

Abstract
Molecular details of epidermal growth factor receptor (EGFR) targeting by nimotuzumab, a therapeutic anti-cancer antibody, have been largely unknown. The current study delineated a functional map of their interface, based on phage display and extensive mutagenesis of both the target antigen and the Fv antibody fragment. Five residues in EGFR domain III (R353, S356, F357, T358, and H359T) and the third hypervariable region of nimotuzumab heavy chain were shown to be major functional contributors to the interaction. Fine specificity differences between nimotuzumab and other anti-EGFR antibodies were revealed. Mapping information guided the generation of a plausible in silico binding model. Knowledge about the epitope/paratope interface opens new avenues for the study of tumor sensitivity/resistance to nimotuzumab and for further engineering of its binding site. The developed mapping platform, also validated with the well-known cetuximab epitope, allows a comprehensive exploration of antigenic regions and could be expanded to map other anti-EGFR antibodies.
Keywords: epitope mapping, molecular modeling, paratope, phage display, site-directed mutagenesis
Introduction
Sustained proliferative signaling is one of the hallmarks of cancer.1 Targeted therapies have focused on growth factors and their receptors, which support tumor progression.2 Deregulated signaling mediated by epidermal growth factor (EGF) and EGF receptor (EGFR) promotes tumor growth and invasion, and is associated with poor prognosis of human malignancies. This pathway has been targeted by two main classes of therapeutic agents: low molecular weight tyrosine-kinase inhibitors that block intracellular signaling,3 and neutralizing monoclonal antibodies (mAbs) against the EGFR extracellular region (erEGFR), which are able to disrupt ligand binding and receptor activation. A recent survey4 describes three anti-EGFR mAbs as currently marketed for cancer therapy: cetuximab (first approved for clinical use in US and Europe in 2004), panitumumab (approved in 2006 and 2007, respectively), and nimotuzumab (the humanized version of R3 murine mAb5,6), which has been approved in Cuba since 2002 and is marketed in over 20 countries, including Brazil, India and China. Its use is supported by a wide clinical experience, showing efficacy to treat patients with head and neck cancer,7 high grade glioma8 and esophageal tumors.9 Additional clinical trials are being performed.10,11 One distinctive feature of nimotuzumab among anti-EGFR therapeutic agents is its low toxicity.12
Nimotuzumab's clinical success has been accompanied by consistent in vitro and in vivo evidences of anti-tumor effects.13,14 A deeper understanding of the molecular bases of these phenomena has been hampered by the unknown identity of the epitope recognized by the antibody. Previous studies addressing this point have been limited to in silico docking simulations.15 This lack of information is in sharp contrast with the plethora of data (both structural and functional) regarding other anti-EGFR mAbs,16-21 mainly cetuximab, which is starting to delineate a connection between mutations affecting the target epitopes and sensitivity/resistance to mAb therapy.22 Linking the observed biological and clinical outcomes with recognition of a particular antigenic determinant by nimotuzumab would thus contribute to our understanding of how anti-EGFR agents impinge on the complex interplay between tumors and the whole organism. Differences/similarities with other anti-EGFR mAbs in terms of fine specificity would complete a picture of their usefulness in order to optimize clinical benefit for every patient.
We developed a phage display-based platform for comprehensive mutagenesis scanning of the functional interface between nimotuzumab and EGFR. Its use allowed the definition of critical residues involved in binding within both the epitope and the paratope. Nimotuzumab fine specificity was thus shown to differ from the ones exhibited by other anti-EGFR mAbs. The experimental data were integrated into a new interaction model for the antibody, which could be the starting point for further engineering of its binding site. Beyond providing a novel molecular view of tumor targeting by nimotuzumab, our functional mapping strategy offers advantages for the exploration of multiple interactions involving the EGFR and should be an optimal complement for other mapping approaches focused on this target.
Results
Nimotuzumab was shown to recognize a conformational epitope on EGF receptor
Enzyme-linked immunosorbent assay (ELISA) on native and denatured erEGFR recombinant protein23 showed that disrupting the antigen (Ag) disulfide bonds drastically decreased nimotuzumab reactivity (by more than 90%), indicating that its epitope is conformation-sensitive (Fig. 1). Cetuximab, already known to recognize a conformational epitope,17 also lost its binding ability upon Ag denaturation. Recognition by a polyclonal antiserum ruled out any non-specific ELISA effect of the treatment. The conformational nature of nimotuzumab epitope, together with the structural complexity of the target (a protein with a large extracellular region, 105 kDa, comprising four domains and 25 disulfide bonds) led to the design of a functional mapping strategy based on the manipulation of properly folded EGFR domains.
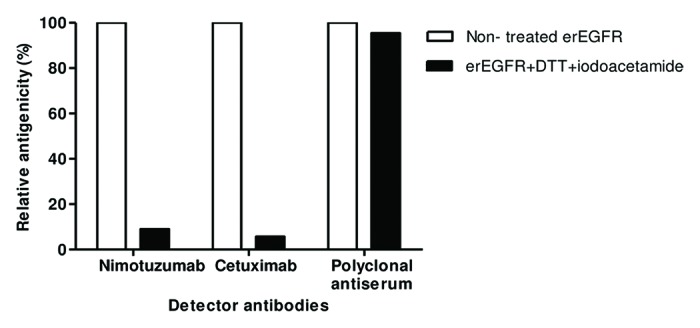
Figure 1. Conformation-sensitivity of the epitope recognized by nimotuzumab. Coating extracellular region EGFR recombinant protein was sequentially treated with DTT and iodoacetamide to disrupt disulfide bonds. Binding of nimotuzumab to either treated or non-treated antigen was detected with anti-human IgG antibodies labeled with horseradish peroxidase. Relative antigenicity (%) of denatured erEGFR was calculated taking the reactivity toward the unmodified antigen as the reference. Cetuximab and polyclonal anti-erEGFR monkey antiserum were used as control antibodies.
Both the target antigenic region and the nimotuzumab paratope itself were successfully displayed on filamentous phages
Phage display was the chosen platform to manipulate the Ag. Competition between nimotuzumab and cetuximab15 (the latter already known to recognize an epitope on erEGFR domain III24) highlighted this domain as the putative nimotuzumab target. An EGFR fragment (residues 311–514) comprising domain III plus the N-terminal end of the adjacent domain IV (Dom III+482–514) was displayed on phages, as shown with 9E10 mAb (recognizing the c-myc tag fused to foreign proteins in our display system) (Fig. 2A). The design of this Ag fragment took into account previous experience attaching C-terminal extensions for proper folding of recombinant domain III.17 Recognition by nimotuzumab and cetuximab (conformation-sensitive antibodies) confirmed its correct folding (Fig. 2A). The presence of the epitopes recognized by both mAbs clearly showed the usefulness of the phage-displayed Dom III+482–514 to study their interactions. While the current work was aimed at exploring the nimotuzumab epitope for the first time, characterization of the cetuximab epitope (which has been extensively studied16,17,21) would validate the accuracy of our mapping procedures.

Figure 2. Phage display of EGR domain III and a nimotuzumab-derived antibody fragment. Purified phages displaying either the nimotuzumab target antigenic region (human EGFR Dom III+482–514) or the nimotuzumab binding site itself (a single chain Fv antibody fragment comprising humanized R3 VH and VL regions connected by a linker peptide) were tested by ELISA on microtiter plates coated with different molecules. The anti-tag 9E10 mAb recognizing the c-myc tag fused to all foreign proteins in our system was used to detect the presence of both phage-displayed proteins. Bound phages were detected with an anti-M13 mAb labeled with horseradish peroxidase. A) Phage-displayed EGFR domain III (Dom III+482–514) was tested on nimotuzumab/cetuximab-coated plates. An unrelated antibody was used as negative control. B) Phage-displayed single chain Fv antibody fragment was tested on plates coated with either erEGFR recombinant protein or an unrelated antigen (BSA).
Parallel characterization of the nimotuzumab paratope would result in a comprehensive functional picture of the Ag-Ab complex from both sides. A phage-displayed single chain Fv (scFv) antibody fragment comprising the humanized R3 heavy and light chain variable regions resembled the original mAb in terms of specificity toward EGFR (Fig. 2B) and affinity (scFv KD = 22 nmol/l, compared with 21 nmol/l for the whole mAb15). Such a phage-displayed variant was thus suitable for manipulating nimotuzumab paratope.
Inter-species mutagenesis scanning of EGFR domain III revealed a critical residue contributing to nimotuzumab epitope
Since both nimotuzumab and cetuximab25 recognize human EGFR without cross-reacting with mouse EGFR, their target epitopes should include one or more residues that differ between the two species. Our mapping strategy started with ELISA screening of a panel of phage-displayed Dom III+482–514 variants harboring individual replacements of each solvent-exposed human residue by its mouse counterpart. All variants were properly displayed on filamentous phages, as shown by their reactivity with the anti-tag 9E10 mAb, and also reactive with either nimotuzumab or cetuximab, ruling out gross folding defects. Most of them were indeed recognized by both anti-EGFR antibodies. The variant containing the replacement H359R specifically lost recognition by nimotuzumab compared with wild-type (wt) Dom III+482–514 (Fig. 3A), indicating a critical involvement of H359 in the nimotuzumab epitope as a first clue to identify its location. The replacement I467M abolished recognition by cetuximab (Fig. 3B), which was consistent with the energetic contribution reported for I467,16 and confirmed the accuracy of inter-species mutagenesis scanning to detect critical residues within EGFR domain III epitopes.

