

Abstract
Tumor expression of certain chemokine receptors is associated with resistance to apoptosis, migration, invasiveness and metastasis. Because CCR9 chemokine receptor expression is very restricted in healthy tissue, whereas it is present in tumors of distinct origins including leukemias, melanomas, prostate and ovary carcinomas, it can be considered a suitable candidate for target-directed therapy. Here, we report the generation and characterization of 91R, a mouse anti-human CCR9 IgG2b monoclonal antibody that recognizes an epitope within the CCR9 N-terminal domain. This antibody inhibits the growth of subcutaneous xenografts from human acute T lymphoblastic leukemia MOLT-4 cells in immunodeficient Rag2−/− mice. Tumor size in 91R-treated mice was reduced by 85% compared with isotype-matched antibody-treated controls. Tumor reduction in 91R-treated mice was concomitant with an increase in the apoptotic cell fraction and tumor necrotic areas, as well as a decrease in the fraction of proliferating cells and in tumor vascularization. In the presence of complement or murine natural killer cells, 91R promoted in vitro lysis of MOLT-4 leukemia cells, indicating that this antibody might eliminate tumor cells via complement- and cell-dependent cytotoxicity. The results show the potential of the 91R monoclonal antibody as a therapeutic agent for treatment of CCR9-expressing tumors.
Keywords: human acute T lymphoblastic leukemia, xenograft, therapeutic antibody, chemokine receptor, targeted cancer therapy, complement-mediated cytotoxicity, cell-mediated cytotoxicity
Introduction
Chemokines and their receptors have an essential role in organogenesis and lymphocyte trafficking, in both homeostatic and inflammatory conditions.1 There is a strong association between aberrant tumor cell expression of chemokine receptors such as CXCR4 or CCR7 and cancer progression, organ-selective metastasis, and poor prognosis.2-4 Although data are scarce for the chemokine receptor 9 (CCR9), its expression on tumor cells correlates with the tumor ability to metastasize in the small intestine.5-7
CCR9 expression is restricted to lymphoid cells in the thymus,8-11 infiltrating cells in small bowel,12 a small subset of circulating memory T lymphocytes (CCR9+α4β7hi),13 IgA-secreting plasma cells1 and plasmacytoid dendritic cells.14 The only known CCR9 ligand, the chemokine TECK (thymus-expressed chemokine, CCL25),8,15 is secreted by epithelial and dendritic cells from the thymus10,16 and the small intestinal crypt epithelium.12 The CCL25-CCR9 interaction is a critical regulator of thymocyte migration within the thymus and of cell homing to the intestinal tract.13
CCR9 overexpression in acute and chronic T cell lineage leukemia is linked to disease aggressiveness.17 Aberrant CCR9 expression in prostate cancer,18 breast cancer19 and melanomas7 is correlated with in vitro invasiveness in response to CCL25. CCR9 provides competitive advantages to tumor cells. CCL25 engagement enhances cell survival and resistance to apoptosis via the phosphatidylinositide 3-kinase (PI3K)/Akt pathway in prostate, breast and ovarian carcinomas;19-21 in leukemia cells, it activates the JNK1 anti-apoptotic pathway22 and participates in Notch1-mediated cell proliferation.23
Specific tools to inhibit growth of human CCR9-positive tumors in xenogenic models are limited to toxin-coupled ligand (CCL25-PE38 fusion protein)24 or ligand-specific antibodies, alone or combined with the cytotoxic agent etoposide.20 These strategies eliminate tumor cells by targeting the CCL25-CCR9 interaction; although results are limited, they provide evidence that CCR9 is a potential target for cancer immunotherapy.
Clinical treatments able to discriminate between normal and tumor cells have safety advantages over non-specific cytotoxic agents, and their use for cancer treatment is in constant growth.25,26 Examples of such specific agents include therapeutic monoclonal antibodies (mAbs) that recognize tumor-associated cell surface antigens or other molecules necessary for cancer cell survival and proliferation, such as human epidermal growth factor receptor-2 (HER-2) and epidermal growth factor receptor (EGFR).27
Here, we report the generation and characterization of 91R, an anti-CCR9 mAb able to selectively inhibit growth of human acute T lymphoblastic leukemia (T-ALL) cells in vivo transplanted into immunodeficient Rag2−/− mice. This antibody has therapeutic potential for the targeted elimination of CCR9+ tumor cells, and could be used alone or in combination with other therapies.
Results
91R mAb specifically recognizes the human chemokine receptor CCR9
The murine anti-hCCR9 mAb (IgG2b) was generated after gene gun immunization with the full-length hCCR9 coding sequence inserted in a eukaryotic expression vector. Specific binding was assessed by flow cytometry on HEK293 cells stably expressing hCCR9 or HEK293 cells stably transfected with the empty vector or with molecules closely related to CCR9. Although human and mouse CCR9 share 86% amino acid sequence identity, the 91R mAb only recognized cells expressing human CCR9 (Fig. 1A); we observed no appreciable binding to cells expressing murine CCR9. 91R did not cross-react with stable HEK293 transfectants expressing hCCR4, hCCR5, hCCR6 or hCCR8 chemokine receptors (Fig. 1A), which have 33–39% identity with hCCR9, further demonstrating 91R specificity.

Figure 1. 91R mAb is specific for human chemokine receptor CCR9. (A) HEK293 cells stably transfected with hCCR9, mCCR9, hCCR4, hCCR5, hCCR6, hCCR8 (open histograms) or the empty pCIneo vector (filled histograms) were stained with 91R mAb and analyzed by flow cytometry. (B) Human leukemia MOLT-4 and Jurkat cells were stained with anti-human CCR9 mAb 91R and 112509 (open histograms) or isotype-matched control mAbs (filled histograms) and analyzed by flow cytometry. (C) Representative flow cytometry analysis of MOLT-4 staining with different doses (0.1–10 μg/ml) of 91R (filled histograms), 112509 (open histograms), or isotype-matched mAb (gray lines) (n = 5). (D) Flow cytometry analysis of human thymocytes using anti-CD4, -CD8 and –CCR9 91R antibodies. Percentages of positive cells in gates of the CD4/CD8 plot are indicated; hCCR9 expression is shown for each subpopulation. (E) Flow cytometry shows a FS/SS dotplot for total human peripheral blood cells and 91R/anti-CD3 staining in the lymphocyte gate. (F) Representative western blot of membrane-enriched fractions of hCCR9- or pCIneo-transfected HEK293 cells, MOLT-4, and Jurkat cells incubated with 91R; the same membrane was probed with anti-CD71 Ab as loading control (n = 3).
91R recognized endogenous human CCR9 on the MOLT-4 T-ALL cell line, but did not stain the negative control Jurkat T-ALL cells (Fig. 1B).13 We also observed a clear reduction in 91R binding to MOLT-4 cells in CCR9-silenced cells (Fig. S1). The staining patterns of 91R and a commercial anti-hCCR9 (112509 mAb) were compared (Fig. 1B); at the same mAb concentration (10 μg/ml), 91R showed 13-fold higher mean fluorescence intensity than 112509 mAb on MOLT-4 cells (MFI; 49.7 vs. 3.8). At a suboptimal concentration (0.1 μg/ml), 91R stained 100% of MOLT-4 cells, with a MFI of 10.9, higher than the signal at saturating concentrations of 112509 (Fig. 1C); this indicates higher 91R mAb affinity for CCR9 or recognition of a more accessible epitope. 91R also detected endogenous CCR9 on human primary cells, since it stained thymocytes at all stages of T cell maturation, with the highest levels in CD4+CD8+ double positive cells (Fig. 1D). In addition, 2–3% of peripheral blood lymphocytes, mainly CD3+ cells, were stained by 91R (Fig. 1E).
