmeterol (SAL) 50mcg BID and Fluticasone Propionate (FP) 500mcg BID individually and in combination as Salmeterol 50mcg/Fluticasone Propionate 500mcg BID (SFC 50/500) compared to placebo in COPD subjects. GlaxoSmithKline Clinical Trials Register (http:ctr.gsk.co.uk) 2005.
- Spencer M, Wire P, Lee B, Chang CN, Darken P, Horstman D. Patients with COPD using salmeterol/fluticasone propionate combination therapy experience improved quality of life. European Respiratory Journal 2003;22 Suppl 45:51s. [Google Scholar]
- Spencer MD, Anderson JA. Salmeterol/fluticasone combination produces clinically important benefits in dyspnea and fatigue [Abstract]. American Thoracic Society 2005 International Conference; May 20‐25; San Diego, California. 2005:B93 [Poster 308].
- Spencer MD, Karia N, Anderson J. The clinical significance of treatment benefits with the salmeterol/fluticasone propionate 50/500mcg combination in COPD. European Respiratory Journal 2004;24 Suppl 48:290s. [Google Scholar]
O'Donnell 2006 {published and unpublished data}
- O'Donnell DE, Sciurba F, Celli B, Mahler DA, Webb KA, Kalberg CJ, et al. Effect of fluticasone propionate/salmeterol on lung hyperinflation and exercise endurance in COPD. Chest 2006;130(3):647‐56. [DOI] [PubMed] [Google Scholar]
- SCO40030. A randomized, double‐blind, placebo‐controlled, parallel group clinical trial evaluating the effect of the fluticasone propionate/salmeterol combination product 250/50mcg bid via DISKUS and salmeterol 50mcg bid via DISKUS on lung hyperinflation in subjects with chronic obstructive pulmonary disease (COPD). GlaxoSmithKline Clinical Trials Register (http:ctr.gsk.co.uk) 2005.
Rennard 2009 {published data only}
- Rennard SI, Tashkin DP, McElhattan J, Goldman M, Ramachandran S, Martin UJ, et al. Efficacy and tolerability of budesonide/formoterol in one hydrofluoroalkane pressurized metered‐dose inhaler in patients with chronic obstructive pulmonary disease. Results from a 1‐year randomized controlled clinical trial. Drugs 2009;69(5):549‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
SCO100470 {unpublished data only}
- SCO100470. A multicentre, randomised, double‐blind, parallel group, 24‐week study to compare the effect of the salmeterol/fluticasone propionate combination product 50/250mcg, with salmeterol 50mcg both delivered twice daily via the DISKUS/ACCUHALER inhaler on lung function and dyspnoea in subjects with Chronic Obstructive Pulmonary Disease (COPD). GlaxoSmithKline Clinical Trials Register (http:ctr.gsk.co.uk) 2006.
Szafranski 2003 {published and unpublished data}
- Anderson P. Budesonide/formoterol in a single inhaler (Symbicort) provides early and sustained improvement in lung function in moderate to severe COPD [Abstract]. Thorax 2002;57 Suppl III:iii43. [Google Scholar]
- Borgstrom L, Asking L, Olsson H, Peterson S. Lack of interaction between disease severity and therapeutic response with budesonide/formoterol in a single inhaler [Abstract]. American Thoracic Society 100th International Conference, May 21‐26. 2004:C22 [Poster 505].
- Calverley P, Pauwels R, Lofdahl CG, Svensson K, Higenbottam T, L‐G Carlsson, et al. Relationship between respiratory symptoms and medical treatment in exacerbations of COPD. European Respiratory Journal 2005;26(3):406‐13. [DOI] [PubMed] [Google Scholar]
- Calverley PMA. Effect of budesonide/formoterol on severe exacerbations and lung function in moderate to severe COPD. Thorax. BTS Winter meeting 2002:S145.