Figure 3. Recognition of phage-displayed EGFR domain III mutated variants. Phages displaying human EGFR Dom III+482–514 mutated variants (where each solvent-exposed residue differing between human and mouse EGFR has been replaced by the aa found in the latter) were produced at a 50 ml scale. Phage-displayed wt Dom III+482–514 was included as a control. Purified phages (1012 viral particles/ml) were incubated on microtiter plates coated with either anti-EGFR mAbs (nimotuzumab [A] and cetuximab [B]) or the anti-c-myc tag 9E10 mAb. Bound phages were detected with an anti-M13 mAb conjugated to horseradish peroxidase. Normalized reactivity for each variant was estimated by dividing the signal obtained with each mAb by the reference signal (measured with the anti-tag mAb). Relative reactivity (%) was calculated as the ratio between normalized reactivity of each variant and that of wt domain III. Arrows indicate lack of recognition of individual variants by a given anti-EGFR antibody.
Comprehensive randomization of a broader antigenic area delineated a detailed functional map of the epitope recognized by nimotuzumab
Solvent-exposed Dom III+482–514 residues (>20% relative solvent accessibility, RSA) in the neighborhood of the critical H359 (<12 Å) were individually replaced by random amino acid (aa) mixtures and evaluated by ELISA on nimotuzumab-coated plates (Fig. S1A). The profile of tolerance to mutations (Table 1), resulted in a functional map of the nimotuzumab epitope (Fig. 4A). A prerequisite to include mutated variants in the analysis was their proper display on filamentous phages (more than 75% of the wt Dom III+482–514 display level as assessed with the anti-c-myc tag 9E10 mAb). Two residues (S356 and H359) were absolutely required for epitope formation. Multiple replacements at these positions, including the conservative substitutions S356T and H359R, abolished recognition (Table 1). Three other residues could only be replaced by aa sharing some properties with them, indicating their functional contributions. F357 and T358 were only substituted by the aromatic Tyr and the hydroxyl-containing Ser, respectively. Partial tolerance to several substitutions at position 358 indicated that the original Thr contributes to a lesser extent than the other residues. R353 could be replaced by Lys (a very similar residue), but also by hydrophobic aa (Leu, Met, Trp), highlighting the ability to establish hydrophobic interactions as a critical feature at this position. Both Arg and Lys are also capable to do so through their long aliphatic moieties.26 The presence of other residues at this position resulted in partial/total loss of recognition. The above described effects were specific, since almost all the mutated variants were well recognized by cetuximab (Fig. S1B). The few changes at these positions resulting in cetuximab reactivity loss predominantly involved replacements by Pro and Cys, which can disturb protein folding through structural backbone constraints and undesired disulfide bonds, rather than modifying a specific epitope (Table 1).
Table 1. Effects of individual replacements within phage-displayed EGFR domain III on recognition by anti-EGFR mAbs.
| R3 binding | Cetuximab binding | ||||||
|---|---|---|---|---|---|---|---|
| Targeted residue | Non-tolerated | Partially tolerated | Tolerated | Non-tolerated | Partially tolerated | Tolerated | |
| Nimotuzumab antigenic region | D323 | I, L, M, V | P, R, S | N | I, L, M, V | P, R, S | N |
| L325 | - | - | A, K, P, R, S, V | - | - | A, K, P, R, S, V | |
| T330 | - | C | D, L. Q, S, W | - | C | D, L, Q, S, W | |
| K333 | C | I, V | L, M, Q, R, S | C | I, V | L, M, Q, R, S | |
| R353** | P | H, Q, S, T | K*, L, M, W | P | - | H, K*, L, M, Q, S, T, W | |
| S356 | I, L, P, Q, R, T | - | - | - | - | I, L, P, Q, R, T | |
| F357 | C, K, L, M, Q, R, S, T, V | - | Y | - | C | K, L, M, Q, R, S, T, V, Y | |
| T358 | F, G, P | E, H, Q, R, W | S | - | H, P, W | E, F, G, Q, R, S | |
| H359 | L, P, Q, R*, S, T, Y | - | - | - | L, P | Q, R*, S, T, Y | |
| P361 | F, G, H, V | Q | A, R | F, H | G, Q, V | A, R | |
| P362 | H, S, T | - | G | H | S, T | G | |
| Cetuximab structural epitope | Q384 | P | - | E, H, K, L, R, T | K, P, R | - | E, H, L, T |
| Q408 | - | - | A, F, L, P, S, T, V | F, P, S, T | - | A, L, V | |
| S418 | C | - | A, G*, K, L, M, V | C, K, L, M, V | - | A, G* | |
| S440 | Y | - | F, H, L, Q, R, T, V | F, H, L, Q, R, T, V, Y | - | - | |
| K443 | - | L, W | D, E, H, N, Q, R*, S | D, E, H, L, N, Q, S, W | - | R* | |
| K465 | - | - | E, L, Q, R, S, T, Y | E, Q, S, T | Y | L, R | |
| S468 | - | C | A, L, N, P, R, V | C, P, R | - | A, L, N, V | |
| I467 | - | - | K, L, M*, N, R, S | K, M*, N, R, S | L | - | |
| N473 | - | - | E, H, K*, L, P, T, V, W | - | - | E, H, K*, L, P, T, V, W | |
Solvent-exposed residues (> 20% RSA) of EGFR domain III surrounding by less than 12 Å the first identified critical aa contributing to nimotuzumab epitope formation (H359), as well as residues comprised in the cetuximab structural epitope, were individually randomized. * indicate replacements by the residue found at the equivalent position in mouse EGFR. Residue R353 (**) is included in both antigenic regions. The resulting phage-displayed mutated Ag variants were tested by ELISA on microplates coated with either anti-EGFR antibodies or the anti-tag 9E10 antibody (recognizing the c-myc tag fused to every protein in our display system). Display levels assessed with the anti-tag mAb were taken as a reference to normalize the reactivity of the different variants toward anti-EGFR antibodies. Relative reactivities (%), compared with the normalized reactivity obtained for wt EGFR domain III, were calculated for each of them. Each mutated variant was evaluated twice by ELISA in two independent experiments with separately produced phage preparations. Replacements resulting in mean relative reactivities below 50% and between 50 and 75% were classified as non-tolerated and partially tolerated, respectively. Tolerated substitutions were those contained in mutated variants keeping more than 75% relative reactivitiy. Profiles of tolerated, non-tolerated and partially tolerated mutations for nimotuzumab (left panel) and cetuximab (right panel, shaded in gray) binding are shown. Residues contributing to the formation of nimotuzumab functional epitope are contained in the box.
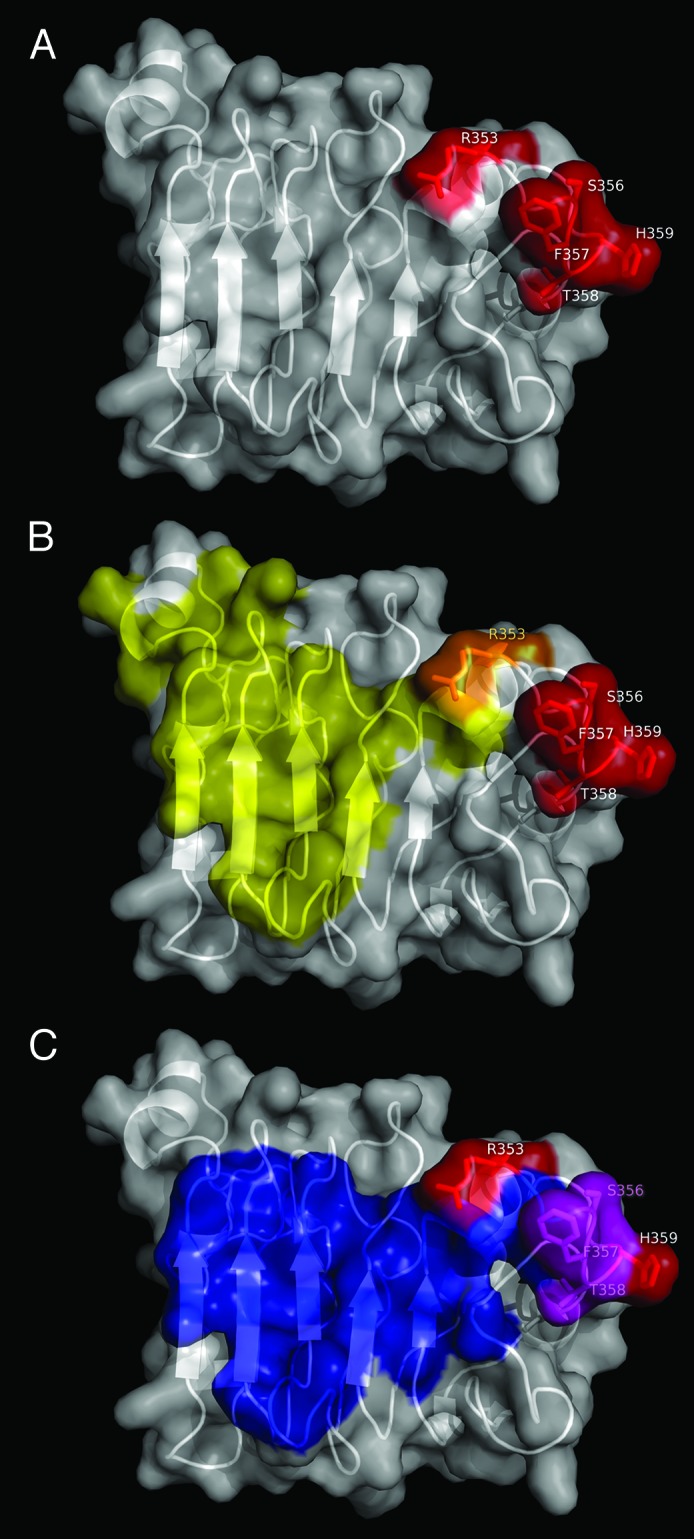
Figure 4. Functional epitope recognized by nimotuzumab. EGFR domain III is shown as a white cartoon with semi-transparent surface. Side chains of the residues contributing to nimotuzumab epitope formation according to mutagenesis studies are represented in red in (A). Relative locations of nimotuzumab and cetuximab epitopes are shown in (B). Domain III residues buried in the interface with cetuximab Fab in the complex (PDB code 1YY9) are colored yellow, while R353, the only residue belonging to both nimotuzumab functional epitope and cetuximab structural epitope, is highlighted in orange. The remaining nimotuzumab epitope residues are colored red. Overlapping between nimotuzumab epitope and ligand binding site is represented in panel (C). The interface between EGFR domain III and EGF and/or TGF-α (residues within 5Å of EGF and TGF-α in receptor/ligand complexes, PDB structures 1IVO and 1MOX respectively) is colored blue. Residues S356, F357, and T358 (highlighted in magenta) are involved in binding of both ligands and nimotuzumab. The rest of the nimotuzumab epitope is shown in red. The figures were generated with Pymol.
