

Abstract
Novel treatments are necessary to reduce the burden of cardiovascular disease (CVD). Alamandine binds to MrgD and is reported to induce vasodilation via stimulation of endothelial nitric oxide synthase (eNOS), but its role in atherogenic blood vessels is yet to be determined. To determine the vasoactive role of alamandine and its precursor AngA in diseased aorta, New Zealand White rabbits were fed a diet containing 1% methionine + 0.5% cholesterol + 5% peanut oil for 4 weeks (MC, n = 5) or control (n = 6). In abdominal aorta, alamandine (1 μM) was added 30 min before a dose–response curve to angiotensin II or AngA (1 nM–1 μM), and immunohistochemistry was used to identify MrgD receptors and eNOS. The thoracic aorta, renal, carotid and iliac arteries were mounted in organ baths. Rings were precontracted with phenylephrine, then a bolus dose of alamandine (1 μM) was added 10 min before a dose–response curve to acetylcholine (0.01 μM–10 μM). The MrgD receptor was localized to normal and diseased aorta and colocalized with eNOS. In control but not diseased blood vessels, alamandine enhanced acetylcholine-mediated vasodilation in the thoracic aorta and the iliac artery (P < 0.05) and reduced it in the renal artery (P < 0.05). In control abdominal aorta, AngA evoked less desensitization than AngII (P < 0.05) and alamandine reduced AngA-mediated vasoconstriction (P < 0.05). In MC, AngA constriction was markedly reduced vs. control (P < 0.05). The vasoactivity of alamandine and AngA are reduced in atherogenesis. Its role in the prevention of CVD remains to be validated.
Keywords: alamandine, angiotensin A, atherosclerosis, endothelial dysfunction
The renin–angiotensin system (RAS) is a major regulator of cardiovascular function and is believed to be overactivated in cardiovascular disease (CVD). The main effector octapeptide, angiotensin II (AngII) is a powerful constrictor of isolated human and rabbit blood vessels. The success of AngII receptor blockers and inhibitors of AngII production in the clinic in reduction of CVD mortality and morbidity has cemented the role of AngII in the pathophysiology of atherosclerosis and hypertension. Moreover, the breakdown product of AngII, Ang(1-7), is believed to counter the effects of AngII, and plasma levels are increased in patients receiving RAS inhibitors (Volpe et al. 2001). This suggests that the beneficial effects of RAS inhibitors might be due to, at least in part, Ang(1-7). Additionally, Ang(1-7) has been shown to enhance acetylcholine-induced vasodilation in the aorta of a rat model of bile duct ligation (Paizis et al. 2005).
Recently a novel peptide of the RAS was discovered in humans, AngA, which differs from AngII by having an Ala in the first amino acid position rather than Asp (Jankowski et al. 2007). AngA is synthesized by enzymatic decarboxylation of Asp1 of the AngII sequence. AngA is a powerful vasoconstrictor similar to AngII (Coutinho et al. 2013) and acts primarily through the AngII type 1 receptor (Yang et al. 2011). In addition, alamandine, which is formed by the deletion of the phenylalanine residue (Phe8) of AngA, binds to a specific receptor, termed MrgD (Lautner et al. 2013). Alamandine has been recently shown to have vasodilatory properties in the mouse vasculature, suggesting a signalling cascade linked to the stimulation of endothelial nitric oxide synthase (eNOS) (Duran et al. 2010).
There is no evidence in the literature regarding the role of AngA and alamandine in an atherosclerotic model, and whether or not MrgD colocalizes in cells with eNOS. In addition, it is unknown whether there is cross-activity between these peptides. Thus we sought to further understand the role of these peptides on vasoconstriction and vasodilation in the rabbit model of atherosclerosis.