- Calverley PMA, Szafranski W, Andersson A. Budesonide/formoterol is a well‐tolerated long term maintenance therapy for COPD. European Respiratory Journal 2005;26 Suppl 49:Poster 1917. [Google Scholar]
- Calverley PMA, Thompson NC, Olsson H. Budesonide/formoterol in a single inhaler sustains lung function improvements in COPD [Abstract]. European Respiratory Journal 2003;22 Suppl 45:P435. [Google Scholar]
- Campbell LM, Szafranski W. Budesonide/formoterol in a single inhaler (Symbicort) provides sustained relief from symptoms in moderate to severe COPD. Thorax. BTS Winter meeting 2002:S143.
- Campell LW, Szafranski W. Budesonide/Formoterol in a single inhaler (Symbicort) reduces severe exacerbations in patients with moderate‐severe COPD. Thorax. BTS Winter meeting 2002:S141.
- Dahl R, Cukier A, Olsson H. Budesonide/formoterol in a single inhaler reduces severe and mild exacerbations in patients with moderate to severe COPD. European Respiratory Journal 2002;20 Suppl 38:242 [P1575]. [Google Scholar]
- Egede F, Menga G. Budesonide/formoterol in a single inhaler provides sustained relief from symptoms and night‐time awakenings in moderate‐severe COPD: results from symptoms and night‐time awakenings in moderate to severe COPD: results from a 1‐year study. European Respiratory Journal 2002;20 Suppl 38:242 [P1574]. [Google Scholar]
- Halpin D, Stahl E, Lundback B, Anderson F, Peterson S. Treatment costs and number needed to treat (NNT) with budesonide/formoterol to avoid one exacerbation of COPD [Abstract]. American Thoracic Society 100th International Conference, May 21‐26. 2004:D22 [Poster 525].
- Jones PW, Stahl E, Svensson K. Improvement in health status in patients with moderate to severe COPD after treatment with budesonide/formoterol in a single inhaler. European Respiratory Journal 2002;20 Suppl 38:250 [P1613]. [Google Scholar]
- Korsgaard J, Sansores R. Budesonide/formoterol (single inhaler) provides sustained relief from shortness of breath and chest tightness in a 1‐year study of patients with moderate to severe COPD. European Respiratory Journal 2002;20 Suppl 38:242 [P1577]. [Google Scholar]
- Lange P, Saenz C. Budesonide/formoterol in a single inhaler is well tolerated in patients with moderate to severe COPD: results of a 1 year study. European Respiratory Journal 2002;20 Suppl 38:242 [P1573]. [Google Scholar]
- Lofdahl CG. Reducing the impact of COPD exacerbations: Clinical efficacy of budesonide/formoterol. European Respiratory Review 2004;13(88):14‐21. [Google Scholar]
- Milanowski J, Nahabedian S. Budesonide/formoterol in a single inhaler acts rapidly to improve lung function and relieve symptoms in patients with moderate to severe COPD. European Respiratory Journal 2002;20 Suppl 38:242 [P1576]. [Google Scholar]
- Szafranski W, Cukier A, Ramirez A, Menga G, Sansores R, Nahabedian S, et al. Efficacy and safety of budesonide/formoterol in the management of chronic obstructive pulmonary disease. European Respiratory Journal 2003;21(1):74‐81. [DOI] [PubMed] [Google Scholar]
Tashkin 2008 {published data only}
- Tashkin DP, Rennard SI, Martin P, Ramachandran S, Martin UJ, Silkoff PE, et al. Efficacy and safety of budesonide and formoterol in one pressurized metered‐dose inhaler in patients with moderate to very severe chronic obstructive pulmonary disease. Results of a 6‐month randomized clinical trial. Drugs 2008;68(14):1975‐2000. [DOI] [PubMed] [Google Scholar]
TORCH {published and unpublished data}
- Calverley PMA, Anderson JA, Celli B, Ferguson GT, Jenkins C, Jones PW, et al. Salmeterol and fluticasone propionate and survival in chronic obstructive pulmonary disease. New England Journal of Medicine 2007;356(8):775‐89. [DOI] [PubMed] [Google Scholar]
- Calverley PMA, Celli B, Ferguson G, Jenkins C, Jones PW, Pride NB, et al. Baseline characteristics of the first 5,000 COPD patients enrolled in the TORCH survival study. European Respiratory Journal 2003;22 Suppl 45:578s. [Google Scholar]
- Celli B, Calverley PMA, Anderson JA, Ferguson GT, Jenkins C, Jones PW, et al. The TORCH (TOwards a Revolution in COPD Health) study: salmeterol/fluticasone propionate (SFC) improves health status, reduces exacerbations and improves lung function over three years. European Respiratory Journal 2006;28 Suppl 50:34s. [Google Scholar]
- Ferguson GT, Calverley PMA, Anderson JA, et al. The TORCH (TOwards a Revolution in COPD Health) study: salmeterol/fluticasone propionate (SFC) improves survival in COPD over three years. European Respiratory Journal 2006;28 Suppl 50:34s. [Google Scholar]
- Jenkins CR, Calverley PMA, Celli B, Ferguson G, Jones PW, Pride N, et al. Seasonal Patterns of Exacerbation Rates in the TORCH Survival Study. http://www.abstracts2view.com 2007:A839.