Some changes at other positions (323, 333, 361, and 362) affected recognition by both nimotuzumab and cetuximab (Table 1), despite being outside of the cetuximab structural epitope, indicating non-specific effects on domain III antigenicity. The original aa at these positions were thus not considered to belong to any of the functional epitopes. Remarkably, two of them were Pro residues (P361 and P362), presumably playing structural roles. Other positions accepted a wide range of replacements without losing antigenicity, ruling out a major contribution of the corresponding residues to epitope formation.
The functional nimotuzumab epitope (Fig. 4A) comprised a stretch of contiguous amino acids (S356-H359) and a non-contiguous, but close, residue (R353). The involvement of R353 (located within the cetuximab structural epitope) indicated some degree of overlapping between both epitopes (Fig. 4B) and explains competition between the antibodies.15 However, their fine specificity is not the same, as shown by the divergent effects of most mutations on reactivity toward each antibody. Residues directly involved in the interaction with EGF and transforming growth factor α (TGF-α) are also in close proximity to those mediating nimotuzumab binding (Fig. 4C). S356, F357, and T358 are indeed involved in the interactions with both ligands27,28 and with nimotuzumab. Overlapping thus explains the inhibitory effect of nimotuzumab on ligand-induced EGFR signaling.13
Identification of residues contributing to cetuximab binding validated our mapping strategy and expanded the functional map of cetuximab epitope
The whole structural cetuximab epitope (Ag residues in contact with the paratope in the complex17) was also explored by randomization (Fig. S2A). Lack of tolerance to multiple replacements (Table 1) confirmed the critical role of K443, K465, and l467, already known to contribute to epitope formation.16,17,21 I467 could not be replaced by any other residue. K443 could only be substituted by Arg (a very similar positively charged aa), while K465 was functionally replaced by both Arg and Leu, highlighting the contribution of hydrophobic interactions at this position. Successful reproduction of previous cetuximab functional mapping data16,21 validated our phage-based approach. The current study included several replacements of each critical residue, providing a more complete characterization of their roles. Two additional relevant residues (S418 and S440) were identified. Their contributions had remained elusive, probably due to the lower local sequence space coverage of previous mutagenesis studies. S418 could only be substituted by smaller residues (Gly and Ala). Larger side chains abrogated recognition, presumably through steric hindrance. Cetuximab mapping experiments, besides providing a reliable validation based on a well-known epitope, revealed new information that expanded the functional map of this antigenic determinant.
Three additional residues (Q384, Q408, and S468) showed a mixed pattern of response to mutations. While tolerance to diverse replacements (including some non-conservative ones) ruled out major individual energetic contributions of the original aa, the effects of other changes indicated an influence on the Ag/ paratope interface. Positions 384 and 468 were not tolerant to the introduction of Cys and Pro (probably disrupting Ag folding in the epitope vicinity), but also of Lys/Arg, presumably due to non-favorable electrostatic interactions. The replacement S468R deserves particular attention because it has been found in cetuximab-resistant tumor cells.22 The replacement of other residues within the cetuximab structural epitope had no functional consequences. The above described effects were specific, since almost all these variants were properly recognized by nimotuzumab (Fig. S2B). Divergent effects of most mutations within the structural cetuximab epitope on reactivity toward both antibodies provided further support to the notion that their target epitopes, although adjacent and partially overlapped, are different.
Combinatorial scanning highlighted the dominant role of heavy chain CDR3 residues within nimotuzumab paratope
The antibody paratope (in the form of a phage-displayed nimotuzumab-derived scFv) was also scanned by mutagenesis. Due to the frequent predominant role of the heavy chain in antibody recognition, the heavy chain variable region (VH) was initially targeted, while the light chain variable region (VL) was left unaltered. Twenty-five positions within VH complementarity-determining regions (CDRs) were diversified in a synthetic library of 8 x 107 members. Residues within protruding segments of VH CDR1 (Y27-Y33), CDR2 (N52-N58), and CDR3 (Q95-G100E) were soft-randomized. CDR1 F29 (keeping the canonical CDR conformation) was only replaced by a small set of hydrophobic residues (F, I, M, L, V), presumably able to fulfill the required structural role. P52A (CDR2), was not modified in the library due to its structural relevance.
A single round of phage selection on erEGFR recombinant protein allowed the segregation of nimotuzumab-derived scFv variants that lost or retained the ability to bind the target Ag. The set of Ag-binding positive variants (25 unique sequences) showed total conservation of some original residues (F29, Y33, Q95, G96, L97, W98, S100A, D100B, G100C, and G100E) and a marked over-representation of others (Y32, N52, F99 and D100), only replaced by a single similar aa (Table 2). The strong conservation of VH CDR3 residues pointed to a dominant functional role of this region. The remaining targeted positions accommodated several aa with diverse physicochemical properties among positive variants (up to eight different residues per position). Although these findings did not rule out cooperative contributions of the corresponding original residues to recognition, library screening was useful to define a surface patch formed by the majority of the CDR3 residues and the adjacent CDR2 N52 (also conserved among positive variants) that plays a crucial role in shaping the paratope interaction (Fig. 5). This patch could be used as the starting region for a more detailed exploration in the search for individually critical residues (see below). R100D was the only targeted residue within VH CDR3 that could be replaced by diverse aa (i.e., Asp and Gln) without affecting binding, probably due to the fact that its side chain points away from the previously defined surface patch (Fig. 5). Conserved residues within CDR1 (F29, Y32, Y33) were partially buried in the 3D structure, so their contribution is not necessarily restricted to direct interactions with the Ag, but also associated with effects on VH domain folding.
Table 2. Exploration of nimotuzumab paratope through combinatorial and site-directed mutagenesis.
| Targeted original residue | CDR | Frequencies of residues among the EGFR-positive variants retrieved from the library | Effect of individual replacements | ||
|---|---|---|---|---|---|
| Tolerated | Partially tolerated | Non-tolerated | |||
| Phe 29 | H1 | F (100%) | ND | ||
| Tyr 32 | H1 | Y (96%), F (4%) | ND | ||
| Tyr 33 | H1 | Y (100%) | ND | ||
| Asn 52 | H2 | N (84%), D (16%) | - | - | A, F, I, K, L, R, T, V, Y |
| Gln 95 | H3 | Q (100%) | - | - | C, E, F, K, L, P, R, S, T, V |
| Gly 96 | H3 | G (100%) | - | - | L, M, N, P, Q, R, T, V, W, Y |
| Leu 97 | H3 | L (100%) | - | - | F, K, N, P, Q, R, S, V, W |
| Trp 98 | H3 | W (100%) | - | F | A, C, E, L, M, N, P, Q, R, V |
| Phe 99 | H3 | F (96%), Y (4%) | W, Y | - | A, I, L, M, P, Q, R, S, T, V |
| Asp 100 | H3 | D (96%), N (4%) | - | - | A, C, E, G, L, P, Q, R, S, T, W |
| Ser 100A | H3 | S (100%) | - | - | A, C, D, F, G, I, K, L, N, R, T, V |
| Asp 100B | H3 | D (100%) | - | - | A, E, F, G, L, P, R, S, T, V, W |
| Gly 100C | H3 | G (100%) | - | - | A, C, D, I, L, N, P, R, S, V, W |
| Gly 100E | H3 | G (100%) | - | - | A, D, E, L, P, R, T, V, W |
A scFv antibody fragment derived from nimotuzumab (comprising its VH and VL regions connected by a linker peptide) was phage-displayed. VH CDRs were diversified in a phage-displayed library by simultaneous soft-randomization of 25 solvent-exposed residues. Phage selection on recombinant erEGFR was useful to pick a set of Ag-binding positive variants, keeping more than 75% relative reactivity compared with the original non-mutated scFv, which was composed by 25 unique sequences. While some of the targeted positions showed a high variability among this set, positive scFv molecules kept the original residues at 14 positions (shown in the table), either exclusively or only replaced by a single additional aa, sharing chemical properties and/or shape with them. Eleven of these positions (harboring solvent-exposed side chains) were separately randomized in order to assess the individual contributions of the corresponding original residues. Since conservation of other non-exposed original residues could be related with their structural roles, they were not targeted (ND = non-determined). The relative reactivity of each single mutated scFv variant against erEGFR was calculated, taking Ag recognition by the non-mutated scFv as the reference. Those mutations resulting in relative reactivities below 50%, between 50 and 75%, and higher than 75%, were classified as non-tolerated, partially tolerated and tolerated, respectively.