91R specificity was further assessed in western blot using membrane extracts from hCCR9 and mock transfectants; 91R specifically recognized a 43 kDa band, in accordance with the estimated molecular weight of hCCR9 (369 amino acids; estimated MW 42,016 Da, ExPASy Server). A 47 kDa band was similarly detected in MOLT-4, but not in Jurkat cell samples (Fig. 1F). The apparent change in molecular weight between hCCR9 bands in HEK293 and MOLT-4 cells might be due to glycosylation differences.
The hCCR9 N-terminal domain is necessary for 91R mAb epitope recognition
CCR9 is comprised of seven transmembrane domains, an extracellular N-terminal (Nt), three intracellular, three extracellular and an intracellular C-terminal domain (Fig. 2A). Extracellular regions of human and mouse CCR9 differ in only 33 amino acid residues. To map the hCCR9 domain recognized by 91R, we generated an expression vector in which the hCCR9 Nt was replaced by the murine sequence (mNt/hCCR9) (Fig. 2A) and expressed in HEK293 cells. Flow cytometry analyses showed that whereas the K629 anti-mouse CCR9 N-terminal domain Ab recognized the chimeric receptor, 91R did not (Fig. 2A), indicating that the hCCR9 Nt domain was required for 91R epitope recognition.

Figure 2. 91R mAb recognizes the human CCR9 N-terminal domain. (A) Diagram of human and mouse CCR9 and the chimeric CCR9 bearing the human CCR9 sequence with the N-terminal domain (Nt) replaced by the murine sequence (mNt/hCCR9); flow cytometry analyses with 91R mAb (anti-hCCR9; open histograms) and isotype-matched control mAb (filled histograms), rabbit polyclonal K629 (anti-mCCR9; open histograms) and rabbit control Ab (filled histograms). (B) Membrane-enriched lysates from pCIneo-, hCCR9-HEK and MOLT-4 cells were used for western blot with 91R; anti-CD71 Ab was used as loading control. Where indicated, cell lysates were PNGase-treated to remove N-glycosylated residues. A representative experiment is shown (n = 2).
As the hCCR9 Nt domain has a putative N-glycosylation site at Asn32, we tested whether 91R recognized a glycosidic epitope or the peptide backbone. MOLT-4 and hCCR9-HEK293 cell lysates were treated with PNGase, followed by electrophoresis and western blot with 91R. In CCR9-transfected HEK293 cell extracts, 91R detected a 43 kDa band in untreated cells and a 39 kDa band in PNGase-treated cell lysates. In untreated MOLT-4 cell extracts, we detected a predominant protein band of 47 kDa and a minor band of 43 kDa; PNGase-treated lysates had a single 39 kDa band (Fig. 2B). These results confirm hCCR9 as an N-glycosylated protein and that this glycosyl moiety is not a part of the epitope recognized by 91R, suggesting that this mAb binds the hCCR9 peptide backbone.
The CCR9 ligand CCL25 partially blocks 91R binding to MOLT-4 cells
To study whether CCL25 affects CCR9 recognition by 91R, MOLT-4 cells were pre-incubated alone or with hCCL25, at 4 °C in the presence of sodium azide to inhibit receptor internalization, followed by incubation with 91R and flow cytometry analysis. 91R binding was partially inhibited by CCL25, but not by the control chemokine CXCL12 (Fig. 3). This result suggests that the chemokine binding site on CCR9 and the epitope recognized by 91R could overlap partially or are in close proximity. Alternatively, CCL25 binding to CCR9 could affect the conformation of the epitope recognized by 91R or inhibit its binding due to steric hindrance.
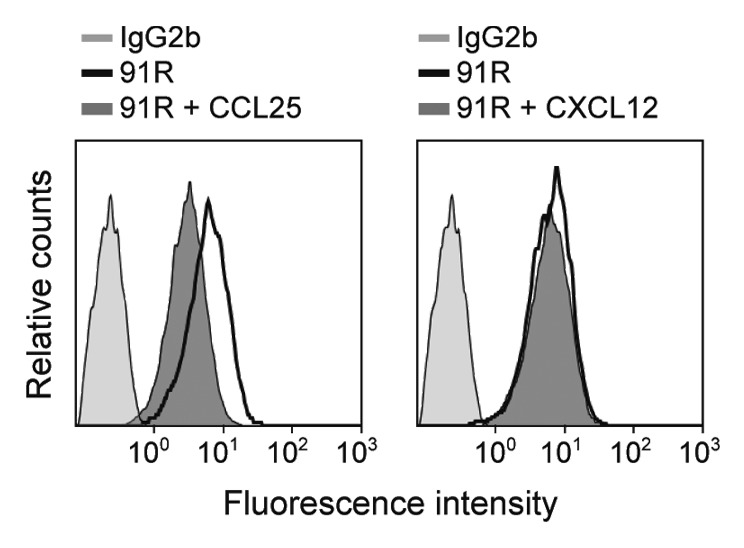
Figure 3. Human CCL25 partially competes with 91R mAb for binding to MOLT-4 cells. Representative flow cytometry analysis of human MOLT-4 cells, preincubated alone or with 10 μg/ml hCCL25 or hCXCL12 (40 min, 4 °C), stained with 91R or isotype-matched mAb (n = 3).
91R mAb inhibits in vivo growth of human tumors in xenografts
CCR9+ MOLT-4 cells, which grow as xenografts when implanted in immunodeficient mice, were used to assess the anti-tumor potential of 91R. In the first experiment, two groups of mice were inoculated subcutaneously (s.c.) with MOLT-4 cells in both dorsal flanks (d0); they then received four weekly intraperitoneal (i.p.) injections of 91R or an isotype-matched mAb (P-020, IgG2b), starting the day after cell implant (Fig. 4A). The size of developing tumors was measured until d56, when mice were sacrificed. Significantly smaller tumors were apparent in 91R-treated mice from d39 (P = 0.0024; Figure 4B). At d56, tumors were removed and weighed; total tumor burden, measured as the mean of tumor weights for each group, was reduced by 84 ± 18% in the 91R-treated group compared with controls (tumor burden per mouse 63.3 ± 30.3 mg vs 397 ± 65 mg; P = 0.0009; Figure 4C). The largest individual tumor from 91R-treated mice was smaller than any of the tumors from controls. All control mice developed tumors, whereas two 91R-treated mice were tumor-free (n = 6 mice/group) (Fig. 4D).

Figure 4. Leukemia xenograft growth is reduced in mice treated with 91R mAb. For xenograft analyses, MOLT-4 cells were inoculated s.c. in Rag2−/− mice on day 0 (d0). Experimental groups received four i.p. doses of 91R or irrelevant IgG2b mAb (first and second, 4 mg/kg; third and fourth, 2 mg/kg). Tumor growth was measured with a caliper every three days. After mice were sacrificed, tumors were removed and weighed. (A) Antibody administration schedule on days 1, 7, 14 and 21 for mice bearing tumor cells injected in each flank. (B) Tumor growth kinetics. Tumor volume was measured at times indicated and calculated as V = [axial diameter length, mm] x [(rotational diameter, mm)2/2] (6 mice/group). (C) Tumor weight (%) relative to IgG2b treatment on d56. Mean ± SEM (n = 6 mice/group). (D) Images of tumors from IgG2b- and 91R-treated mice at the time of sacrifice (day 56). Bar = 1 cm. (E) Antibody administration schedule on days 7, 14, 21, and 28 in mice injected only in one flank. (F) Tumor volume was calculated as in B (10 mice/group). (G) Percentage of tumor weight relative to IgG2b treatment on d69. Results show mean ± SEM (n = 10 mice/group). (H) Images of tumors from IgG2b- and 91R-treated mice at the time of sacrifice (day 69). Bar = 1 cm. *** P < 0.001, ** P < 0.01, * P < 0.05.