Materials and methods
Male New Zealand White rabbits (n = 6) at 3 months of age received either a normal rabbit chow diet or a diet supplemented with 0.5% cholesterol + 1% methionine + 5% peanut oil for 4 weeks (MC, n = 5). Animals were then sacrificed, and the thoracic and abdominal aorta, carotid, renal and iliac arteries were removed, cleaned of connective tissue and fat, cut into 3-mm rings and immediately placed in organ baths (OB8, Zultek Engineering, Melbourne, Australia) for vasoactivity studies. The abdominal aorta was used for vasoconstrictive studies and immunohistochemistry, whereas all other vessels were used to assess the vasodilative properties of alamandine on phenylephrine preconstriction followed by an acetylcholine dose–response curve. The experiments were carried out according to the National Health and Medical Research Council “Australian Code of Practice for the Care and Use of Animals for Scientific Purposes” (8th Edition, 2013) and approved by the institutional animal ethics committee.
Immunohistochemistry
Segments of abdominal aorta were fixed overnight in 4% paraformaldehyde solution in 1× PBS, pH7.4. All arteries were then processed, embedded in one paraffin block and serially sectioned all in one batch to minimize variability. MrgD (#sc-138439; Santa Cruz Biotechnology, Dallas, TX, USA), eNOS (#610297; BD Bioscience, North Ryde, NSW, Australia), macrophage (RAM-11, #M0633; Dako Australia Pty. Ltd., North Sydney, NSW, Australia) and SMC (HHF-35, M0635; Dako Australia Pty. Ltd) were purchased. All primary antibodies were diluted 1:100. For negative control, 100× peptide was added (for MrgD, #sc-138439-P; Santa Cruz Biotechnology) or isotype-matched IgG (#X0931; Dako Australia Pty. Ltd). Immunohistochemistry was performed as previously described in our laboratory (Zulli et al. 2003, 2006) using the ‘Envision’ commercially available immunohistochemistry kit (#K4007) purchased from Dakocytomation.
Aortic reactivity studies
The baths were filled with Krebs solution and kept at a constant temperature of 37 °C and continuously bubbled with 95% O2 /5% CO2 for 1 h. The rings were then stretched to 2.5 g of tension for aorta and 1 g for all other arteries. After 30 min, rings were restretched to their original tension and allowed to stabilize for 2 h. After this, all rings were subjected to a high-potassium physiological salt solution (124 mM K+) to induce maximal constriction and after the rings reached plateau (approximately 15 min), the rings were repeatedly flushed with Krebs solution until vessels reached baseline and then allowed to rest for 1 h, with Krebs solution changed every 20 min (Zulli et al. 2003).
In the abdominal aorta, Ang(1-7) or alamandine was added to the organ bath (10−6 M), and 20 min later, an AngII or AngA concentration curve was performed (Zulli et al. 2008) (10−9–10−6 M, half log units). In thoracic aorta and all other arteries, rings were precontracted to 50–80% of maximum (as determined by 124 mM K+ response) with phenylephrine (10−6–10−5 M). After plateau (45 min), a bolus dose of alamandine (10−6 M) was added, and 10 min later, rings were subjected to an acetylcholine dose–response curve (10−8–10−5 M).
Statistical analysis
Control rings were compared with MC rings via two-way analysis of variance (anova) using graphpad prism, San Diego, CA, USA followed by a Bonferroni multiple comparison test. Results are presented as mean ± SEM. Significance was taken at P < 0.05 in all cases.
Results
Immunohistochemistry
The MrgD receptor was localized to normal and diseased aorta. In diseased aorta, positive immunoreactivity was identified in the endothelial layer, within atherosclerotic plaques and adventitia. In plaques, MrgD-positive immunoreactivity was clearly identified within cells, which also expressed HHF-35 (smooth muscle cell) but not RAM-11 (macrophages, black arrows) (Figure 1, 1st row). Within the plaque, some cells did not express MrgD R even though the cells expressed RAM-11 and HHF-35 (Figure 1, 2nd row, designated cells A, B, C).
Figure 1.