Figure 5. Functional map of the nimotuzumab paratope emerging from combinatorial mutagenenesis scanning. The structure of nimotuzumab Fv (PDB code 3GKW) is represented as a cartoon with semi-transparent surface. Heavy chain is shown in white, while light chain is represented in gray. Residues within protruding segments of complementarity determining regions of both chains (which were diversified through combinatorial mutagenesis in phage-displayed libraries) are highlighted in different colors. Targeted VL residues, for which a regular pattern of responsiveness to mutations was not found, are colored yellow. Those VH residues that could be replaced by multiple aa without affecting EGFR recognition are shown in green, while critical functional residues within VH CDR1, CDR2 and CDR3 that were conserved among EGFR-binding positive variants selected on the antigen are colored cyan, purple and dark blue, respectively. The figure was generated with Pymol.
Combinatorial screening also showed a contribution of nimotuzumab light chain to antigen recognition
Once the functional role of nimotuzumab VH domain was established, the contribution of the VL domain was explored. Sixteen protruding residues within VL CDRs were totally randomized, keeping VH domain unaltered (Fig. 5). Because total randomization would have resulted in a large theoretical diversity (difficult to be covered in a single library), four independent CDR-shuffling libraries were constructed. Two libraries (containing 1.77 × 109 and 6.5 × 108 members) targeted two different VL CDR1 segments (S26-N27A and V27C-N30). The usefulness of the second library was limited due to the frequent emergence of variants with low or negligible display levels (assessed with the anti-tag mAb). Two other libraries (having sizes of 2.5 × 107 and 1.35 × 109, respectively) included modifications at CDR2 (K50, S52-R54) and CDR3 residues (H93-P95).
Phage selection from these libraries on erEGFR did not result in the enrichment of variants keeping the original residue at any VL position (Fig. 5). Position 95, despite accepting several aa with different properties, had a high abundance of the original Pro among Ag-binding variants (74%). Tolerance to multiple mutations (without a regular sequence pattern) suggested a limited functional role of VL residues. However, the emergence of phage-displayed Ag-binding negative variants having particular combinations of VL mutations showed that this domain has a definite contribution to nimotuzumab reactivity.
Individual mutagenesis of selected VH CDR positions refined the functional map of the nimotuzumab paratope
Even though combinatorial scanning delineated a general functional picture of the paratope, this approach did not provide reliable information about the role of each particular aa, due to the mixed effects of simultaneous mutations. Site-directed mutagenesis, focused on accessible VH residues conserved under the pressure of the selector Ag, confirmed the critical role of Q95, G96, L97, W98, F99, D100, S100A, D100B, G100C, G100E (VH CDR3), as well as of the neighbor VH CDR2 N52 (Table 2). ScFv immunoreactivity was fully abolished by multiple independent mutations targeting them. Partial and total tolerance to replacements W98F and F99Y/F99W showed the contribution of aromatic rings at both positions. The above described results rendered a refined functional map of the explored surface patch within the nimotuzumab paratope and revealed a subset of residues that are critically required for binding. It is important to remark that such a comprehensive mutagenesis scanning was restricted to a functionally dominant region previously highlighted through library screening, and additional cooperative effects from the remaining VH CDR residues and from nimotuzumab VL are likely to contribute to binding.
Information from functional mapping guided the generation of an in silico model of binding between nimotuzumab and EGF receptor
The first step toward the generation of a binding model was a blind docking simulation between nimotuzumab Fv and EGFR domain III. Solutions were filtered not only by typical docking quality criteria, but also by using the involvement of the already identified functional epitope/paratope in the putative binding interfaces to distinguish those that were likely to resemble the real complex structure. Those solutions engaging regions on either the antibody or the antigen that are located far apart from the ones already identified as relevant in mutagenesis studies were discarded. The lowest energy solution emerging as plausible from this analysis was the starting point for a wide perturbation dock and a second filtering round according to functional mapping data, using the same criterion to eliminate those solutions centered in the wrong locations. Four poses were thus selected for a subsequent focused perturbation dock aimed at evaluating funnel formation in order to confirm their properties as local energy minima. The structure with the lowest energy within each funneling group was challenged by in silico mutagenesis. Both funneling quality and the correspondence between the effects of mutations on binding in virtual and experimental settings were used to choose the best model.
The resulting molecular model showed a good docking funnel, with a high likelihood of being a global energy minimum (Fig. 6A). More than 70% of the in silico replacements within both the paratope and the antigen on this model reproduced the effects of the same mutations in the experimental screening (having the highest coincidence rate among the four models compared in the final step). The predicted epitope/paratope contact area was 1404 Å2. According to this model, the cluster of residues belonging to the functional epitope forms the major part of the predicted structural epitope (Fig. 6B), which is semi-docked in a shallow cavity formed mainly by VH CDR3, but also by VL CDR3 and to a lesser extent by VL CDR1 (Fig. 6B). Strikingly, although VH CDR3 lies in close proximity to the EGFR epitope, a detailed analysis of the predicted interface (Fig. 6C) did not reveal many direct interactions involving VH CDR3 side chains. The most notable exception is L97 (VH CDR3), which interacts with the EGFR backbone. If this model is correct, the role of VH CDR3 could be to create a complementary surface to accommodate the epitope in the right position to establish a network of cooperative interactions with the paratope as a whole. EGFR S356 appears to form an H-bond with the backbone of VH CDR3 G96. Additional interactions of EGFR residues within nimotuzumab functional epitope (S356, F357, T358, H359) involve paratope residues within VL CDR1, VH CDR1 and VH CDR2.
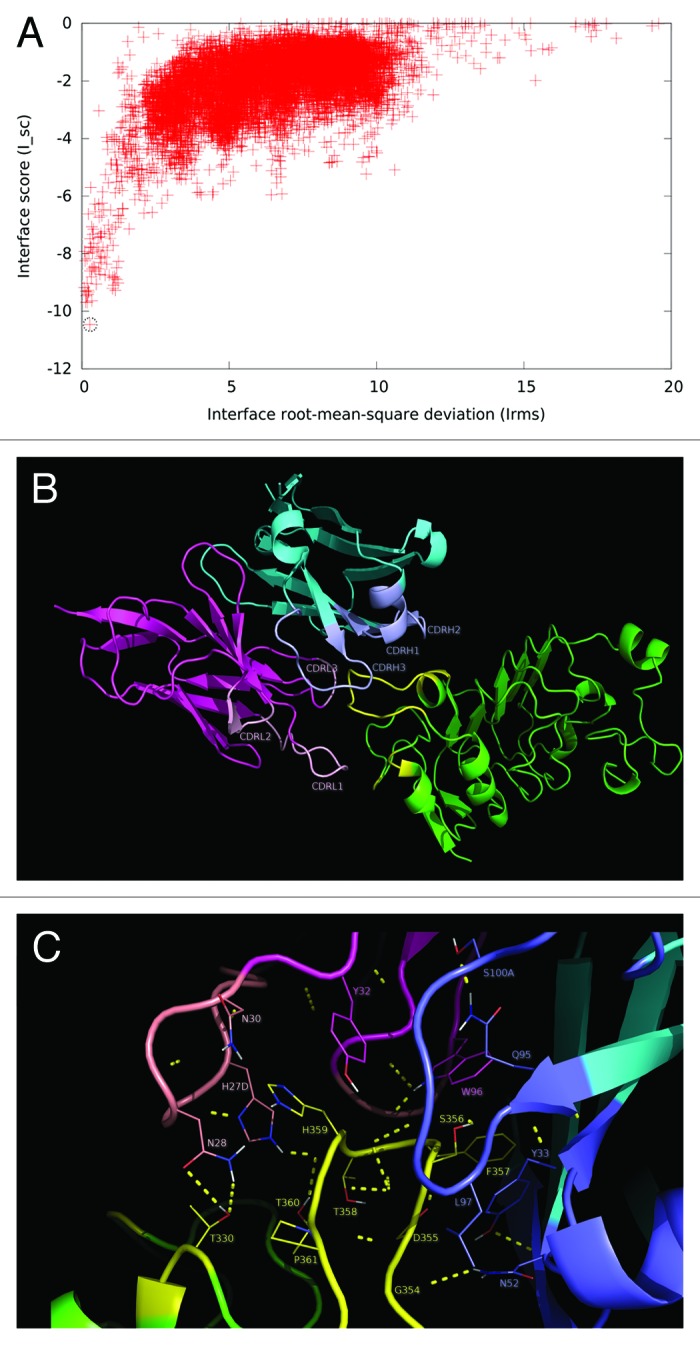
Figure 6. Structural model of nimotuzumab/EGFR complex. (A) Energy landscape of the perturbation analysis from which the best docking model (solution indicated by the dotted line circle) was obtained. (B) Cartoon representation of the structure of the nimotuzumab/EGFR complex according to the selected model. Nimotuzumab heavy and light chains, and erEGFR are colored cyan, magenta and green, respectively. The structural epitope is in yellow while complementarity determining regions from heavy and light chains are represented in blue and red respectively. (C) Interaction interface. Side chains of residues in the interface are shown as lines and hydrogen bonds are shown as discontinuous yellow lines. Residues and chains are colored as in (B).