To test the ability of the 91R mAb to inhibit tumor growth in more stringent conditions, we initiated treatment at 7 d post-MOLT-4 cell implant, with four doses at weekly intervals (Fig. 4E). For these experiments, MOLT-4 cells were injected into one flank only and tumor size measured until d69, when mice were sacrificed. Significant differences in tumor size between the two mouse groups were apparent by d48 (P = 0.012; Figure 4F), and tumor burden data showed a 64 ± 29% reduction in mice administered 91R compared with control-treated mice (163 ± 56 mg vs 451 ± 117 mg; P = 0.039; Figure 4G). In this experiment, two control mAb- and four 91R-treated mice were tumor-free, and the size of the largest tumor from 91R-treated mice was comparable to the smallest tumor from controls (Fig. 4H).
To evaluate tumor growth at early stages when direct caliper measurement was not possible, we injected MOLT-4 cells expressing luciferase (MOLT-4-luc) into the dorsal flanks of Rag2−/− mice. To determine the effect of reducing dose number and antibody amount, we administered 91R and control antibodies on d1 (4 mg/kg) and d6 (2 mg/kg) (Fig. 5A). Implanted tumors were monitored by luminescence imaging (Fig. 5B), and mice were sacrificed on d62. Luminescence analyses showed tumor growth from d2, which was significantly inhibited in 91R-treated mice from d12 (P = 0.032; Figure 5B, C). 91R treatment resulted in a total reduction in tumor burden of 85 ± 11% relative to controls (Fig. 5D). Three of the seven 91R-treated mice were tumor-free, and tumors from the remaining four mice were smaller than those of controls, as determined by relative luminescence (Fig. 5C) and by weight (223 ± 103 mg vs 1,478 ± 262 mg; P = 0.001; Figure 5E). These data support a role for 91R in blocking the in vivo xenograft progression of acute leukemia tumor growth.

Figure 5. Short-term kinetics of 91R-induced reduction of leukemia xenograft growth. (A) Treatment schedule using luminescent MOLT-4 cells (MOLT-4-luc) inoculated s.c. into each flank of Rag2−/− mice on d0. Experimental groups received i.p. inoculations of 91R or control IgG2b mAb on d1 (4 mg/kg) and d6 (2 mg/kg). Luminescence imaging was analyzed from days 1 to 28; mice were sacrificed on d62 and tumors removed. (B) Images of a representative mouse from each group at indicated times post-cell inoculation. (C) Tumor growth kinetics after tumor implant. Relative bioluminescence units are shown as mean ± SEM (D) Percentage of tumor weight relative to IgG2b treatment at d62. Results show mean ± SEM (E) Tumor weights per mouse; data show mean ± SEM. C-E, n = 7 mice/group. *** P < 0.001, ** P < 0.01, * P < 0.05.
91R-treated tumors show increased necrosis and apoptosis, and reduced angiogenesis and cell proliferation
We examined the effect of 91R treatment on MOLT-4 tumors by histochemical analysis. Sections from tumor xenografts treated with 91R or control mAb and collected at necropsy were hematoxylin/eosin-stained and the necrotic area relative to total area was calculated for each tumor section; the necrotic region was defined as that devoid of cells and surrounded by areas with dense accumulation of purple-stained nuclei (Fig. 6A). Tumors were classified into three categories, based on the extent of necrotic areas: low (< 1%), medium (1–30%) and high (> 30%). High necrosis levels were detected only in 91R-treated mice (40% of tumors); medium levels were observed in 50% of 91R-treated and 20% of control mouse tumors. Differences in necrotic area distribution for each antibody treatment were significant (P < 0.0001; Figure 6B).

Figure 6. 91R promotes apoptosis and necrosis and reduces cell proliferation and angiogenesis in tumor xenografts. (A-D) Histological analysis of xenografted MOLT-4 tumors (n = 5 mice/group). (A) Hematoxylin/eosin-stained sections from xenografted MOLT-4 tumors treated with 91R or control IgG2b mAb; bar = 2 mm. Right, images at higher magnification; bar = 25 μm. (B) Graph shows percentage of tumors classified by necrotic stage (< 1%, 1–30% and > 30%). Chi-square test, ***P < 0.0001. (C) Apoptosis level in tumors was analyzed by TUNEL assays. Proliferation levels were determined by PCNA immunostaining. Blood vessels were detected by CD31 staining. Tissue sections were DAPI-counterstained. Bar = 50 μm. (D) Quantitative analyses of TUNEL- and PCNA-positive nuclei and vessels per optical field. Mann-Whitney test, *** P < 0. 001, ** P < 0. 01, * P < 0.05.
TUNEL assays were used to determine degree of apoptosis, which precedes cell clearance and could lead to necrotic acellular areas. Compared with controls, 91R-treated tumors showed a significant increase in apoptotic cell density (1.93-fold; P < 0.0001; Figure 6C, D left). Staining of paraffin-embedded 91R-treated tumor sections with anti-PCNA (proliferating cell nuclear antigen) mAb showed a significant decrease in the fraction of proliferating cells compared with control-treated tumors (40%; P < 0.0001; Figure 6C, D center). Tumor growth is also associated with the extent of intratumor neovascularization.28-30 Evaluation of tumor angiogenesis by immunohistochemical detection of the endothelial marker CD31 showed a reduction in microvessel density in 91R-treated compared with control-treated tumors (50.7%; P < 0.0001; Figure 6C, D right).
These data indicate that 91R interferes with tumor growth by increasing apoptotic cell death and necrosis levels, and reducing cell proliferation and intratumor microvessel density.
91R mAb mediates complement-dependent cytotoxicity
Since complement-dependent cytotoxicity (CDC) is one of the main in vivo mechanisms for tumor cell elimination by therapeutic antibodies,25,26 we tested the in vitro ability of 91R to induce lysis of MOLT-4 leukemia cells by complement fixation. MOLT-4 cells were pre-incubated with 91R, 112509 or appropriate isotype-matched mAb, followed by complement addition (1 h, 37 °C). Specific cell death was evaluated by flow cytometry analyses of 7-AAD incorporation. 91R (40 μg/ml) promoted complement-specific lysis of MOLT-4 cells (49 ± 2%; P < 0.0001; Figure 7A) at a level higher than 112509 mAb (18 ± 2%; P = 0.03; Figure 7A). 91R-mediated CDC was antibody concentration-dependent, as demonstrated in dose-response experiments that showed a sigmoidal response. The response was detectable from 0.4 μg/ml 91R, and showed 40% cell lysis with 4 μg/ml mAb (Fig. 7B); it was also dependent on time (71.7 ± 4.4% lysis after 5 h; Figure 7C) and on complement concentration (Fig. 7D). These results suggest that CDC might be one of the mechanisms by which 91R reduces MOLT-4 tumor xenografts in mice.

Figure 7. 91R mAb promotes in vitro complement-dependent cytotoxicity in human leukemic MOLT-4 cells. MOLT-4 cells were opsonized with 91R or isotype-matched mAb (40 μg/ml, 30 min, 37 °C), washed, and incubated (1 h) with 25% active (37 °C) or inactive (56 °C) baby rabbit complement (BRC); cell viability was evaluated in a flow cytometer by 7-AAD staining. (A) Specific complement lysis in the absence of antibody or with 91R, 112509, or isotype-matched mAb (IgG2a or IgG2b). Each condition was analyzed in triplicate. Data show mean ± SEM for four independent experiments. (B) Dose-response curve for specific complement lysis using 91R and a control IgG2b mAb at indicated concentrations. Data show mean ± SEM for one representative experiment of four. (C) Effect of time exposure to BRC. Data show mean ± SEM for triplicates from one representative experiment of two. (D) Specific complement lysis in a dose response curve for BRC. Data show percent mean ± SEM for triplicates from one representative experiment of two. *** P < 0.001, ** P < 0.01, * P < 0.05.