- Jones PW, Calverley P, Celli B, Ferguson G, Jenkins C, Pride N. Trans‐Regional Validity of the SGRQ in the TORCH Survival Study. http://www.abstracts2view.com 2007:A122.
- McGarvey LP, John M, Anderson JA, Zvarich MT, Wise RA. Ascertainment of cause‐specific mortality in COPD : operations of the TORCH Clinical Endpoint Committee. Thorax 2007;62:411‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCO30003. A multicentre, randomised, double‐blind, parallel group, placebo‐controlled study to investigate the long‐term effects of salmeterol/fluticasone propionate (SERETIDE®/VIANI®/ADVAIR®) 50/500mcg bd, salmeterol 50mcg bd and fluticasone propionate 500mcg bd, all delivered via the DISKUS®/ACCUHALER® inhaler, on the survival of subjects with chronic obstructive pulmonary disease (COPD) over 3 years of treatment. www.ctr.gsk.co.uk 2006.
- Vestbo J, Calverley P, Celli B, Ferguson G, Jenkins C, Jones P, et al. The TORCH (TOwards a Revolution in COPD Health) survival study protocol. European Respiratory Journal 2004;24(2):206‐10. [DOI] [PubMed] [Google Scholar]
- Wise RA, McGarvey LP, John M, Anderson JA Zvarich MT. Reliability of cause‐specific mortality adjudication in a COPD clinical trial. http://www.abstracts2view.com 2007:A120.
TRISTAN {published and unpublished data}
- Calverley P, Pauwels R, Vestbo J, Jones P, Pride N, Gulsvik A, et al. Combined salmeterol and fluticasone in the treatment of chronic obstructive pulmonary disease: a randomised controlled trial. Lancet 2003;361(9356):449‐56. [DOI] [PubMed] [Google Scholar]
- Calverley PMA, Pauwels RA, Vestbo J, Jones PW, Pride NB, Gulsvik A, et al. Clinical improvements with salmeterol / fluticasone propionate combination in differing severities of COPD. http://www.abstracts2view.com 2003:A035 [Poster D50].
- Calverley PMA, Pauwels RA, Vestbo J, Jones PW, Pride NB, Gulsvik A, et al. Salmeterol/fluticasone propionate combination for one year provides greater clinical benefit than its individual components. Proceedings of the 98th International American Thoracic Society Conference http://www.abstracts‐on‐line.com/abstracts/ATS. 2002:A98 [Poster 306].
- Calverly PMA, Pauwels R, Vestbo J, Jones P, Pride N, Gulsvik A, et al. Safety of salmeterol/fluticasone propionate combination in the treatment of chronic obstructive pulmonary disease. European Respiratory Journal 2002;20 Suppl 38:242 [P1572]. [Google Scholar]
- Hunjan MK, Chandler F. Numbers needed to treat (NNT) to avoid an exacerbation in patients with chronic obstructive pulmonary disease (COPD) using salmeterol/fluticasone propionate combination (SFC) and associated costs [Abstract]. American Thoracic Society 100th International Conference, May 21‐26. 2004:D22 [Poster 503].