Immunohistochemical localization of MrgD receptors (MrgD R) clearly shows positive immunostaining within atherosclerotic plaques and the endothelial layer. The first row shows 3-μm serially adjacent sections stained for RAM-11 (Macrophage), MrgD R and HHF-35 (smooth muscle cells). Inserted cropped sections show little or no staining for macrophage/MrgD R +/+ cells (arrows), yet HHF-35/MrgD R +/+ cells were abundant (cropped sections in 2nd row attached by dashed line as well as identified by letters A, B and C). In other plaques, cells expressing eNOS also expressed MrgD R (3rd row, letters D, E and F), and the endothelial layer also showed co-expression of both proteins (cropped sections, 3rd row). Small arterioles in the vaso vasorum showed positive endothelial MrgD R (cropped sections, 4th row), and negative controls also shown (−). All sections were photographed at an original 400× magnification.
In atherosclerotic plaques, MrgD immunopositive staining was also present in cells which expressed positive eNOS immunoreactivity (3rd row, designated cells D, E, F), and the endothelial layer also expressed both proteins (inserted magnified boxes).
The receptor was also localized to the endothelial layer of small adventitial arterioles (4th row, magnified boxes show endothelial layer). Negative control showed no binding [4th row, designated (−)].
Vasoactivity studies
In control blood vessels, the bolus dose of alamandine did not cause direct vasodilation in any artery tested per se, yet it enhanced acetylcholine-mediated vasodilation in the thoracic aorta (Figure 2a, 64 ± 5.4% vs. 79 ± 1.6%, P < 0.05) and the iliac artery (Figure 2b, 79 ± 8% vs. 94 ± 4%, P < 0.05). It did not affect the carotid artery (Figure 2c), but reduced acetylcholine-mediated vasodilation in the renal artery (Figure 2d, 85 ± 6.6% vs. 70 ± 10%, P < 0.05). Interestingly, in the MC group, alamandine failed to have any significant effect on acetylcholine-induced relaxation in the thoracic aorta (Figure 2e), the iliac artery (Figure 2f), the carotid artery (Figure 2g) or the renal artery (Figure 2h). Similarly, there was no evidence of direct vasodilation after the bolus (10−6 M) dose (data not shown).
Figure 2.

In control rings, alamandine enhanced endothelium-dependent relaxation to acetylcholine in the (a) thoracic aorta and (b) iliac artery. Alamandine did not affect the carotid artery (c), but reduced endothelium-dependent relaxation to acetylcholine in the renal artery (d). In rings from animals fed the atherogenic diet (MC), alamandine failed to significantly affect endothelium-dependent relaxation to acetylcholine (e–h) (V = vehicle, Alam. = alamandine). *P < 0.05.
In control abdominal aorta, AngA evoked less desensitization than AngII (Figure 3a, 43 ± 8% vs. 24 ± 2%, P < 0.05). Alamandine reduced AngA-mediated vasoconstriction (Figure 3b, 53 ± 4% vs. 33 ± 4%, P < 0.05), yet alamandine and Ang(1-7) had no effect on AngII-mediated vasoconstriction (Figure 2c).
Figure 3.
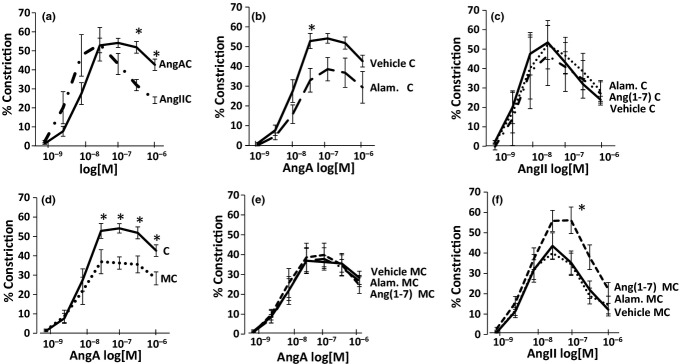
In control rings, AngA evoked less desensitization than AngII (a) and almandine reduced AngA-mediated vasoconstriction (b). Neither alamandine nor Ang(1-7) affected AngII-mediated vasoconstriction (c). In MC, AngA vasoconstriction was reduced compared with control (d), and neither almandine nor Ang(1-7) affected this constriction (e). Interestingly, Ang(1-7) but not alamandine increased AngII-mediated vasoconstriction (f). *P < 0.05.