91R mediates antibody-dependent cytotoxicity by natural killer cells
Antibody-dependent cell-mediated cytotoxicity (ADCC) is an important anti-tumor mechanism of many therapeutic mAbs such as trastuzumab and rituximab.31 Fc receptors on the natural killer (NK) cell surface allow killing of antibody-coated target cells.32 NK cell cytolytic function was measured by the percentage of killing of MOLT-4 cells pre-coated with anti-hCCR9 91R mAb, 112509 mAb, control antibody or sera from MOLT-4-immunized mice (M4). Primary NK cells from BALB/c mice spleens were activated with IL-2-supplemented medium for 6–7 d. Target cancer cells labeled with a green fluorescent dye (CFSE) and coated with the indicated antibody were incubated with murine NK cells (4 h, 37 °C). We then evaluated 7-AAD incorporation by green cells to determine specific NK-mediated MOLT-4 cell death.
The 91R mAb promoted 57.73 ± 1.1% specific cell lysis (20 μg/ml; Figure 8A), whereas 112509 antibody promoted 20.57 ± 0.8% (P < 0.0001), and the M4 pool of sera raised against MOLT-4 cells (used as a MOLT-4-specific inducer of NK cell cytotoxicity) promoted 91.13 ± 1.1% specific lysis. The 91R mAb concentration needed for a detectable response was 0.004 μg/ml, and 0.4 μg/ml 91R still showed 45.89% specific cell lysis (Fig. 8B). Our results suggest a role for NK cells in the in vivo reduction of tumor growth observed in the xenograft models.
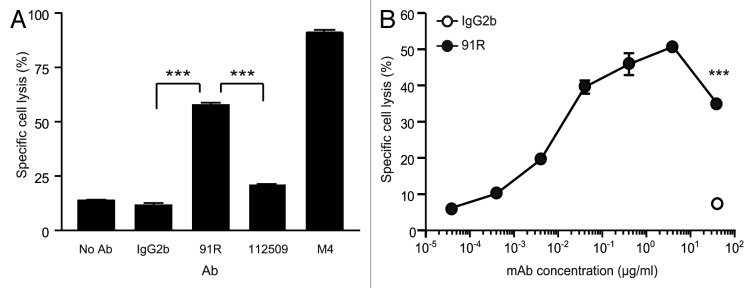
Figure 8. NK cell-mediated cytotoxicity promoted by 91R mAb in human MOLT-4 leukemia cells. (A) Specific NK-dependent cytotoxicity mediated by 91R, 112509, negative control mAb or positive control M4 serum. NK cells were isolated from BALB/c spleens and cultured for 6–7 d in medium containing IL-2. CFSE-labeled MOLT-4 target cells were preincubated with 91R, 112509 or control mAb (20 μg/ml), or M4 pooled sera (1:1000) (30 min, 37 °C). NK cells and labeled target cells were then co-cultured at a 20:1 ratio (4 h, 37 °C). Specific lysis was determined by staining dead cells with 7-AAD and analyzing the number of 7-AAD+ green cells by flow cytometry. Each condition was analyzed in triplicate. Data show mean ± SEM (n = 5 independent experiments). (B) Dose-response curve for specific complement lysis using 91R and a control IgG2b mAb at indicated concentrations. Data show mean ± SEM for duplicates from one representative experiment of four. *** P < 0.001, ** P < 0.01, * P < 0.05.
Discussion
Tumor cell overexpression of certain homeostatic chemokine receptors is linked to cancer progression, metastasis and poor prognosis.2 Aberrant expression of the chemokine receptor CCR9 is detected in some solid tumors.21,33,34 CCR9 is also associated with leukemia aggressiveness17 and mediates organ-selective metastasis of melanoma to small intestine,5-7 suggesting its potential as a target for cancer treatment. We report here the generation and characterization of a specific anti-human CCR9 mAb (91R) that inhibits tumor growth in an in vivo xenograft model of human acute lymphoblastic leukemia.
The 91R antibody was raised using the complete CCR9 receptor as immunogen. The antibody recognizes neither the murine ortholog, which shares 86% identity with hCCR9,8 nor four related human chemokine receptors. 91R mAb detects CCR9 in cell types that express this receptor endogenously, such as human thymocytes8 and human T-ALL cells.8,13 Since even very low concentrations of this mAb stain MOLT-4 cells intensely, 91R appears to have high affinity for CCR9 or to recognize a well-exposed CCR9 epitope. 91R identified an epitope in the hCCR9 N-terminal domain, as shown using chimeric human/mouse constructs. CCL25 partially competed for 91R binding to hCCR9, suggesting that this ligand and the mAb recognize receptor regions that overlap partially or are in close proximity. The results are consistent with involvement of the receptor N-terminal domain in chemokine binding.35,36
Several authors suggested that N-terminal domain-specific antibodies and small molecular weight antagonists for chemokines have potential as therapeutic tools;35 we therefore tested whether 91R inhibits in vivo growth of CCR9+ human T-ALL cells when implanted in immunodeficient Rag2−/− mice. Xenograft experiments showed potent 91R anti-tumor activity in vivo; 91R treatment initiated one day after tumor cell inoculation reduced tumor burden by 85% at the end of the experiment. 91R had a strong inhibitory effect, even when mAb treatment was initiated 7 d post-MOLT-4 cell inoculation or when mice received fewer doses of 91R. Using bioluminescent MOLT-4 cells, we detected very early 91R anti-tumor effects, which were significant by d12 post-MOLT-4 cell injection. These data highlight the potent anti-tumor activity of 91R in all conditions tested. This activity was systemic, as dorsal subcutaneous tumors responded to i.p. administration of the antibody. 91R treatment-induced xenograft tumor reduction was concomitant with an increase in apoptotic cell number and tumor necrotic area, as well as with a decrease in the proliferative cell fraction and in tumor vascularization, as determined by expression of the angiogenic marker CD31.28-30
The anti-tumor effect of 91R might be due to initiation of a complement- or effector cell-mediated response, as shown for most anti-tumor therapeutic antibodies.25 In vitro results for complement- and NK-mediated antibody-dependent MOLT-4 cell lysis by 91R suggest that this mAb promotes its in vivo anti-tumor effect by a combination of both mechanisms; this would be compatible with the fact that although Rag2−/− mice lack T and B lymphocytes, they have an intact innate immune system and complement pathways.37 We cannot rule out a role for other effector mechanisms such as direct induction of apoptosis38 or the disruption of proliferative signals;20,21,39 studies are currently under way to determine whether the 91R mAb alters CCR9 signaling. Therapeutic agents that attack a tumor in different ways increase the possibility of a response, which would render the drug more effective, allowing the use of lower mAb doses and reducing the likelihood of adverse secondary effects. This is the case with anti-CD20 rituximab, which kills B cells by several mechanisms, including CDC and ADCC.40
The use of anti-CCR9 mAb as a tool to immunotarget CCL25-CCR9 interactions in CCR9+ tumors could have several advantages over other strategies such as the use of anti-CCL25 mAb20 or CCL25-PE38 fusion protein.24 A cell surface-restricted receptor molecule such as CCR9 is targeted more efficiently than its soluble ligand (in this case CCL25) by equivalent amounts of a similar-affinity mAb. In addition, anti-receptor mAb strategies can use the potential of the immune system to kill target cells (CDC, ADCC); moreover, these strategies combine high specificity and reduced toxicity with the advantage of long mAb half-life in the blood stream. The systemic, long-term anti-tumor activity of 91R also makes it potentially useful for treatment and imaging diagnosis of metastatic cancers.
Current anticancer research endeavors to develop more effective therapies with improved discrimination between tumor and normal cells. A direct link between the expression level of the targeted molecule on the tumor cell and more efficient cell killing has been established for B cell leukemias.41,42 Given the elevated CCR9 levels on most human T-ALL cells,17,22 similar to those on MOLT-4 cells, CCR9+ tumor cells might be targeted efficiently. Since only a small number of lymphocytes express CCR9, including memory T cells that infiltrate the intestinal mucosa and some circulating T cells (< 3%);13 bystander cytotoxic effects on healthy cells are thus likely to be negligible compared with the benefits of killing CCR9+-expressing tumor cells.