- Hunjan MK, Williams DT. Costs of avoiding exacerbations in patients with chronic obstructive pulmonary disease (COPD) treated with salmeterol/ fluticasone propionate combination (seretide) and salmeterol. European Respiratory Journal 2004;24 Suppl 48:291s. [Google Scholar]
- Hunjan MK, Williams DT. Salmeterol/ fluticasone propionate combination is clinically effective in avoiding exacerbations in patients with moderate/severe COPD. European Respiratory Journal 2004;24 Suppl 48:513s. [Google Scholar]
- Jones PW, Edin HM, Anderson J. Salmeterol/fluticasone propionate combination improves health status in COPD patients. Proceedings of the 98th International American Thoracic Society Conference http://www.abstracts‐on‐line.com/abstracts/ATS. 2002:A39 [Poster K39].
- Jones PW, Ståhl E. Budesonide /formoterol sustains clinically relevant improvements in health status in COPD [Abstract]. European Respiratory Journal 2005;26 Suppl 49:Abstract 1352. [Google Scholar]
- Jones PW, Vestbo J, Pauwels RA, Calverley PMA, Anderson JA, Spencer MD. Informative drop out in COPD studies. Investigation of health status of withdrawals in the TRISTAN study. 13th ERS Annual Congress, 27 Spetember 2003, Vienna. 2003:P1593.
- Nitschmann S. Inhalational combination therapy in chronic obstructive lung disease. Tristan study. German Internist 2004;45(6):727‐8. [DOI] [PubMed] [Google Scholar]
- Pauwels R, Vestbo J, Calverley PMA, Jones PW, Pride NB, Gulsvik A. Characterization of exacerbations in the TRISTAN study of salmeterol/fluticasone propionate (SFC) combination in moderate to severe COPD. http://www.abstracts2view.com 2003.
- Pauwels RA, Calverly PMA, Vestbo J, Jones PW, Pride N, Gulsvik A, et al. Reduction of exacerbations with salmeterol/fluticasone combination 50/500 mcg bd in the treatment of chronic obstructive pulmonary disease. European Respiratory Journal 2002;20 Suppl 38:240 [P1569]. [Google Scholar]
- SFCB3024. A multicentre, randomised, double‐blind, parallel group, placebo‐controlled study to compare the efficacy and safety of the salmeterol/FP combination product at a strength of 50/500mcg bd with salmeterol 50mcg bd alone and FP 500mcg bd alone, delivered via the DISKUS™/ACCUHALER™, in the treatment of subjects with chronic obstructive pulmonary disease (COPD) for 12 months. GlaxoSmithKline Clinical Trials Register (http:ctr.gsk.co.uk) 2005.
- Spencer M, Briggs AH, Grossman RF, Rance L. Development of an economic model to assess the cost effectiveness of treatment interventions for chronic obstructive pulmonary disease. Pharmacoeconomics 2005;23(6):619‐37. [DOI] [PubMed] [Google Scholar]
- Spencer MD, Karia N, Anderson J. The clinical significance of treatment benefits with the salmeterol/fluticasone propionate 50/500mcg combination in COPD. European Respiratory Journal 2004;24 Suppl 48:290s. [Google Scholar]
- Vestbo J, Calverley PMA, Pauwels R, Jones P, Pride N, Gulsvik A, et al. Absence of gender susceptibility to the combination of salmeterol and fluticasone in the treatment of chronic obstructive pulmonary disease. European Respiratory Journal 2002;20 Suppl 38:240 [P1570]. [Google Scholar]
- Vestbo J, Pauwels R, Anderson JA, Jones P, Calverley P. Early onset of effect of salmeterol and fluticasone propionate in chronic obstructive pulmonary disease. Thorax 2005;60(4):301‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vestbo J, Pauwels RS, Calverley PMA, Jones PW, Pride NB, Gulsvik A. Salmeterol / fluticasone propionate combination produces improvement in lung function detectable within 24 Hours in moderate to severe COPD. http://www.abstracts2view.com 2003.