A detailed inspection of in silico mutagenesis results predicted a critical role for four EGFR residues (S356, H359, P361, and P362). This was in agreement with the previously assigned functional role of S356 and H359, and with the influence of P361 and P362 in domain III antigenicity toward both nimotuzumab and cetuximab (Table 1). Paratope in silico mutagenesis rendered results consistent with a major role of nimotuzumab VH region in recognition. Multiple VH residues (13 in total) were predicted to be critical for binding. Among them, F29, Y32, N52, Q95, L97, S100A, D100B, and G100C had been previously shown to be functionally relevant (Table 2). Only three VL residues were predicted to be critical. One of them was P95 (highly prevalent among phage-displayed Ag-binding positive variants). The absence of a 100% match between in silico and experimental mutagenesis results could be related to the inability of computational tools to properly handle backbone flexibility and water-mediated interactions.
Discussion
Both binding affinity and fine specificity dictate the outcome of antibody therapy. Different antibodies against the same target can have diverse, and even opposite, biological effects, which have been associated to recognition of particular epitopes,29,30 and to the creation or disruption of different interactions.31,32 The identity of the epitopes can be deciphered through either structural33 or functional methods.34 The latter procedures are based on the identification of energetically critical residues contributing to epitope formation. Large functional mapping efforts have been done to characterize anti-EGFR antibodies (e.g., cetuximab, panitumumab).16-21 The initial goal of understanding neutralizing ability on ligand-mediated activation has subsequently expanded to the design of antibody combinations targeting different epitopes20 with synergic effects.35 This application requires quick mapping of multiple epitopes, before crystal structures of the Ag/Ab complexes can be solved.
Selection of mimotopes resembling EGFR epitopes from large random phage-displayed libraries has produced controversial results. While some peptides selected on cetuximab resemble the array of side chains forming its epitope,36 other immunological mimics do not show an obvious similarity to EGFR.37 Some attempts to find the correspondence between selected peptides and Ag sequences have resulted in the identification of antigenic regions totally different from the actual epitope(s).38
In contrast with peptide-based mapping approaches, site-directed mutagenesis of EGFR, either produced as a soluble molecule17-20 or displayed at the surface of mammalian cells,21 allowed the identification of epitope location through the definition of mutational hot spots disrupting the Ag/Ab interaction. Membrane display overcomes the need for expression and purification of multiple recombinant mutated Ag variants, and places the desired replacements in the more natural context, the full-length receptor anchored on the plasma membrane. The number of mutated Ag variants that can be individually screened in these experimental settings is usually limited by the complexities of mammalian cell manipulation.
Due to its higher efficiency, EGFR epitope mapping has been dominated by yeast display of mutated Ag variants generated through error-prone PCR.16,35 Even though this approach has multiple advantages (e.g., antigen synthesis in an eukaryotic environment, no requirement for prior epitope knowledge, the possibility to screen hundreds of variants by flow cytometry), the phage-based strategy described here keeps the high throughput potential and incorporates valuable features. While sequence space coverage by error-prone PCR can be very extensive, site-directed randomization results in more comprehensive local exploration of a given region. In the current case, such scanning reached every solvent-exposed residue within two antigenic regions: the structural cetuximab epitope and the putative nimotuzumab contact region. Several mutations (4–10) at each targeted position were screened (7.3 replacements per position on average), allowing the definition of the molecular properties underlying each interaction. Our study included 163 EGFR mutated variants (less than those characterized by yeast display), but replacements were concentrated in functionally relevant regions, and useless mutations, like those targeting Cys residues, were avoided.
Two pre-requisites need be fulfilled to use this mapping strategy. The first one is that the phage-displayed form of the Ag keeps its interaction(s). Successful display of EGFR Dom III+482–514 (a complex protein fragment having five disulfide bonds) extended the scope of previous experiences involving interleukin-2 as the target.29,30 Despite these and other examples of display of complex mammalian proteins on filamentous phages,39 the inability to display a given target in an appropriate antigenic form would preclude our mapping strategy to be used as described here. Yeast or ribosome display could be valuable alternatives in such cases. The second requirement is some previous knowledge about the recognized antigenic region. The current work took advantage of the lack of cross-reactivity of both nimotuzumab and cetuximab with mouse EGFR to obtain a relatively small collection of mutated variants (23) useful to scan the EGFR domain III surface in the search for critical residue(s). Both this collection (useful to characterize any non-cross-reactive anti-EGFR antibody) and the remaining 140 mutated variants can be stored (as Ag-displaying phages and as DNA) for new mapping experiments. Tailor-made additional collections designed to map other antibodies (taking into account the available information about their interactions) could be easily constructed and screened.
The only previous attempt to map the nimotuzumab epitope relied on in silico docking simulations and led to the prediction that the antigenic determinant overlaps with the one recognized by cetuximab, but is displaced toward its C-terminal end. The antibody would thus inhibit EGF binding (as cetuximab does), but allow the free receptor to dimerize spontaneously.15 This low level ligand-independent activation was postulated to determine the observed favorable clinical profile of nimotuzumab compared with cetuximab (less toxicity). According to our mapping results, nimotuzumab binding should preclude receptor self-dimerization through the recognition of an epitope partially overlapping with the one recognized by cetuximab and displaced toward the domain III N-terminal end. This is consistent with the recently reported ability of nimotuzumab to inhibit ligand-independent EGFR activation.40 Alternative explanations addressing the nimotuzumab low toxicity, like its moderate binding affinity,41,42 its particular therapeutic regime, and its fine specificity (now known to differ from cetuximab), thus deserve further exploration.
Beyond the implications of epitope identification to elucidate the mechanism of action of antibodies, resistance-related issues are attracting increasing attention. While failure of cetuximab and panitumumab therapy is frequently associated with mutations affecting the signaling cascade downstream of EGFR,43,44 the target epitope itself can evolve to escape antibody recognition. The replacement S468R in the erEGFR arises during cetuximab treatment, both in vitro and in vivo,22 resulting in recognition abrogation and targeting failure. Unlike mutations affecting downstream signaling pathways, epitope variations have specific effects for each antibody, and define unique resistance profiles. Such molecular specificity is likely to be behind the effectiveness of panitumumab after disease progression under cetuximab therapy.45,46 The clinical importance of mutations affecting epitopes adds further relevance to functional mapping mutagenesis-based studies. The current work is the first attempt to decipher the influence of erEGFR mutations on recognition by nimotuzumab. Detailed knowledge of the contribution of five critical residues and their neighbors allows an accurate prediction of the significance of potential antigen modifications and opens a new avenue to study sensitivity/resistance. Divergent effects of multiple mutations on reactivity with nimotuzumab and cetuximab predict different resistance profiles. The same is expected with respect to panitumumab (having a different set of critical residues).21 The information about target epitopes improves our understanding of EGFR-directed therapies, and increases the chances of achieving clinical benefit through personalized strategies based on the available therapeutic agents and the molecular heterogeneity of patients and their tumors.
The mutagenesis study of the paratope, besides completing the functional picture of the interaction from the antibody side, revealed critical regions (mainly the VH CDR3) that dictate binding ability of the antibody. The relevance of the whole CDR3 contrasts with the finding of a few individually critical residues within other paratopes.47 Such knowledge is essential for any further binding site engineering.18 In the same way that the target antigen can evade therapy effects, the therapeutic agent can be optimized (through directed evolution) to improve clinical efficacy. Novel paratopes arising from these procedures conserve the critical functional features, together with modifications aimed at incorporating/improving biological functions. Such approaches have led to affinity48 and specificity optimization,47 and even to the incorporation of dual specificities within the same paratope.49
While a deeper mechanistic understanding of antibody biological effects would require knowing not only the identity of the epitope/paratope, but also the geometry of the Ag/Ab complex (which would only be revealed through experimental structural studies), in silico docking simulations were able to predict a plausible picture of nimotuzumab interactions. The major drawback of these techniques is the large number of possible solutions, resembling or not the real binding modes. Almost 40% of the complexes cannot be correctly predicted by docking procedures.50 This limitation is particularly serious when studying complexes with protein antigens, usually involving large interfaces, in sharp contrast with small antigens accommodated inside a paratope cavity.51 Our functional mapping results allowed substantial restriction of the three-dimensional space in the search for realistic binding models. The results were thus strongly biased toward identifying models that involve the functionally relevant regions, unlike the previously reported relatively blind surface scanning.15 The use of information about the residues involved in epitope/paratope formation to filter possible solutions has been used to guide docking.52 The model we propose here for nimotuzumab/EGFR interaction is consistent with most experimental evidence and could be a useful tool for further exploration and engineering of the binding site.