Research and development of anti-chemokine receptor mAbs as potential anti-cancer agents is increasing rapidly. A human anti-CXCR4 mAb (MDX-1338) has been developed to treat acute myelogenous leukemia (AML),43,44 and a humanized afucosylated antibody to CCR4 (mogamulizumab, KW-0761; Poteligeo®) was approved in Japan for treatment of relapsed or refractory adult T cell leukemia/lymphoma (ATLL).45 91R, the mAb anti-human CCR9 reported here, represents a potential therapeutic anti-cancer antibody for primary as well as metastatic CCR9+ tumors.
Materials and Methods
Ethics Statement
Animal maintenance followed European Union and national guidelines for animal experimentation and treatment protocols were approved by the CNB/CSIC Ethics Committee for Animal Research. Human cell samples were obtained after informed patient consent in accordance with the Helsinki Declaration. Research was approved by the CNB/CBMSO/CSIC Ethics Committee for Joint Research.
Cells and reagents
Human embryonic kidney 293 (HEK293) cells and HEK293 cells stably transfected with chemokine receptors hCCR6 or hCCR8 were cultured as described.8,46,47 pCIneo, hCCR4, hCCR5, hCCR9 and mCCR9 stable transfectants were a kind gift of A. Zaballos (ISCIII, Madrid, Spain). MOLT-4 (CRL-182) and Jurkat (TIB-152) human T-ALL cell lines were from the American Type Culture Collection. Bioluminescent MOLT-4 cells (MOLT-4-luc) were generated by infection with a recombinant lentivirus encoding enhanced green fluorescent protein (EGFP) and red luciferase (Promega). Infected cells expressing high EGFP levels were isolated by fluorescence activated cell sorting, cloned, expanded, and used for in vivo bioluminescence assays. MOLT-4-luc cell growth kinetics was similar to parental MOLT-4 cells and they retained surface CCR9 expression. Cells were cultured in Dulbecco's modified Eagle's medium (Lonza) supplemented with 10% fetal bovine serum (FBS, GE Healthcare), 2 mM L-glutamine, 50 U/ml penicillin, and 50 μg/ml streptomycin (complete medium). Neomycin-resistant stable HEK293 transfectants were cultured in the presence of 1 mg/ml G418 (Sigma) and periodically tested for chemokine receptor expression (not shown). Thymocytes from thymic fragments removed during corrective cardiac surgery and human peripheral blood lymphocytes were obtained from donors. 48
Recombinant human CCL25 and CXCL12 were from Peprotech. We used the following antibodies: anti-hCCR9 (112509, mouse mAb IgG2a; R&D), -mCCR9 (K629; rabbit Ab),11 -hCD3-FITC (UCHT1), -hCD4-PCy5 (13B8.2), -hCD8-PE (B9.11) (all three mouse mAb; Beckman), -hCD71 transferrin receptor (H300; rabbit Ab), -PCNA (PC10; mouse mAb) (both from Santa Cruz Biotech), -mCD31 (MEC 13.3; rat mAb; BD Biosciences); -mCD49b-PE (DX5), -mCD45-PCy5 (30-F11) (both rat mAb; Biolegend), and -mCD3-FITC (2C11; hamster mAb; Beckman). Control antibodies were P-020 (mouse mAb IgG2b; our laboratory) and MPC-11 (mouse mAb IgG2a; BD Biosciences). The M4 serum pool was generated by immunizing three BALB/c mice with three intraperitoneal injections of 107 MOLT-4 cells in PBS (days 1, 25 and 50); sera were collected on day 60.
Generation of human CCR9-specific mAb
Murine anti-human CCR9 mAb were raised by gene gun (Bio-Rad) particle-mediated DNA administration to BALB/c mice of the pCIneo plasmid bearing the human CCR9 cDNA. DNA was coated onto 1.6 nm gold particles and the DNA/gold complex (2 μg/day) was delivered to each mouse; mice were boosted on days 30 and 60 with the same amount of plasmid. Sera were collected 7–10 d after the last boost and tested for specific antibodies by flow cytometry using stably transfected hCCR9-HEK293 cells, and pCIneo-HEK293 cells as negative controls. Selected mice were boosted intravenously with 107 hCCR9-HEK293 cells 3 and 2 d prior to splenocyte fusion.49 Two weeks post-fusion, culture supernatants were screened by flow cytometry for CCR9-specific antibodies using hCCR9-HEK293 cells. Positive hybridomas were cloned, mAb purified from culture supernatants, and antibody isotype determined by enzyme-linked immunosorbent assay.49
Generation of chimeric CCR9
pCIneo expression vectors bearing human or mouse CCR9 cDNA inserted into NheI-EcoRI sites were used to generate chimeric CCR9. Vectors were digested with NheI and BspLI. The fragment containing 62 N-terminal amino acids of the murine CCR9 sequence was cloned into the digested human CCR9 plasmid to generate the mNt/hCCR9 expression vector, which was transiently transfected in HEK293 cells. Chimeric CCR9 expression was evaluated by flow cytometry.
Flow cytometry
For staining, 2 x 105 cells/well were centrifuged in V-bottom 96-well plates and washed with phosphate-buffered saline (PBS) containing 0.5% bovine serum albumin (BSA), 1% FBS and 0.1% sodium azide (PBSst). Non-specific binding was blocked by pre-incubating cells with 40 μg/ml rat IgG (Sigma; 100 μl final volume, 20 min, 4 °C). Cells were incubated with primary Ab (30 min, 4 °C), washed, and incubated with FITC- or PE-goat F(ab´)2 anti-mouse IgG (H+L) antibody (Beckman Coulter; 30 min, 4 °C). Samples were analyzed on an Epics XL or a Cytomics cytometer (Beckman Coulter).
For competition studies, cells were incubated with 50 μl hCCL25 or hCXCL12 (10 μg/ml, 40 min, 4 °C), followed by 50 μl 91R or isotype-matched mAb (0.5 μg/ml, 30 min, 4 °C). After washing, the FITC-goat anti-mouse IgG antibody was added (30 min, 4 °C). CCR9 expression was evaluated by flow cytometry.
Western blot assays
To extract the membrane fraction, cell pellets (5 x 106 cells) were resuspended in hypotonic buffer (5 mM Tris/HCl pH 7.4, 50 mM NaCl, 1 mM MgCl2, 2 mM ethylene glycol tetraacetic acid), subjected to four freeze-thaw cycles and centrifuged (750 x g, 2 min, 4 °C). Supernatants were centrifuged (18,000 x g, 30 min, 4 °C). Membrane pellets were resuspended in PBS with Halt Protease Inhibitor Cocktail (Thermo Scientific), solubilized in 20% SDS, 100 mM dithiothreitol (30 min, room temperature (RT)) and resolved by SDS- PAGE. Proteins were transferred to Immobilon polyvinylidene difluoride membranes (Millipore), blocked (5% BSA, 5% non-fat dry milk and 0.05% Tween 20 in PBS), immunoblotted with 91R mAb (1 μg/ml, 2 h, RT) and incubated with a peroxidase-coupled goat anti-mouse IgG antibody. Membranes were reprobed with anti-hCD71 Ab (0.4 μg/ml, 2 h, RT) as loading control. Blots were developed using enhanced chemiluminescence western blot detection reagents (GE Healthcare). Where indicated, samples were N-deglycosylated using peptide-N-glycosidase F (PNGase F, New England Biolabs; 1 h, 37 °C).
Xenograft assays
BALB/c Rag2−/− mice (Taconic Farms) were bred in the CNB animal facility and used at 8 to 22 wk of age.
For in vivo experiments, MOLT-4 cells (2 × 106) were inoculated s.c. on day 0 in each flank of Rag2−/− mice. In these experimental conditions, 80–90% of the cell inoculations gave rise to tumors. Two groups (6 mice each) were inoculated (i.p.) with anti-hCCR9 91R or an IgG2b control mAb on days 1, 7, 14 and 21 (4 mg/kg on d1 and 7; 2 mg/kg on d14 and 21). In a second experiment, MOLT-4 cells (2 × 106) were inoculated s.c. into one flank of female Rag2−/− mice; two groups (10 mice each) were inoculated i.p. with anti-hCCR9 91R or an IgG2b control mAb on days 7, 14, 21 and 28 (4 mg/kg on d7 and 14; 2 mg/kg on d21 and 28). Tumor size was measured with a Vernier caliper (Mitutoyo) and tumor volume (mm3) calculated as V = [axial diameter length, mm] x [(rotational diameter, mm)2/2]. Tumor burden is expressed as percent tumor volume relative to that of IgG2b-treated mice. Mice were sacrificed on day 56 or 69, depending on the experiment; mice and tumors were weighed and processed for histology.