- Vestbo J, Soriano JB, Anderson JA, Calverley P, Pauwels R, Jones P. Gender does not influence the response to the combination of salmeterol and fluticasone propionate in COPD. Respiratory Medicine 2004;98(11):1045‐50. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Aaron 2007 {published data only}
- Aaron SD, Vandemheen KL, Fergusson D, Maltais F, Bourbeau J, Goldstein R, et al. Tiotropium in combination with placebo, salmeterol, or fluticasone‐salmeterol for treatment of chronic obstructive pulmonary disease: a randomized trial. Annals of Internal Medicine 2007;146(8):545‐55. [DOI] [PubMed] [Google Scholar]
- Kaplan A. Effects of tiotropium combined with either salmeterol or salmeterol/fluticasone in moderate to severe COPD. Primary Care Respiratory Journal 2007;16(4):258‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bourbeau 2007 {published data only}
- Bourbeau J, Christodoulopoulos P, Maltais F, Yamauchi Y, Olivenstein R, Hamid Q. Effect of salmeterol/fluticasone propionate on airway inflammation in COPD: A randomised controlled trial. Thorax 2007;62(11):938‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
Cukier 2007 {published data only}
- Cukier A, Ferreira CAS, Stelmach R, Ribeiro M, Cortopassi F, Calverley PMA. The effect of bronchodilators and oxygen alone and in combination on self‐paced exercise performance in stable COPD. Respiratory Medicine 2007;101(4):743‐53. [DOI] [PubMed] [Google Scholar]
Golabi 2006 {published data only}
- Golabi P, Topaloglu N, Karakurt S, Celikel T. Effects of tiotropium and salmeterol/fluticasone combination on lung hyperinflation dyspnea and exercise tolerance in COPD [Abstract].. European Respiratory Journal 2006;28 Suppl 50:33s. [Google Scholar]
Haque 2006 {published data only}
- Haque RA, Torrego A, Essilfie‐Quaye S, Kharitonov SA, Johnson M, Adcock IM, et al. Effect of salmeterol and fluticasone on glucocorticoid receptor translocation in sputum macrophages and peripheral blood mononuclear cells from patients with chronic obstructive pulmonary disease. Proceedings of the American Thoracic Society. 2006:A848.
INSPIRE {published data only}
- GlaxoSmithKline (SCO40036). Multicentre, randomised, double‐blind, double‐dummy, parallel group, 104‐week study to compare the effect of the salmeterol/fluticasone propionate combination product (SERETIDE*) 50/500mcg delivered twice daily via the DISKUS*/ACCUHALER* inhaler with tiotropium bromide 18 mcg delivered once daily via the HandiHaler inhalation device on the rate of health care utilisation exacerbations in subjects with severe chronic obstructive pulmonary disease (COPD). http://ctr.gsk.co.uk (accessed 8th April 2008).
- Seemungal T, Stockley R, Calverley P, Hagan G, Wedzicha JA. Investigating new standards for prophylaxis in reduction of exacerbations ‐ The INSPIRE study methodology. Journal of Chronic Obstructive Pulmonary Disease 2007;4(3):177‐83. [DOI] [PubMed] [Google Scholar]
- Wedzicha J, Stockley R, Seemungal T, Hagan G, Calverley P. The INSPIRE study: effect of salmeterol/fluticasone propionate versus tiotropium bromide on COPD exacerbations. Respirology 2007;12 Suppl 4:A112. [Google Scholar]
Lindberg 2007 {published data only}
- Lindberg A, Szalai Z, Pullerits T, Radeczky E. Fast onset of effect of budesonide/formoterol versus salmeterol/fluticasone and salbutamol in patients with chronic obstructive pulmonary disease and reversible airway obstruction. Respirology 2007;12(5):732‐9. [DOI] [PubMed] [Google Scholar]
- Lindberg A, Szalai Z, Pullertis T, Radeczky El. Budesonide/formoterol (B/F) has an onset of action that is similar to salbutamol and faster than salmeterol/fluticasone in patients with COPD. European Respiratory Journal 2006;28 Suppl 50:214s. [Google Scholar]
Schermer 2007 {published data only}
- Schermer TR, Albers JM, Verblackt HW, Costongs RJ, Westers P. Lower inhaled steroid requirement with a fluticasone/salmeterol combination in family practice patients with asthma or COPD. Family Practice 2007;24(2):181‐8.. [DOI] [PubMed] [Google Scholar]
Sethi 2006 {published data only}
- Sethi S, Grove L, Wrona C, Maloney J. Prevalence of bacterial colonization in COPD is not altered by fluticasone/salmeterol. Proceedings of the American Thoracic Society. 2006:A115.