In the MC group, AngA constriction was markedly reduced vs. control diet (Figure 2d, 28 ± 3.3% vs. 43 ± 8%, P < 0.05), and almandine or Ang(1-7) failed to affect this response (Figure 2e). Ang(1-7) enhanced AngII-mediated vasoconstriction (10−7 M, 56 ± 8% vs. 35 ± 5.7%, P < 0.05), but alamandine did not have this effect (Figure 2f). The effect of AngA was abolished by candesartan (data not shown).
Discussion
The major findings of this study are that in the atherosclerotic rabbit model: (i) the MrdG receptor was localized to cells in atherosclerotic plaques also expressing eNOS, (ii) AngA-mediated vasoconstriction and the enhanced vasodilatory role of alamandine are reduced and that (iii) alamandine and Ang(1-7) have opposing effects on AngII (but not AngA)-mediated vasoconstriction.
In this study, the MrdG receptor was localized mainly in cells which also expressed HHF-35, a marker for smooth muscle cells, and rarely in cells which also expressed RAM-11, a marker for macrophages. This indicates that the MrdG receptor could be involved in the regulation of smooth muscle cell proliferation in atherogenesis. Indeed, the MrdG receptor was also localized in cells which were immunoreactive to eNOS, the enzyme responsible for the production of nitric oxide, a potent inhibitor of smooth muscle cell proliferation (Sarkar et al. 1996). In addition, although the MrdG receptor was rarely localized to cells which also expressed RAM-11, the receptor could regulate the oxidation of low-density lipoprotein via nitric oxide release, thus reducing atherogenesis (Leitinger et al. 1995). Lastly, the receptor and eNOS were localized to the endothelium, indicating a role in the regulation of endothelial function. This role is supported by Lautner et al. (2013) and the vasoactive results presented in this study. Taken together, these results suggest that the MrdG receptor could be involved in the pathogenesis of atherosclerosis by modification of the three key steps in atherogenesis, smooth muscle cell proliferation, low-density lipoprotein oxidation and endothelial function.
To provide further evidence of a vasodilatory role for alamandine in the vascular bed, we incubated phenylephrine-preconstricted thoracic aorta, carotid, renal and iliac arteries with a bolus dose (10−6 M) of alamandine for 10 min, and then proceeded to determine whether this enhanced vasodilatory response to acetylcholine, occurs, as previously shown for Ang(1-7) in diseased rat aorta (Paizis et al. 2005). A dose–response curve to acetylcholine was performed. In the control group, we show enhanced dilation in the thoracic aorta and iliac artery but not in the carotid, and reduced dilation in the renal artery. However, these effects were not observed after 4 weeks of an atherogenic diet, and the effects in the thoracic aorta failed to reach significance. These data clearly point to differential effects in conduit blood vessels and reduced effects in disease. Further investigations in resistance arteries (mesenteric arteries and renal interlobar arteries) are underway.
AngA and AngII are reported to have similar vasoconstrictive effects. For example, in the non-diseased rat model, IV administration of either AngII or AngA had virtually identical effects on mean arterial pressure, which were inhibited by AngII AT1 receptor blockade with losartan (Coutinho et al. 2013). Yet, in the perfused non-diseased rat kidney, AngA was 10× less potent than AngII in increasing kidney perfusion pressure, and this was also inhibited by AngII receptor blockade (Jankowski et al. 2007). The authors also showed using human embryonic kidney cells (HEK293) that this effect is due to reduced intrinsic activity, and not affinity, at the AngII AT1 receptor. We show that, in the normal and diseased rabbit abdominal aorta, AngA shows less desensitization than AngII, but no shift in response curves. This indicates a probable normal affinity for AngII AT1 receptors in this model.