For bioluminescence xenograft assays, female Rag2−/− mice were inoculated s.c. in each flank with 2 × 106 MOLT-4-luc cells on d0. After 24 h, mice were anesthetized and D-luciferin (150 mg/kg; Biosynth) was administered to allow imaging and luminescence quantification for balanced assignment to experimental groups. Mice (8/group, one mouse in the 91R-treated group died at week 2 post-treatment after anesthesia injection) were inoculated i.p. with 91R or an IgG2b control mAb on d1 (4 mg/kg) and d6 (2 mg/kg). Luminescence imaging was repeated at six time points until mice were sacrificed on d62. Mice were anesthetized with ketamine (2 ml/Kg Imalgene 500; Merial Laboratories) and 2% xylazine (0.6 ml/Kg Xilagesic; Calier Laboratories) 10 min before analysis. Imaging was done with a 1394 ORCA II ERG camera (Hamamatsu) in a lightproof chamber for 100 s; Wasabi software (Hamamatsu) was used to quantify the data and produce pseudocolor images.
Histology and quantification of necrotic area
Dissected tumors were divided in half; one half was fixed overnight in 4% paraformaldehyde (pH 7.4), washed in PBS, paraffin-embedded and sectioned (5 μm). The other half was embedded in OCT (Leica Microsystems), snap-frozen in dry ice-chilled isopentane, stored at -80 °C, and later sectioned (8 μm).
Paraffin-embedded sections were hematoxylin/eosin-stained by standard procedures and mounted in Micromount media (Leica Microsystems). Images were captured on a Zeiss Axiophot microscope (Carl Zeiss) with a Digital Sight camera (Nikon). Total tumor section and necrotic areas were quantified with NIH ImageJ software.
Terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end labeling (TUNEL)
To measure cell death by TUNEL, paraffin-embedded sections were dewaxed and rehydrated, then permeabilized (PBS, 0.5% Triton X-100; 10 min, RT). Slides were pre-incubated (15 min, RT in the dark, humidified chambers) with TdT buffer pH 6.6 (Sigma) and 1 mM CoCl2. Sections were incubated with reaction mix (1 h, 37 °C; recombinant terminal transferase and biotin-16-dUTP; Roche) and washed in preincubation buffer (10 min, RT). The reaction was terminated with PBS and 0.01% Tween 20, and slides were incubated with streptavidin-Cy5 (Jackson ImmunoResearch; 1 h, RT).
Immunohistochemistry (IHC)
Cell proliferation was quantified by labeling proliferating cell nuclear antigen (PCNA) in dewaxed and rehydrated paraffin-embedded sections. Antigens were exposed by steaming in sodium citrate buffer pH 6.5 (15 min), stained with the MaxFluor Mouse on Mouse Fluorescence Detection Kit (Max Vision Biosciences) using anti-PCNA (overnight, 4 °C, 2 μg/ml). Tumor blood vessels were stained with anti-mCD31 mAb. OCT-embedded sections were fixed in 100% acetone (-20 °C, 10 min), air-dried and washed with Tris-buffered saline (TBS). Samples were blocked with 2.5% goat serum and 0.5% BSA in TBS (2 h, RT), stained with anti-CD31 (overnight, 4 °C, 80 ng/ml), incubated and washed, and Alexa Fluor 647-labeled goat anti-rat IgG was added. Nuclei were stained with 4,6-diamidino-2-phenylindole (DAPI; Sigma) and slides mounted with Fluoromount-G (Southern Biotech) for all microscopy preparations.
Tissue imaging and analysis
Digital images were acquired on a Leica laser scanning multispectral confocal microscope (TCS SP5, Leica Microsystems). Image stacks consisted of five image planes acquired through a 20 × lens (calculated optimal zoom factor 2, z-step 1.39 μm). At least three sections and 12 to 30 random, non-contiguous, non-overlapping fields per section were acquired and examined for each tumor. Density per optical field of TUNEL- and PCNA-positive nuclei and of CD31-positive vessel fragments was quantified and the estimated mean number of positive structures calculated using data from non-necrotic regions. Quantification was performed with zoomed images using the Adobe Photoshop Count Tool.
Complement-dependent cytotoxicity (CDC)
MOLT-4 cells (105 target cells/100 μl) were plated in a 96-well V-bottom plate, incubated with indicated concentrations of anti-hCCR9 or an isotype-matched control mAb (30 min, 37 °C), centrifuged and washed. Active or 56 °C heat-inactivated baby rabbit complement (25%; AbD Serotec) was added in serum-free Dulbecco's modified Eagle's medium with 1% BSA (1 h, 37 °C). Cells were stained with the viability exclusion marker 7-aminoactinomycin (7-AAD; BD Biosciences; 10 min, 4 °C) and the number of non-viable cells evaluated by flow cytometry; each condition was analyzed in triplicate. Specific lysis was calculated as 100x (% dead cells with active complement - % dead cells with inactive complement)/(100% - % dead cells with inactive complement).
Antibody-dependent cell-mediated cytotoxicity (ADCC)
Murine natural NK were isolated from BALB/c spleens using the Auto Macs Pro negative selection system (Miltenyi). Purified cells were analyzed for mCD3, mCD45 and mCD49b expression on a flow cytometer. All preparations contained at least 90% NK cells defined as CD3-CD45+CD49b+. Cells were cultured for 6–7 d in RPMI 1640 (Lonza)-10% FBS with 1000 U/ml murine recombinant IL-2 (Peprotech). For cytotoxicity assays, target MOLT-4 cells labeled with Cell Trace CFSE (Invitrogen) were pre-incubated (30 min) with indicated mAb concentrations. NK and target cells were co-cultured (4 h) at a 20:1 ratio in RPMI-10% FBS, then stained with 7-AAD (10 min, 4 °C) and analyzed by flow cytometry. Gating on 7-AAD-positive cells within the CFSE+ population indicated the proportion of dead target cells. Specific killing was calculated as 100 × [(% dead target cells in sample – % spontaneous dead target cells)/(100 – % spontaneous dead target cells)]. Target cells incubated without effector cells were used to assess spontaneous cell death.
Statistical analyses
Statistical analyses were performed using GraphPad Prism 4 software. Statistical significance was established at P < 0.05 as evaluated by Student’s t test, unless otherwise indicated. Results are shown as mean ± SEM.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr. José A. García-Sanz (CIB-CSIC) for helpful suggestions in experimental strategy and for critical reading of the manuscript, Dr. María Luisa Toribio for advice in the use of human cells, Tamara Rueda and Mercedes Llorente for antibody purification, Silvia Gutiérrez Erlandsson for help with confocal microscopy, Vanesa Cadenas for excellent technical help, Sara Escudero and M. Carmen Moreno-Ortíz for help with flow cytometry analyses, and Catherine Mark for editorial assistance. This work was supported by the Instituto de Salud Carlos III (grant PI10/00594) and the CSIC (grants 200920I016 and 201120E007), Ministerio de Economía y Competitividad to L.K. M.V. and A.F. were supported by the CSIC grant 201120E007.
Dedication
Dedicated to the memory of Sonia Chamorro, who died on 17 March 2014.