Sutherland 2006 {published data only}
- Sutherland ER, Moss TA, Stevens AD, Pak J, Martin RJ. Modulation of sputum gene expression in COPD by fluticasone /salmeterol. European Respiratory Journal 2006;28 Suppl 50:662s. [Google Scholar]
Trofimenko 2006 {published data only}
- Trofimenko IN, Chernyak BA. The efficacy of salmeterol/fluticasone (SF) for 6 month's therapy at severe COPD patients. European Respiratory Journal 2006;28 Suppl 50:30s. [Google Scholar]
Zheng 2006 {published data only}
- Zheng J, Zhong N, Yang L, Wu Y, Chen P, Wen Z, et al. The efficacy and safety of fluticasone propionate 500 mg/salmeterol 50 mg combined via diskus/accuhaler in Chinese patients with chronic obstructive pulmonary disease (COPD). Chest 2006;130(4):182s. [Google Scholar]
- Zhong N, Zheng J, Yang L, Wu Y, Chen P, Wen Z, et al. The efficacy and safety of salmeterol 50µg/fluticasone propionate 500µg combined via accuhaler in Chinese patients with chronic obstructive pulmonary disease [Abstract]. Respirology 2006;11 Suppl 5:A150. [Google Scholar]
Additional references
Appleton 2006
- Appleton S, Poole P, Smith B, Veale A, Lasserson TJ, Chan MM, et al. Long‐acting beta2‐agonists for poorly reversible chronic obstructive pulmonary disease. Cochrane Database of Systematic Reviews 2006, Issue 3. [DOI: 10.1002/14651858.CD001104] [DOI] [PMC free article] [PubMed] [Google Scholar]
Burge 2000
- Burge PS, Calverley PM, Jones PW, Spencer S, Anderson JA, MaslenTK. Randomised, double blind, placebo controlled study of fluticasone propionate in patients with moderate to severe chronic obstructive pulmonary disease: the ISOLDE trial. BMJ 2000;320:1297‐303. [ISOLDE] [DOI] [PMC free article] [PubMed] [Google Scholar]
Calverley 2005
- Calverley P, Pauwels R, Lofdahl CG, Svensson K, Higenbottam T, Carlsson LG, et al. Relationship between respiratory symptoms and medical treatment in exacerbations of COPD. European Respiratory Journal 2005;26(3):406‐13. [DOI] [PubMed] [Google Scholar]
Cazzola 2008
- Cazzola M, MacNee W, Martinez FJ, Rabe KF, Franciosi LG, Barnes PJ, et al. Outcomes for COPD pharmacological trials: from lung function to biomarkers. European Respiratory Journal 2008;31:416‐68. [DOI] [PubMed] [Google Scholar]
Celli 2008
- Celli BR, Thomas NE, Anderson JA, Ferguson GT, Jenkins CR, Jones PW, et al. Effect of pharmacotherapy on rate of decline of lung function in chronic obstructive pulmonary disease: results from the TORCH study. American Journal of Respiratory and Critical Care Medicine 2008;178(4):332‐8. [DOI] [PubMed] [Google Scholar]
Dahl 2001
- Dahl R, Greefhorst LA, Nowak D, Nonikov V, Byrne AM, Thomson MH, et al. Inhaled formoterol dry powder versus ipratropium bromide in chronic obstructive pulmonary disease. American Journal of Resiratory Critical Care Medicine 2001;164(5):778–84. [DOI] [PubMed] [Google Scholar]
GOLD 2010
- Global Strategy for Diagnosis, Management, Prevention of COPD. Global Initiative for Chronic Obstructive Lung Disease. Global strategy for the diagnosis, management, and prevention of chronic obstructive lung disease. http://www.goldcopd.org 2010.