Interestingly, we now show that AngA, but not AngII-mediated vasoconstriction, is markedly reduced in the abdominal aorta of the rabbit fed the atherogenic diet. The reasons for this reduction in constriction by AngA require further investigation, as it could be due to several interrelated mechanisms within the renin–angiotensin system. However, in humans with stage 5 chronic kidney disease, the plasma AngA:AngII ratio was reported to be increased (Jankowski et al. 2007), indicating that in disease changes in circulating AngA:AngII can occur, which could affect vasoconstriction in the vascular tree. Taken together, it is intriguing to speculate that circulating AngA is increased in disease to compensate for the reduction in AngA-mediated vasoconstriction.
The heptapeptide Ang(1-7) has been shown to impair AngII-mediated vasoconstriction in human arteries (Roks et al. 1999) as well as inhibition of AngII-mediated signalling in the rat model (Tao et al. 2014). Thus, we wanted to determine whether alamandine or Ang(1-7) also affected AngA- or AngII-mediated vasoconstriction. We found that in control aorta, both alamandine and Ang(1-7) inhibited AngA (but not AngII)- mediated vasoconstriction, yet the heptapeptides have similar effects. This is clearly a species variation as Ang(1-7) has been reported to inhibit AngII effects in human arteries (Roks et al. 1999) and rat cell lines (Freeman et al. 1996).
Unexpectedly, in diseased aorta, the heptapeptides had no effect on AngA-mediated vasoconstriction, but Ang(1-7) (but not alamandine) enhanced AngII-mediated vasoconstriction. These results provide a clear distinction between Ang(1-7) and alamandine on AngII (but not AngA)-mediated vasoconstriction in diseased blood vessels. As AngA and alamandine are believed to be produced by aspartate decarboxylation of AngII and Ang(1-7), respectively (Jankowski et al. 2007; Lautner et al. 2013), it is intriguing to speculate that enhancing the activity of aspartate decarboxylation to remove Ang(1-7) from the local milieu could minimize AngII-mediated vasoconstriction/AngIIA-mediated vasoconstriction during disease, and it could suggest an alternative mechanisms for the benefits observed with chronic in vivo Ang(1-7) treatment (Iusuf et al. 2008; Katovich et al. 2008). This view is supported by a most recent study by Lautner et al. (2013), who clearly showed similar protective effects of in vivo alamandine administration. Whether or not the in vivo benefits of Ang(1-7) treatment are due to its conversion to alamandine will require further investigation.
In conclusion, we report that (i) the MrdG receptor is present in atherosclerotic plaques and (ii) alamandine and AngA have a vasoactive role in the vasculature of the rabbit, yet its function is diminished in by an atherogenic diet and (iii) the effects are not equal to that evoked by AngII and Ang(1-7). Whether or not alamandine can be exploited as a novel therapeutic target to treat CVD requires further study.
References
- Coutinho DC, Foureaux G, Rodrigues KD, et al. Cardiovascular effects of angiotensin A: a novel peptide of the renin-angiotensin system. J. Renin Angiotensin Aldosterone Syst. 2013 doi: 10.1177/1470320312474856. doi: 10.1177/1470320312474856. [DOI] [PubMed] [Google Scholar]
- Duran WN, Breslin JW, Sanchez FA. The NO cascade, eNOS location, and microvascular permeability. Cardiovasc. Res. 2010;87:254–261. doi: 10.1093/cvr/cvq139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman EJ, Chisolm GM, Ferrario CM, Tallant EA. Angiotensin-(1-7) inhibits vascular smooth muscle cell growth. Hypertension. 1996;28:104–108. doi: 10.1161/01.hyp.28.1.104. [DOI] [PubMed] [Google Scholar]