References
- 1.Cyster JG. Lymphoid organ development and cell migration. Immunol Rev. 2003;195:5–14. doi: 10.1034/j.1600-065X.2003.00075.x. [DOI] [PubMed] [Google Scholar]
- 2.Zlotnik A, Burkhardt AM, Homey B. Homeostatic chemokine receptors and organ-specific metastasis. Nat Rev Immunol. 2011;11:597–606. doi: 10.1038/nri3049. [DOI] [PubMed] [Google Scholar]
- 3.Sun X, Cheng G, Hao M, Zheng J, Zhou X, Zhang J, Taichman RS, Pienta KJ, Wang J. CXCL12 / CXCR4 / CXCR7 chemokine axis and cancer progression. Cancer Metastasis Rev. 2010;29:709–22. doi: 10.1007/s10555-010-9256-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ben-Baruch A. Organ selectivity in metastasis: regulation by chemokines and their receptors. Clin Exp Metastasis. 2008;25:345–56. doi: 10.1007/s10585-007-9097-3. [DOI] [PubMed] [Google Scholar]
- 5.Letsch A, Keilholz U, Schadendorf D, Assfalg G, Asemissen AM, Thiel E, Scheibenbogen C. Functional CCR9 expression is associated with small intestinal metastasis. J Invest Dermatol. 2004;122:685–90. doi: 10.1111/j.0022-202X.2004.22315.x. [DOI] [PubMed] [Google Scholar]
- 6.Richmond A. CCR9 homes metastatic melanoma cells to the small bowel. Clin Cancer Res. 2008;14:621–3. doi: 10.1158/1078-0432.CCR-07-2235. [DOI] [PubMed] [Google Scholar]
- 7.Amersi FF, Terando AM, Goto Y, Scolyer RA, Thompson JF, Tran AN, Faries MB, Morton DL, Hoon DS. Activation of CCR9/CCL25 in cutaneous melanoma mediates preferential metastasis to the small intestine. Clin Cancer Res. 2008;14:638–45. doi: 10.1158/1078-0432.CCR-07-2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zaballos A, Gutiérrez J, Varona R, Ardavín C, Márquez G. Cutting edge: identification of the orphan chemokine receptor GPR-9-6 as CCR9, the receptor for the chemokine TECK. J Immunol. 1999;162:5671–5. [PubMed] [Google Scholar]
- 9.Youn BS, Kim CH, Smith FO, Broxmeyer HE. TECK, an efficacious chemoattractant for human thymocytes, uses GPR-9-6/CCR9 as a specific receptor. Blood. 1999;94:2533–6. [PubMed] [Google Scholar]
- 10.Wurbel MA, Philippe JM, Nguyen C, Victorero G, Freeman T, Wooding P, Miazek A, Mattei MG, Malissen M, Jordan BR, et al. The chemokine TECK is expressed by thymic and intestinal epithelial cells and attracts double- and single-positive thymocytes expressing the TECK receptor CCR9. Eur J Immunol. 2000;30:262–71. doi: 10.1002/1521-4141(200001)30:1<262::AID-IMMU262>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 11.Carramolino L, Zaballos A, Kremer L, Villares R, Martín P, Ardavín C, Martínez-A C, Márquez G. Expression of CCR9 beta-chemokine receptor is modulated in thymocyte differentiation and is selectively maintained in CD8(+) T cells from secondary lymphoid organs. Blood. 2001;97:850–7. doi: 10.1182/blood.V97.4.850. [DOI] [PubMed] [Google Scholar]
- 12.Kunkel EJ, Campbell JJ, Haraldsen G, Pan J, Boisvert J, Roberts AI, Ebert EC, Vierra MA, Goodman SB, Genovese MC, et al. Lymphocyte CC chemokine receptor 9 and epithelial thymus-expressed chemokine (TECK) expression distinguish the small intestinal immune compartment: Epithelial expression of tissue-specific chemokines as an organizing principle in regional immunity. J Exp Med. 2000;192:761–8. doi: 10.1084/jem.192.5.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zabel BA, Agace WW, Campbell JJ, Heath HM, Parent D, Roberts AI, Ebert EC, Kassam N, Qin S, Zovko M, et al. Human G protein-coupled receptor GPR-9-6/CC chemokine receptor 9 is selectively expressed on intestinal homing T lymphocytes, mucosal lymphocytes, and thymocytes and is required for thymus-expressed chemokine-mediated chemotaxis. J Exp Med. 1999;190:1241–56. doi: 10.1084/jem.190.9.1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wendland M, Czeloth N, Mach N, Malissen B, Kremmer E, Pabst O, Förster R. CCR9 is a homing receptor for plasmacytoid dendritic cells to the small intestine. Proc Natl Acad Sci U S A. 2007;104:6347–52. doi: 10.1073/pnas.0609180104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wurbel MA, Malissen M, Guy-Grand D, Meffre E, Nussenzweig MC, Richelme M, Carrier A, Malissen B. Mice lacking the CCR9 CC-chemokine receptor show a mild impairment of early T- and B-cell development and a reduction in T-cell receptor gammadelta(+) gut intraepithelial lymphocytes. Blood. 2001;98:2626–32. doi: 10.1182/blood.V98.9.2626. [DOI] [PubMed] [Google Scholar]
- 16.Vicari AP, Figueroa DJ, Hedrick JA, Foster JS, Singh KP, Menon S, Copeland NG, Gilbert DJ, Jenkins NA, Bacon KB, et al. TECK: a novel CC chemokine specifically expressed by thymic dendritic cells and potentially involved in T cell development. Immunity. 1997;7:291–301. doi: 10.1016/S1074-7613(00)80531-2. [DOI] [PubMed] [Google Scholar]
- 17.Qiuping Z, Qun L, Chunsong H, Xiaolian Z, Baojun H, Mingzhen Y, Chengming L, Jinshen H, Qingping G, Kejian Z, et al. Selectively increased expression and functions of chemokine receptor CCR9 on CD4+ T cells from patients with T-cell lineage acute lymphocytic leukemia. Cancer Res. 2003;63:6469–77. [PubMed] [Google Scholar]
- 18.Singh S, Singh UP, Stiles JK, Grizzle WE, Lillard JW., Jr. Expression and functional role of CCR9 in prostate cancer cell migration and invasion. Clin Cancer Res. 2004;10:8743–50. doi: 10.1158/1078-0432.CCR-04-0266. [DOI] [PubMed] [Google Scholar]
- 19.Johnson-Holiday C, Singh R, Johnson E, Singh S, Stockard CR, Grizzle WE, Lillard JW., Jr. CCL25 mediates migration, invasion and matrix metalloproteinase expression by breast cancer cells in a CCR9-dependent fashion. Int J Oncol. 2011;38:1279–85. doi: 10.3892/ijo.2011.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sharma PK, Singh R, Novakovic KR, Eaton JW, Grizzle WE, Singh S. CCR9 mediates PI3K/AKT-dependent antiapoptotic signals in prostate cancer cells and inhibition of CCR9-CCL25 interaction enhances the cytotoxic effects of etoposide. Int J Cancer. 2010;127:2020–30. doi: 10.1002/ijc.25219. [DOI] [PubMed] [Google Scholar]
- 21.Johnson EL, Singh R, Johnson-Holiday CM, Grizzle WE, Partridge EE, Lillard JW, Jr., Singh S. CCR9 interactions support ovarian cancer cell survival and resistance to cisplatin-induced apoptosis in a PI3K-dependent and FAK-independent fashion. J Ovarian Res. 2010;3:15. doi: 10.1186/1757-2215-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qiuping Z, Jei X, Youxin J, Wei J, Chun L, Jin W, Qun W, Yan L, Chunsong H, Mingzhen Y, et al. CC chemokine ligand 25 enhances resistance to apoptosis in CD4+ T cells from patients with T-cell lineage acute and chronic lymphocytic leukemia by means of livin activation. Cancer Res. 2004;64:7579–87. doi: 10.1158/0008-5472.CAN-04-0641. [DOI] [PubMed] [Google Scholar]