Higgins 2008
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.0.1 [updated September 2008]. The Cochrane Collaboration, 2008. Available from www.cochrane‐handbook.org.
Jones 2005
- Jones PW. St George’s Respiratory Questionnaire: MCID. COPD: Journal of Chronic Obstructive Pulmonary Disease 2005;2:75‐9. [DOI] [PubMed] [Google Scholar]
Karner 2011
- Karner C, Cates CJ. Long‐acting beta2‐agonist in addition to tiotropium versus either tiotropium or long‐acting beta2‐agonist alone for chronic obstructive pulmonary disease. Cochrane Database of Systematic Reviews 2011, Issue 2. [DOI: 10.1002/14651858.CD008989] [DOI] [PMC free article] [PubMed] [Google Scholar]
Karner 2011a
- Karner C, Cates CJ. The effect of adding inhaled corticosteroids to tiotropium and long‐acting beta2‐agonists for chronic obstructive pulmonary disease. Cochrane Database of Systematic Reviews 2011, Issue 9. [DOI: 10.1002/14651858.CD009039.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Karner 2011b
- Karner C, Cates CJ. Combination inhaled steroid and long‐acting beta2‐agonist in addition to tiotropium versus tiotropium or combination alone for chronic obstructive pulmonary disease. Cochrane Database of Systematic Reviews 2011, Issue 3. [DOI: 10.1002/14651858.CD008532.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Karner 2012
- Karner C, Chong J, Poole P. Tiotropium versus placebo for chronic obstructive pulmonary disease. Cochrane Database of Systematic Reviews 2012, Issue 7. [DOI: 10.1002/14651858.CD009285.pub2] [DOI] [PubMed] [Google Scholar]
Leidy 2003
- Leidy NK, Schmier JK, Jones MKC, et al. Evaluating symptoms in chronic obstructive pulmonary disease: validation of the Breathlessness, Cough and Sputum Scale. Respiratory Medicine 2003;97 Suppl A:59‐70. [PubMed] [Google Scholar]
Mahler 1999
- Mahler DA, Donohue JF, Barbee RA, Goldman MD, Gross NJ, Wisniewski ME, et al. Efficacy of salmeterol xinafoate in the treatment of COPD. Chest 1999;115(4):957‐65. [DOI] [PubMed] [Google Scholar]
Miravitlles 2004
- Miravitlles M, Ferrer M, Pont A, Zalacain R, Alvarez‐Sala JL, Masa F, et al. Effect of exacerbations on quality of life in patients with chronic obstructive pulmonary disease: a 2 year follow up study. Thorax 2004;59(5):387‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
Miravitlles 2007
- Miravitlles M, Anzueto A, Legnani D, Forstmeier L, Fargel M. Patient's perception of exacerbations of COPD ‐ the PERCEIVE study. Respiratory Medicine 2007;101(3):453‐60. [DOI] [PubMed] [Google Scholar]
Nannini 2007a
- Nannini L, Cates CJ, Lasserson TJ, Poole P. Combined corticosteroid and long‐acting beta‐agonist in one inhaler versus placebo for chronic obstructive pulmonary disease. Cochrane Database of Systematic Reviews 2007, Issue 4. [DOI: 10.1002/14651858.CD006826] [DOI] [PMC free article] [PubMed] [Google Scholar]
Nannini 2010
- Nannini LJ, Cates CJ, Lasserson TJ, Poole P. Combined corticosteroid and long‐acting beta‐agonist in one inhaler versus inhaled steroids for chronic obstructive pulmonary disease. Cochrane Database of Systematic Reviews 2010, Issue 1. [DOI: 10.1002/14651858.CD006826] [DOI] [PMC free article] [PubMed] [Google Scholar]
NICE 2010
- National Collaborating Centre for Chronic Conditions. Managament of chronic obstructive pulmonary disease in primary and secondary care. http://www.nice.org.uk 2010;available at http://guidance.nice.org.uk/CG101/Guidance/pdf/English:1‐673. [Google Scholar]
Osman 1997
- Osman IM, Godden DJ, Friend JA, Legge JS, Douglas JG. Quality of life and hospital re‐admission in patients with chronic obstructive pulmonary disease. Thorax 1997;52(1):67‐71. [DOI] [PMC free article] [PubMed] [Google Scholar]
RevMan 5 [Computer program]
- Copenhagen, The Nordic Cochrane Centre: The Cochrane Collaboration. Review Manager (RevMan) Version 5.1. Copenhagen, The Nordic Cochrane Centre: The Cochrane Collaboration, 2008.