In summary, the current work provided for the first time a detailed functional map of the interaction of nimotuzumab with the target antigen EGFR. Besides adding a molecular dimension to our understanding of its therapeutic effects and opening new possibilities for the study and optimization of its sensitivity/resistance profile among human tumors, similar functional mapping procedures could be used to dissect the interactions of other therapeutic agents, particularly those targeting the EGF/EGFR system.
Methods
Assessment of conformation-sensitivity of nimotuzumab epitope
Maxisorp microtiter plates were coated overnight at 4 °C with erEGFR recombinant protein at 10 µg/ml in phosphate buffered saline (PBS). Some wells were sequentially treated with dithiothreitol (DTT) and iodoacetamide (both at 100 mmol/l) in PBS during 1h at room temperature (RT) each in the darkness. Coated untreated wells were simultaneously incubated with PBS. Plates were washed with PBS, and blocked with skim powder milk at 4% (w/v) in PBS (M-PBS) during 1h at RT. Anti-EGFR mAbs (10 µg/ml) were incubated in the plates for 1h at RT. Polyclonal anti-erEGFR monkey antiserum, diluted 1/500 in M-PBS, was used as positive control. Plates were washed with 0.1% (v/v) Tween 20 (PBS-T) and incubated during 1 h at RT with an anti-human IgG antibody conjugated to horseradish peroxidase (HRP), appropriately diluted in M-PBS. After washing the plates with PBS-T, substrate solution (500 µg/ml ortho-phenylenediamine and 0.015% hydrogen peroxide in 0.1 mol/l citrate-phosphate buffer, pH 5.0) was added. The reaction was stopped after 15 min, with 2.5 mol/l sulfuric acid. Absorbance was measured at 490 nm.
Phage display of human EGFR domain III and a nimotuzumab-derived antibody fragment
The gene coding for human EGFR Dom III+482–514 (flanked by ApaLI and NotI restriction sites) was cloned into pHAB phagemid.47 Nimotuzumab VH and VL genes (flanked by ApaLI/SfiI and SalI/NotI restriction sites respectively) were also cloned into pHAB, connected by the gene coding for a linker peptide.TG1 E. coli cells ((K12_(lac-pro), supE, thi, hsdD5/F’ traD36, proA+B+, lacIq, lacZ_M15) were transformed with the resulting genetic constructs and used to rescue phages displaying each protein at a 50 ml scale.53 Binding properties of the phage-displayed proteins were tested by ELISA (see below).
Site-directed mutagenesis and randomization
Mutated variants of phage-displayed proteins were constructed by Kunkel mutagenesis54 using described procedures.55 Briefly, single strand DNA (from either Dom III+482–514 gene-containing or nimotuzumab-scFv gene-containing pHAB phagemid) obtained from phages produced by the CJ236 E. coli strain (dut- ung- thi-1 relA1 spoT1 mcrA/pCJ105 [F’ camr]) was used as the template. Antisense mutagenic oligonucleotides were used to introduce the desired mutations or the degenerate randomization triplet NNK. Phagemid inserts were sequenced by Macrogen.
Construction of combinatorial libraries
The nimotuzumab-scFv-derived library having VH mutations was synthesized and cloned into pHAB phagemid by GENEART. Twenty-four residues at VH CDRs were soft randomized by introducing degenerate triplets containing the original nucleotide (90%) plus the equimolar mixture of the three remaining nucleotides (10%) at each position. The codon coding for F29 was substituted by TTC. Underlined letters represent soft-randomized positions. TG1 E.coli cells were electroporated with library DNA.
Four nimotuzumab-scFv-derived libraries having VL mutations were constructed. Single strand DNA corresponding to stop templates,56 in which each VL CDR had been separately replaced by a segment containing three stop codons (TAA)3, was purified from phages produced by CJ236 E.coli cells, and used for large scale reactions with antisense mutagenic oligonucleotides. Each stop-containing segment was thus replaced by a stretch of NNK triplets resulting in total randomization of the targeted positions.56 TG1 E.coli cells were electroporated with the reaction products to obtain the libraries.
Phage selection
Library phages were rescued with M13KO7 helper phage at a 300 ml scale as described.53 Immunotubes (Nunc) were coated with recombinant erEGFR at 10 µg/ml in PBS overnight at 4 °C. Purified phages (5 × 1012 viral particles) and coated immunotubes were blocked with M-PBS during 1h at RT. Blocked phages were incubated on blocked immunotubes 1h at RT. Tubes were washed 20 times with PBS-T and twice with PBS. Bound phages were eluted with 100 mmol/l triethylamine during 10 min at RT, and neutralized with 1 mol/l Tris, pH 7.5. Exponentially growing TG1 E.coli cells were infected with selected phages and used to rescue phage-displayed nimotuzumab-scFv variants at a 96-well scale.
ELISA screening of the reactivity of EGFR domain III and its mutated variants
Maxisorp microtiter plates were coated overnight at 4 °C with anti-EGFR mAbs, the anti-c-myc tag 9E10 mAb and an unrelated mAb at 10 µg/ml in PBS. Plates were blocked 1 h at RT with M-PBS. Purified phages displaying EGFR Dom III+482–514 or its mutated variants (diluted in M-PBS) were added to the plates and incubated during 1h at RT. After washing with PBS-T, an anti-M13 mAb conjugated to HRP (GE Healthcare, USA), appropriately diluted in M-PBS, was added. Plates were incubated 1h at RT and washed. Substrate solution was added. The reaction was stopped after 15 min with 2.5 mol/l sulfuric acid. The absorbances were measured at 490 nm.
Characterization of phage-displayed nimotuzumab-scFv and its mutated variants
Polyvinyl chloride microtiter plates were coated overnight at 4 °C with recombinant erEGFR, the anti-c-myc tag 9E10 mAb and bovine serum albumin (BSA) at 10 µg/mL in PBS. Plates were blocked for 1 h at RT with M-PBS. Purified phages (1012 viral particles/ml in M-PBS), or phage-containing 96-well supernatants (diluted 1/3 in M-PBS) were added to the plates and incubated during 2h at RT. Bound phages were detected with an anti-M13 mAb conjugated to HRP as described in the previous section.
Phage-displayed nimotuzumab-scFv variants (either recognizing the target Ag or not) were chosen for sequencing. XL1-Blue E. coli cells (recA1 endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac F´ proAB lacIqZ_M15 Tn10 Tetr) were infected with the corresponding phage-containing supernatants and used to purify plasmid DNA with the QIAprep Spin Miniprep kit (Qiagen, USA). Phagemid inserts were sequenced by Macrogen (Korea).
In silico modeling of the interaction between EGFR and nimotuzumab
Docking simulations were performed with Rosetta Dock program57 from Rosetta Commons 3.5 suite. Fv region of nimotuzumab structure (from PDB 3GKW15) was superimposed to cetuximab coordinates in PDB file 1YY9,17 and the coordinates of the resulting Fv/EGFR domain III complex were the input structure for a blind docking procedure. 105 solutions were generated using the flags -randomize1, -randomize2, -spin, which allowed a complete randomization of both docking partners' positions. Solutions with lowest Interface score (I_sc) values were filtered according to the involvement of the already identified functional epitope/paratope. The lowest energy solution was the subject of a wide perturbation dock generating 105 solutions (using the flags -dock_pert 15 25 and -spin). Poses with the lowest I_sc values were filtered again taking into account functional data. The four selected poses were separately submitted to a localized perturbation dock (with the flags -dock_pert 3 8, -spin), resulting in 104 solutions from each one. Graphical representation of I_sc vs. Interface rms (Irms) was used to test funneling formation (indicative of the starting complex being at least a local, and hopefully a global, energy minimum). The lowest energy pose from each funneling group was the subject of an in silico mutagenesis analysis (see below).
In silico mutagenesis
Residues already explored in the experimental mutagenesis study (D323, L325, T330, K333, R353, S356-H359, P361, P362, Q384, Q408, S418, S440, K443, K465, I467, S468, N473 from erEGFR domain III; S26-N30, K50-R54 and H93-P95 from nimotuzumab VL region; and Y27-Y33, N52-N58 and Q95-G100E from VH region) were individually replaced, using the above described docking models as the input coordinates. Each structure was minimized with constraints, using Rosetta Commons 3.5 suite. Substitution of each residue by all the other standard aa was performed with the ddg_monomer application58 and the low resolution protocol. Mutations with ddG values over +1 RU (Rosetta Units) were considered to affect folding. Interfaces of the mutated structures with ddG values lower than +1 RU were analyzed with score_interface application (also in Rosetta Commons 3.5 suite). Mutations with onebody delta energies over +1 RU were considered to affect the Ag/Ab interaction. Critical residues were those which could not be replaced by any other aa (or only by very similar residues) without affecting binding.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Tania Crombet for helpful discussions and Yaquelin Marichal for her excellent technical assistance.