- Iusuf D, Henning RH, van Gilst WH, Roks AJ. Angiotensin-(1-7): pharmacological properties and pharmacotherapeutic perspectives. Eur. J. Pharmacol. 2008;585:303–312. doi: 10.1016/j.ejphar.2008.02.090. [DOI] [PubMed] [Google Scholar]
- Jankowski V, Vanholder R, van der Giet M, et al. Mass-spectrometric identification of a novel angiotensin peptide in human plasma. Arterioscler. Thromb. Vasc. Biol. 2007;27:297–302. doi: 10.1161/01.ATV.0000253889.09765.5f. [DOI] [PubMed] [Google Scholar]
- Katovich MJ, Grobe JL, Raizada MK. Angiotensin-(1-7) as an antihypertensive, antifibrotic target. Curr. Hypertens. Rep. 2008;10:227–232. doi: 10.1007/s11906-008-0043-9. [DOI] [PubMed] [Google Scholar]
- Lautner RQ, Villela DC, Fraga-Silva RA, et al. Discovery and characterization of alamandine: a novel component of the renin-angiotensin system. Circ. Res. 2013;112:1104–1111. doi: 10.1161/CIRCRESAHA.113.301077. [DOI] [PubMed] [Google Scholar]
- Leitinger N, Oguogho A, Rodrigues M, Sinzinger H. The effect of NO/EDRF and monocytes/macrophages on LDL-oxidation. J. Physiol. Pharmacol. 1995;46:385–408. [PubMed] [Google Scholar]
- Paizis G, Tikellis C, Cooper ME, et al. Chronic liver injury in rats and humans upregulates the novel enzyme angiotensin converting enzyme 2. Gut. 2005;54:1790–1796. doi: 10.1136/gut.2004.062398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roks AJ, van Geel PP, Pinto YM, et al. Angiotensin-(1-7) is a modulator of the human renin-angiotensin system. Hypertension. 1999;34:296–301. doi: 10.1161/01.hyp.34.2.296. [DOI] [PubMed] [Google Scholar]
- Sarkar R, Meinberg EG, Stanley JC, Gordon D, Webb RC. Nitric oxide reversibly inhibits the migration of cultured vascular smooth muscle cells. Circ. Res. 1996;78:225–230. doi: 10.1161/01.res.78.2.225. [DOI] [PubMed] [Google Scholar]
- Tao X, Fan J, Kao G, et al. Angiotensin-(1-7) attenuates angiotensin II-induced signaling associated with activation of a tyrosine phosphatase in Sprague-Dawley rats cardiac fibroblasts. Biol. Cell. 2014;106:1–11. doi: 10.1111/boc.201400015. [DOI] [PubMed] [Google Scholar]
- Volpe M, Azizi M, Danser AH, Nguyen G, Ruilope LM. Twisting arms to angiotensin receptor blockers/antagonists: the turn of cancer. Eur. Heart J. 2001;32:19–22. doi: 10.1093/eurheartj/ehq382. [DOI] [PubMed] [Google Scholar]
- Yang R, Smolders I, Vanderheyden P, et al. Pressor and renal hemodynamic effects of the novel angiotensin A peptide are angiotensin II type 1A receptor dependent. Hypertension. 2011;57:956–964. doi: 10.1161/HYPERTENSIONAHA.110.161836. [DOI] [PubMed] [Google Scholar]
- Zulli A, Widdop RE, Hare DL, Buxton BF, Black MJ. High methionine and cholesterol diet abolishes endothelial relaxation. Arterioscler. Thromb. Vasc. Biol. 2003;23:1358–1363. doi: 10.1161/01.ATV.0000080686.39871.54. [DOI] [PubMed] [Google Scholar]
- Zulli A, Buxton BF, Black MJ, Ming Z, Cameron A, Hare DL. The immunoquantification of caveolin-1 and eNOS in human and rabbit diseased blood vessels. J. Histochem. Cytochem. 2006;54:151–159. doi: 10.1369/jhc.5A6677.2005. [DOI] [PubMed] [Google Scholar]
- Zulli A, Ye B, Wookey PJ, Buxton BF, Hare DL. Calcitonin gene-related peptide inhibits angiotensin II-mediated vasoconstriction in human radial arteries: role of the Kir channel. J. Thorac. Cardiovasc. Surg. 2008;136:370–375. doi: 10.1016/j.jtcvs.2007.12.064. [DOI] [PubMed] [Google Scholar]