- 23.Mirandola L, Chiriva-Internati M, Montagna D, Locatelli F, Zecca M, Ranzani M, Basile A, Locati M, Cobos E, Kast WM, et al. Notch1 regulates chemotaxis and proliferation by controlling the CC-chemokine receptors 5 and 9 in T cell acute lymphoblastic leukaemia. J Pathol. 2012;226:713–22. doi: 10.1002/path.3015. [DOI] [PubMed] [Google Scholar]
- 24.Hu Y, Zhang L, Wu R, Han R, Jia Y, Jiang Z, Cheng M, Gan J, Tao X, Zhang Q. Specific killing of CCR9 high-expressing acute T lymphocytic leukemia cells by CCL25 fused with PE38 toxin. Leuk Res. 2011;35:1254–60. doi: 10.1016/j.leukres.2011.01.015. [DOI] [PubMed] [Google Scholar]
- 25.Weiner LM, Surana R, Wang S. Monoclonal antibodies: versatile platforms for cancer immunotherapy. Nat Rev Immunol. 2010;10:317–27. doi: 10.1038/nri2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer. 2012;12:278–87. doi: 10.1038/nrc3236. [DOI] [PubMed] [Google Scholar]
- 27.Reichert JM, Dhimolea E. The future of antibodies as cancer drugs. Drug Discov Today. 2012;17:954–63. doi: 10.1016/j.drudis.2012.04.006. [DOI] [PubMed] [Google Scholar]
- 28.Folkman J. What is the evidence that tumors are angiogenesis dependent? J Natl Cancer Inst. 1990;82:4–6. doi: 10.1093/jnci/82.1.4. [DOI] [PubMed] [Google Scholar]
- 29.Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249–57. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- 30.Meert AP, Paesmans M, Martin B, Delmotte P, Berghmans T, Verdebout JM, Lafitte JJ, Mascaux C, Sculier JP. The role of microvessel density on the survival of patients with lung cancer: a systematic review of the literature with meta-analysis. Br J Cancer. 2002;87:694–701. doi: 10.1038/sj.bjc.6600551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seidel UJ, Schlegel P, Lang P. Natural killer cell mediated antibody-dependent cellular cytotoxicity in tumor immunotherapy with therapeutic antibodies. Front Immunol. 2013;4:76. doi: 10.3389/fimmu.2013.00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med. 2000;6:443–6. doi: 10.1038/74704. [DOI] [PubMed] [Google Scholar]
- 33.Singh R, Stockard CR, Grizzle WE, Lillard JW, Jr., Singh S. Expression and histopathological correlation of CCR9 and CCL25 in ovarian cancer. Int J Oncol. 2011;39:373–81. doi: 10.3892/ijo.2011.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shen X, Mailey B, Ellenhorn JD, Chu PG, Lowy AM, Kim J. CC chemokine receptor 9 enhances proliferation in pancreatic intraepithelial neoplasia and pancreatic cancer cells. J Gastrointest Surg. 2009;13:1955–62, discussion 1962. doi: 10.1007/s11605-009-1002-8. [DOI] [PubMed] [Google Scholar]
- 35.Szpakowska M, Fievez V, Arumugan K, van Nuland N, Schmit JC, Chevigné A. Function, diversity and therapeutic potential of the N-terminal domain of human chemokine receptors. Biochem Pharmacol. 2012;84:1366–80. doi: 10.1016/j.bcp.2012.08.008. [DOI] [PubMed] [Google Scholar]
- 36.Rajagopalan L, Rajarathnam K. Structural basis of chemokine receptor function--a model for binding affinity and ligand selectivity. Biosci Rep. 2006;26:325–39. doi: 10.1007/s10540-006-9025-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shinkai Y, Rathbun G, Lam KP, Oltz EM, Stewart V, Mendelsohn M, Charron J, Datta M, Young F, Stall AM, et al. RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell. 1992;68:855–67. doi: 10.1016/0092-8674(92)90029-C. [DOI] [PubMed] [Google Scholar]
- 38.Shan D, Ledbetter JA, Press OW. Apoptosis of malignant human B cells by ligation of CD20 with monoclonal antibodies. Blood. 1998;91:1644–52. [PubMed] [Google Scholar]
- 39.Johnson-Holiday C, Singh R, Johnson EL, Grizzle WE, Lillard JW, Jr., Singh S. CCR9-CCL25 interactions promote cisplatin resistance in breast cancer cell through Akt activation in a PI3K-dependent and FAK-independent fashion. World J Surg Oncol. 2011;9:46. doi: 10.1186/1477-7819-9-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bezombes C, Fournié JJ, Laurent G. Direct effect of rituximab in B-cell-derived lymphoid neoplasias: mechanism, regulation, and perspectives. Mol Cancer Res. 2011;9:1435–42. doi: 10.1158/1541-7786.MCR-11-0154. [DOI] [PubMed] [Google Scholar]
- 41.Manches O, Lui G, Chaperot L, Gressin R, Molens JP, Jacob MC, Sotto JJ, Leroux D, Bensa JC, Plumas J. In vitro mechanisms of action of rituximab on primary non-Hodgkin lymphomas. Blood. 2003;101:949–54. doi: 10.1182/blood-2002-02-0469. [DOI] [PubMed] [Google Scholar]
- 42.van Meerten T, van Rijn RS, Hol S, Hagenbeek A, Ebeling SB. Complement-induced cell death by rituximab depends on CD20 expression level and acts complementary to antibody-dependent cellular cytotoxicity. Clin Cancer Res. 2006;12:4027–35. doi: 10.1158/1078-0432.CCR-06-0066. [DOI] [PubMed] [Google Scholar]
- 43.Bertolini F, Dell’Agnola C, Mancuso P, Rabascio C, Burlini A, Monestiroli S, Gobbi A, Pruneri G, Martinelli G. CXCR4 neutralization, a novel therapeutic approach for non-Hodgkin’s lymphoma. Cancer Res. 2002;62:3106–12. [PubMed] [Google Scholar]
- 44.Kuhne MR, Mulvey T, Belanger B, Chen S, Pan C, Chong C, Cao F, Niekro W, Kempe T, Henning KA, et al. BMS-936564/MDX-1338: a fully human anti-CXCR4 antibody induces apoptosis in vitro and shows antitumor activity in vivo in hematologic malignancies. Clin Cancer Res. 2013;19:357–66. doi: 10.1158/1078-0432.CCR-12-2333. [DOI] [PubMed] [Google Scholar]
- 45.Ito Y, Miyamoto T, Chong Y, Aoki T, Kato K, Akashi K, Kamimura T. Successful treatment with anti-CC chemokine receptor 4 MoAb of relapsed adult T-cell leukemia/lymphoma after umbilical cord blood transplantation. Bone Marrow Transplant. 2013;48:998–9. doi: 10.1038/bmt.2012.268. [DOI] [PubMed] [Google Scholar]
- 46.Carramolino L, Kremer L, Goya I, Varona R, Buesa JM, Gutiérrez J, Zaballos A, Martínez-A C, Márquez G. Down-regulation of the beta-chemokine receptor CCR6 in dendritic cells mediated by TNF-alpha and IL-4. J Leukoc Biol. 1999;66:837–44. doi: 10.1002/jlb.66.5.837. [DOI] [PubMed] [Google Scholar]
- 47.Goya I, Gutiérrez J, Varona R, Kremer L, Zaballos A, Márquez G. Identification of CCR8 as the specific receptor for the human beta-chemokine I-309: cloning and molecular characterization of murine CCR8 as the receptor for TCA-3. J Immunol. 1998;160:1975–81. [PubMed] [Google Scholar]
- 48.Martín-Gayo E, Sierra-Filardi E, Corbí AL, Toribio ML. Plasmacytoid dendritic cells resident in human thymus drive natural Treg cell development. Blood. 2010;115:5366–75. doi: 10.1182/blood-2009-10-248260. [DOI] [PubMed] [Google Scholar]
- 49.Kremer L, Márquez G. Generation of monoclonal antibodies against chemokine receptors. Methods Mol Biol. 2004;239:243–60. doi: 10.1385/1-59259-435-2:243. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.