Glossary
Abbreviations:
- aa
amino acid
- Ag
antigen
- CDRs
complementarity-determining regions
- DTT
dithiothreitol
- EGF
epidermal growth factor
- EGFR
EGF receptor
- ELISA
enzyme-linked immunosorbent assay
- erEGFR
EGFR extracellular region
- mAbs
monoclonal antibodies
- PBS
phosphate-buffered saline
- RSA
relative solvent accessibility
- RT
room temperature
- scFv
single chain Fv
- TGF-α
transforming growth factor alpha
- VH
heavy chain variable region
- VL
light chain variable region
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2:127–37. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 3.Baselga J. Targeting tyrosine kinases in cancer: the second wave. Science. 2006;312:1175–8. doi: 10.1126/science.1125951. [DOI] [PubMed] [Google Scholar]
- 4.Reichert JM. Marketed therapeutic antibodies compendium. MAbs. 2012;4:413–5. doi: 10.4161/mabs.19931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fernandez A, Spitzer E, Perez R, Boehmer FD, Eckert K, Zschiesche W, Grosse R. A new monoclonal antibody for detection of EGF-receptors in western blots and paraffin-embedded tissue sections. J Cell Biochem. 1992;49:157–65. doi: 10.1002/jcb.240490208. [DOI] [PubMed] [Google Scholar]
- 6.Mateo C, Moreno E, Amour K, Lombardero J, Harris W, Pérez R. Humanization of a mouse monoclonal antibody that blocks the epidermal growth factor receptor: recovery of antagonistic activity. Immunotechnology. 1997;3:71–81. doi: 10.1016/S1380-2933(97)00065-1. [DOI] [PubMed] [Google Scholar]
- 7.Rodríguez MO, Rivero TC, del Castillo Bahi R, Muchuli CR, Bilbao MA, Vinageras EN, Alert J, Galainena JJ, Rodríguez E, Gracias E, et al. Nimotuzumab plus radiotherapy for unresectable squamous-cell carcinoma of the head and neck. Cancer Biol Ther. 2010;9:343–9. doi: 10.4161/cbt.9.5.10981. [DOI] [PubMed] [Google Scholar]
- 8.Solomón MT, Selva JC, Figueredo J, Vaquer J, Toledo C, Quintanal N, Salva S, Domíngez R, Alert J, Marinello JJ, et al. Radiotherapy plus nimotuzumab or placebo in the treatment of high grade glioma patients: results from a randomized, double blind trial. BMC Cancer. 2013;13:299. doi: 10.1186/1471-2407-13-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramos-Suzarte M, Lorenzo-Luaces P, Lazo NG, Perez ML, Soriano JL, Gonzalez CE, Hernadez IM, Albuerne YA, Moreno BP, Alvarez ES, et al. Treatment of malignant, non-resectable, epithelial origin esophageal tumours with the humanized anti-epidermal growth factor antibody nimotuzumab combined with radiation therapy and chemotherapy. Cancer Biol Ther. 2012;13:600–5. doi: 10.4161/cbt.19849. [DOI] [PubMed] [Google Scholar]
- 10.Boland W, Bebb G. The emerging role of nimotuzumab in the treatment of non-small cell lung cancer. Biologics. 2010;4:289–98. doi: 10.2147/BTT.S8617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Strumberg D, Schultheis B, Scheulen ME, Hilger RA, Krauss J, Marschner N, Lordick F, Bach F, Reuter D, Edler L, et al. Phase II study of nimotuzumab, a humanized monoclonal anti-epidermal growth factor receptor (EGFR) antibody, in patients with locally advanced or metastatic pancreatic cancer. Invest New Drugs. 2012;30:1138–43. doi: 10.1007/s10637-010-9619-8. [DOI] [PubMed] [Google Scholar]
- 12.Allan DGP. Nimotuzumab: evidence of clinical benefit without rash. Oncologist. 2005;10:760–1. doi: 10.1634/theoncologist.10-9-760. [DOI] [PubMed] [Google Scholar]
- 13.Crombet-Ramos T, Rak J, Pérez R, Viloria-Petit A. Antiproliferative, antiangiogenic and proapoptotic activity of h-R3: A humanized anti-EGFR antibody. Int J Cancer. 2002;101:567–75. doi: 10.1002/ijc.10647. [DOI] [PubMed] [Google Scholar]
- 14.Diaz Miqueli A, Blanco R, Garcia B, Badia T, Batista AE, Alonso R, Montero E. Biological activity in vitro of anti-epidermal growth factor receptor monoclonal antibodies with different affinities. Hybridoma (Larchmt) 2007;26:423–31. doi: 10.1089/hyb.2007.0516. [DOI] [PubMed] [Google Scholar]
- 15.Talavera A, Friemann R, Gómez-Puerta S, Martinez-Fleites C, Garrido G, Rabasa A, López-Requena A, Pupo A, Johansen RF, Sánchez O, et al. Nimotuzumab, an antitumor antibody that targets the epidermal growth factor receptor, blocks ligand binding while permitting the active receptor conformation. Cancer Res. 2009;69:5851–9. doi: 10.1158/0008-5472.CAN-08-4518. [DOI] [PubMed] [Google Scholar]
- 16.Chao G, Cochran JR, Wittrup KD. Fine epitope mapping of anti-epidermal growth factor receptor antibodies through random mutagenesis and yeast surface display. J Mol Biol. 2004;342:539–50. doi: 10.1016/j.jmb.2004.07.053. [DOI] [PubMed] [Google Scholar]
- 17.Li S, Schmitz KR, Jeffrey PD, Wiltzius JJ, Kussie P, Ferguson KM. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell. 2005;7:301–11. doi: 10.1016/j.ccr.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 18.Li S, Kussie P, Ferguson KM. Structural basis for EGF receptor inhibition by the therapeutic antibody IMC-11F8. Structure. 2008;16:216–27. doi: 10.1016/j.str.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 19.Kamat V, Donaldson JM, Kari C, Quadros MR, Lelkes PI, Chaiken I, Cocklin S, Williams JC, Papazoglou E, Rodeck U. Enhanced EGFR inhibition and distinct epitope recognition by EGFR antagonistic mAbs C225 and 425. Cancer Biol Ther. 2008;7:726–33. doi: 10.4161/cbt.7.5.6097. [DOI] [PubMed] [Google Scholar]
- 20.Koefoed K, Steinaa L, Søderberg JN, Kjær I, Jacobsen HJ, Meijer PJ, Haurum JS, Jensen A, Kragh M, Andersen PS, et al. Rational identification of an optimal antibody mixture for targeting the epidermal growth factor receptor. MAbs. 2011;3:584–95. doi: 10.4161/mabs.3.6.17955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Voigt M, Braig F, Göthel M, Schulte A, Lamszus K, Bokemeyer C, Binder M. Functional dissection of the epidermal growth factor receptor epitopes targeted by panitumumab and cetuximab. Neoplasia. 2012;14:1023–31. doi: 10.1593/neo.121242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Montagut C, Dalmases A, Bellosillo B, Crespo M, Pairet S, Iglesias M, Salido M, Gallen M, Marsters S, Tsai SP, et al. Identification of a mutation in the extracellular domain of the Epidermal Growth Factor Receptor conferring cetuximab resistance in colorectal cancer. Nat Med. 2012;18:221–3. doi: 10.1038/nm.2609. [DOI] [PubMed] [Google Scholar]
- 23.Ramírez BS, Pestana ES, Hidalgo GG, García TH, Rodríguez RP, Ullrich A, Férnandez LE. Active antimetastatic immunotherapy in Lewis lung carcinoma with self EGFR extracellular domain protein in VSSP adjuvant. Int J Cancer. 2006;119:2190–9. doi: 10.1002/ijc.22085. [DOI] [PubMed] [Google Scholar]
- 24.Cochran JR, Kim Y-S, Olsen MJ, Bhandari R, Wittrup KD. Domain-level antibody epitope mapping through yeast surface display of epidermal growth factor receptor fragments. J Immunol Methods. 2004;287:147–58. doi: 10.1016/j.jim.2004.01.024. [DOI] [PubMed] [Google Scholar]
- 25.Sato JD, Kawamoto T, Le AD, Mendelsohn J, Polikoff J, Sato GH. Biological effects in vitro of monoclonal antibodies to human epidermal growth factor receptors. Mol Biol Med. 1983;1:511–29. [PubMed] [Google Scholar]
- 26.Van den Burg B, Dijkstra BW, Vriend G, Van der Vinne B, Venema G, Eijsink VG. Protein stabilization by hydrophobic interactions at the surface. Eur J Biochem. 1994;220:981–5. doi: 10.1111/j.1432-1033.1994.tb18702.x. [DOI] [PubMed] [Google Scholar]
- 27.Ogiso H, Ishitani R, Nureki O, Fukai S, Yamanaka M, Kim JH, Saito K, Sakamoto A, Inoue M, Shirouzu M, et al. Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell. 2002;110:775–87. doi: 10.1016/S0092-8674(02)00963-7. [DOI] [PubMed] [Google Scholar]
- 28.Garrett TP, McKern NM, Lou M, Elleman TC, Adams TE, Lovrecz GO, Zhu HJ, Walker F, Frenkel MJ, Hoyne PA, et al. Crystal structure of a truncated epidermal growth factor receptor extracellular domain bound to transforming growth factor alpha. Cell. 2002;110:763–73. doi: 10.1016/S0092-8674(02)00940-6. [DOI] [PubMed] [Google Scholar]