Roisin 2000
- Rodriguez‐Roisin R. Toward a consensus definition of for COPD exacerbations. Chest 2000;117(5):398s‐401s. [DOI] [PubMed] [Google Scholar]
Singh 2009
- Singh S, Amin AV, Loke YK. Long‐term use of inhaled corticosteroids and the risk of pneumonia in chronic obstructive pulmonary disease: a meta‐analysis. Archives of Internal Medicine 2009;169(3):219‐29. [PUBMED: 19204211] [DOI] [PubMed] [Google Scholar]
Singh 2010
- Singh Sonal, Loke YK. An overview of the benefits and draw backs of inhaled corticosteroids in chronic obstructive pulmonary disease. International Journal of Chronic Obstructive Pulmonary Disease 2010;5:189‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
Spencer 2011
- Spencer S, Karner C, Cates CJ, Evans DJ. Inhaled corticosteroids versus long‐acting beta2‐agonists for chronic obstructive pulmonary disease. Cochrane Database of Systematic Reviews 2011, Issue 12. [DOI: 10.1002/14651858.CD007033.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Suissa 2006
- Suissa S. Statistical treatment of exacerbations in therapeutic trials of chronic obstructive pulmonary disease. American Journal of Respiratory and Critical Care Medicine 2006;173(8):842‐6. [DOI] [PubMed] [Google Scholar]
Sutherland 2003
- Sutherland ER, Allmers H, Ayas NT, Venn AJ, Martin RJ. Inhaled corticosteroids reduce the progression of airflow limitation in chronic obstructive pulmonary disease: a meta‐analysis. Thorax 2003;58:937‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
Visual Rx [Computer program]
- Cates CJ. Visual Rx 2.0. 2003.
Welsh 2011
- Welsh EJ, Cates CJ, Poole P. Combination inhaled steroid and long‐acting beta2‐agonist versus tiotropium for chronic obstructive pulmonary disease. Cochrane Database of Systematic Reviews 2011, Issue 7. [DOI: 10.1002/14651858.CD007891.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Yang 2007
- Yang IA, Fong KM, Sim EHA, Black PN, Lasserson TJ. Inhaled corticosteroids for stable chronic obstructive pulmonary disease. Cochrane Database of Systematic Reviews 2007, Issue 2. [DOI: 10.1002/14651858.CD002991] [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Nannini 2002
- Nannini L, Poole P. Combined corticosteroid and longacting bronchodilator in one inhaler for chronic obstructive pulmonary disease. Cochrane Database of Systematic Reviews 2002, Issue 3. [DOI: 10.1002/14651858.CD003794] [DOI] [PubMed] [Google Scholar]
Nannini 2004
- Nannini L, Cates CJ, Lasserson TJ, Poole P. Combined corticosteroid and long‐acting beta‐agonist in one inhaler for chronic obstructive pulmonary disease. Cochrane Database of Systematic Reviews 2004, Issue 3. [DOI: 10.1002/14651858.CD003794.pub2] [DOI] [PubMed] [Google Scholar]
Nannini 2007
- Nannini L, Lasserson TJ, Poole P. Combined corticosteroid and long‐acting beta‐agonist in one inhaler for chronic obstructive pulmonary disease. Cochrane Database of Systematic Reviews 2007, Issue 4. [DOI: 10.1002/14651858.CD006829] [DOI] [PubMed] [Google Scholar]