- 29.Rojas G, Pupo A, Leon K, Avellanet J, Carmenate T, Sidhu S. Deciphering the molecular bases of the biological effects of antibodies against Interleukin-2: a versatile platform for fine epitope mapping. Immunobiology. 2013;218:105–13. doi: 10.1016/j.imbio.2012.02.009. [DOI] [PubMed] [Google Scholar]
- 30.Rojas G, Cabrera Infante Y, Pupo A, Carmenate T. Fine epitope specificity of antibodies against interleukin-2 explains their paradoxical immunomodulatory effects. MAbs. 2014;6:273–85. doi: 10.4161/mabs.27224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boyman O, Kovar M, Rubinstein MP, Surh CD, Sprent J. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science. 2006;311:1924–7. doi: 10.1126/science.1122927. [DOI] [PubMed] [Google Scholar]
- 32.Klein C, Lammens A, Schafer W, Georges G, Schwaiger M. mossner, E., Hopfner, K.P., Umaña, P. & Niederfellner, G. Epitope interactions of monoclonal antibodies targeting CD20 and their relationships to functional properties. MAbs. 2013;5:1–12. doi: 10.4161/mabs.22771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davies DR, Cohen GH. Interactions of protein antigens with antibodies. Proc Natl Acad Sci U S A. 1996;93:7–12. doi: 10.1073/pnas.93.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Benjamin DC, Perdue SS. Site-directed mutagenesis in epitope mapping. Methods. 1996;9:508–15. doi: 10.1006/meth.1996.0058. [DOI] [PubMed] [Google Scholar]
- 35.Spangler JB, Neil JR, Abramovitch S, Yarden Y, White FM, Lauffenburger DA, Wittrup KD. Combination antibody treatment down-regulates epidermal growth factor receptor by inhibiting endosomal recycling. Proc Natl Acad Sci U S A. 2010;107:13252–7. doi: 10.1073/pnas.0913476107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hartmann C, Müller N, Blaukat A, Koch J, Benhar I, Wels WS. Peptide mimotopes recognized by antibodies cetuximab and matuzumab induce a functionally equivalent anti-EGFR immune response. Oncogene. 2010;29:4517–27. doi: 10.1038/onc.2010.195. [DOI] [PubMed] [Google Scholar]
- 37.Riemer AB, Kurz H, Klinger M, Scheiner O, Zielinski CC, Jensen-Jarolim E. Vaccination with cetuximab mimotopes and biological properties of induced anti-epidermal growth factor receptor antibodies. J Natl Cancer Inst. 2005;97:1663–70. doi: 10.1093/jnci/dji373. [DOI] [PubMed] [Google Scholar]
- 38.Cardó-Vila M, Giordano RJ, Sidman RL, Bronk LF, Fan Z, Mendelsohn J, Arap W, Pasqualini R. From combinatorial peptide selection to drug prototype (II): targeting the epidermal growth factor receptor pathway. Proc Natl Acad Sci U S A. 2010;107:5118–23. doi: 10.1073/pnas.0915146107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith GP, Petrenko VA. Phage Display. Chem Rev. 1997;97:391–410. doi: 10.1021/cr960065d. [DOI] [PubMed] [Google Scholar]
- 40.Berger C, Krengel U, Stang E, Moreno E, Madshus IH. Nimotuzumab and cetuximab block ligand-independent EGF receptor signaling efficiently at different concentrations. J Immunother. 2011;34:550–5. doi: 10.1097/CJI.0b013e31822a5ca6. [DOI] [PubMed] [Google Scholar]
- 41.Crombet T, Osorio M, Cruz T, Roca C, del Castillo R, Mon R, Iznaga-Escobar N, Figueredo R, Koropatnick J, Renginfo E, et al. Use of the humanized anti-epidermal growth factor receptor monoclonal antibody h-R3 in combination with radiotherapy in the treatment of locally advanced head and neck cancer patients. J Clin Oncol. 2004;22:1646–54. doi: 10.1200/JCO.2004.03.089. [DOI] [PubMed] [Google Scholar]
- 42.Garrido G, Tikhomirov IA, Rabasa A, Yang E, Gracia E, Iznaga N, Fernández LE, Crombet T, Kerbel RS, Pérez R. Bivalent binding by intermediate affinity of nimotuzumab: a contribution to explain antibody clinical profile. Cancer Biol Ther. 2011;11:373–82. doi: 10.4161/cbt.11.4.14097. [DOI] [PubMed] [Google Scholar]
- 43.Lièvre A, Bachet JB, Le Corre D, Boige V, Landi B, Emile JF, Côté JF, Tomasic G, Penna C, Ducreux M, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006;66:3992–5. doi: 10.1158/0008-5472.CAN-06-0191. [DOI] [PubMed] [Google Scholar]
- 44.Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, Juan T, Sikorski R, Suggs S, Radinsky R, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:1626–34. doi: 10.1200/JCO.2007.14.7116. [DOI] [PubMed] [Google Scholar]
- 45.Saif MW, Kaley K, Chu E, Copur MS. Safety and efficacy of panitumumab therapy after progression with cetuximab: experience at two institutions. Clin Colorectal Cancer. 2010;9:315–8. doi: 10.3816/CCC.2010.n.046. [DOI] [PubMed] [Google Scholar]
- 46.Sonoda H, Mekata E, Shimizu T, Endo Y, Tani T. Safety and efficacy of panitumumab therapy after metastatic colorectal cancer progression with cetuximab: Experience at a single Japanese institution. Oncol Lett. 2013;5:1331–4. doi: 10.3892/ol.2013.1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rojas G, Pupo A, Gómez S, Krengel U, Moreno E. Engineering the binding site of an antibody against N-glycolyl GM3: from functional mapping to novel anti-ganglioside specificities. ACS Chem Biol. 2013;8:376–86. doi: 10.1021/cb3003754. [DOI] [PubMed] [Google Scholar]
- 48.Lamdan H, Gavilondo JV, Muñoz Y, Pupo A, Huerta V, Musacchio A, Pérez L, Ayala M, Rojas G, Balint RF, et al. Affinity maturation and fine functional mapping of an antibody fragment against a novel neutralizing epitope on human vascular endothelial growth factor. Mol Biosyst. 2013;9:2097–106. doi: 10.1039/c3mb70136k. [DOI] [PubMed] [Google Scholar]
- 49.Bostrom J, Yu SF, Kan D, Appleton BA, Lee CV, Billeci K, Man W, Peale F, Ross S, Wiesmann C, et al. Variants of the antibody herceptin that interact with HER2 and VEGF at the antigen binding site. Science. 2009;323:1610–4. doi: 10.1126/science.1165480. [DOI] [PubMed] [Google Scholar]
- 50.Chaudhury S, Berrondo M, Weitzner BD, Muthu P, Bergman H, Gray JJ. Benchmarking and analysis of protein docking performance in Rosetta v3.2. PLoS One. 2011;6:e22477. doi: 10.1371/journal.pone.0022477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee M, Lloyd P, Zhang X, Schallhorn JM, Sugimoto K, Leach AG, Sapiro G, Houk KN. Shapes of antibody binding sites: qualitative and quantitative analyses based on a geomorphic classification scheme. J Org Chem. 2006;71:5082–92. doi: 10.1021/jo052659z. [DOI] [PubMed] [Google Scholar]
- 52.Vercruysse T, Boons E, Venken T, Vanstreels E, Voet A, Steyaert J, De Maeyer M, Daelemans D. Mapping the binding interface between an HIV-1 inhibiting intrabody and the viral protein Rev. PLoS One. 2013;8:e60259. doi: 10.1371/journal.pone.0060259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marks JD, Hoogenboom HR, Bonnert TP, McCafferty J, Griffiths AD, Winter G. By-passing immunization. Human antibodies from V-gene libraries displayed on phage. J Mol Biol. 1991;222:581–97. doi: 10.1016/0022-2836(91)90498-U. [DOI] [PubMed] [Google Scholar]
- 54.Kunkel TA. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc Natl Acad Sci U S A. 1985;82:488–92. doi: 10.1073/pnas.82.2.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rojas G. Fine epitope mapping based on phage display and extensive mutagenesis of the target antigen. Methods Mol Biol. 2014;1131:447–76. doi: 10.1007/978-1-62703-992-5_27. [DOI] [PubMed] [Google Scholar]
- 56.Fellouse FA, Sidhu SS. Making antibodies in bacteria. In Making and using antibodies. A practical handbook 2007. G.C. Howard, and M.R.Kaser, CRC Press, Boca Raton, Florida, pp 157-177. [Google Scholar]
- 57.Gray JJ, Moughon S, Wang C, Schueler-Furman O, Kuhlman B, Rohl CA, Baker D. Protein-protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. J Mol Biol. 2003;331:281–99. doi: 10.1016/S0022-2836(03)00670-3. [DOI] [PubMed] [Google Scholar]
- 58.Kellogg EH, Leaver-Fay A, Baker D. Role of conformational sampling in computing mutation-induced changes in protein structure and stability. Proteins. 2011;79:830–8. doi: 10.1002/prot.22921. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.