

Abstract
The epithelial cell adhesion molecule (EpCAM) is expressed by a wide range of human carcinomas, making it an attractive diagnostic and therapeutic target in oncology. Its recent identification on cancer stem cells has raised further interest in its use for tumor targeting and therapy. Here, we present the characterization and therapeutic potential of 3–17I, a novel human EpCAM-targeting monoclonal antibody. Strong reaction of 3–17I was observed in all lung, colon, and breast human tumor biopsies evaluated. By flow cytometry and confocal fluorescence microscopy, we demonstrate that 3–17I specifically targets EpCAM-positive cell lines. We also show evidence for mAb-sequestration in endo-/lysosomes, suggesting internalization of 3–17I by receptor-mediated endocytosis. The ribosomal-inactivating toxin saporin was linked to 3–17I, creating the per se non-toxic immunotoxin 3–17I-saporin, a promising candidate for the drug delivery technology photochemical internalization (PCI). PCI is based on a light-controlled destruction of endolysosomal membranes and subsequent cytosolic release of the sequestered payload upon light exposure. EpCAM-positive human cancer cell lines MCF7 (breast), BxPC-3 (pancreas), WiDr (colon), and the EpCAM-negative COLO320DM (colon), were treated with 3–17I-saporin in combination with the clinically relevant photosensitizer TPCS2a (Amphinex), followed by exposure to light. No cytotoxicity was observed after treatment with 3–17I-saporin without light exposure. However, cell viability, proliferation and colony-forming capacity was strongly reduced in a light-dependent manner after PCI of 3–17I. Our results show that 3–17I is an excellent candidate for diagnosis of EpCAM-positive tumors and for development of clinically relevant antibody-drug conjugates, using PCI for the treatment of localized tumors.
Keywords: EpCAM, drug delivery, cancer, antibody drug conjugate, antitumor, monoclonal antibody, antibody therapeutics, photochemical internalization
Introduction
The epithelial cell adhesion molecule (EpCAM; also called CD326, ESA, EGP-2, or TROP-1,) is one of the best-studied target antigens of human tumors.1,2 It is a type 1 membrane glycoprotein with an apparent molecular weight of 40 kDa3 and functions as a homotypic cell adhesion molecule.4
EpCAM is abundantly expressed in primary tumors and has been shown to be involved in metastasis of many epithelial tumors, particularly in adenocarcinoma.5,6 EpCAM is also expressed in the corresponding normal epithelia, albeit more variable and at lower expression levels than found in tumors.7 An increased EpCAM expression is a poor prognostic marker in breast and gallbladder carcinomas,8,9 and EpCAM is overexpressed in cancer stem cells in pancreatic and colorectal adenocarcinomas and in breast carcinomas.10-12 These data underscore the potential utility of EpCAM as an immunotherapeutic target for treatment of the most abundant human cancers.
Several anti-EpCAM therapeutic antibodies have been developed and tested in clinical studies over the past 30 years, as monotherapy or in combination therapy.13 The proposed mechanisms of their antitumor effects include antibody-dependent cell-mediated cytotoxicity (ADCC) and complement-mediated cytotoxicity (CDC).14
So far, few anti-EpCAM monoclonal antibodies (mAbs) have shown clinical potential. MT201 (adecatumumab) is a human mAb that is currently under clinical investigation in patients with prostate and breast cancer, where it is showing promising results.13-15 MOC31 is another anti-EpCAM antibody that has been studied extensively. Its humanized form has showed promising results in vivo, and has been utilized for tumor targeting in various drug delivery systems.15 One significant worry associated with clinical use of anti-EpCAM antibodies is the lack of selectivity for affected tissues and cells. Because EpCAM is expressed on a wide range of normal epithelia, systemic intolerability and damage of normal EpCAM-expressing tissue is seen with high-affinity antibodies. Tolerance studies of antibodies with different affinities have suggested that low affinity binders are indeed better tolerated.13 Hence, there is a clear need for the discovery of new anti-EpCAM antibodies with improved therapeutic potential.
Here, we present a novel human mAb targeting EpCAM that was discovered and patented by Affitech Research AS.16 3–17I shows good affinity, good cross-reactivity profiles and excellent ADCC and CDC activity, and shows improved properties over MT201 and MOC31, as described in Ref. 16. The antibody can potentially be used as a diagnostic or therapeutic, or as a basis for engineering other antibodies or binding molecules for EpCAM, e.g., bispecific antibodies, immunotoxins, antibody-drug conjugates (ADCs).
Photochemical internalization (PCI) is a drug delivery technology for local and light controlled cytosolic release of therapeutics entrapped in endosomes and lysosomes, where the effect will be confined to the illuminated area only. PCI is based on the use of a photosensitizer, as in photodynamic therapy (PDT), which co-localizes in endolysosomal compartments with the drug to be delivered. Optimal PCI photosensitizers are amphiphilic and bind to the plasma membrane with their lipophilic part incorporated into the lipid-layer of the membrane and the hydrophilic part sticking outwards and hence, PCI photosensitizers are taken up into the cells by non-receptor mediated endocytosis. Light or laser-activation of the PCI photosensitizer results in generation of mainly singlet oxygen, but also other reactive oxygen species that induces lipid peroxidation and ruptures of the endolysosomal membranes resulting in escape of the drug into the cytosol.17,18 The PCI principle has been documented in several animal models19,20 and a second generation photosensitizer, TPCS2a/Amphinex®, has been developed for clinical use.21 A Phase 1 study with TPCS2a-PCI with the cytotoxic agent bleomycin has been completed22 and a Phase 2 trial in patients with head and neck cancers was initiated in 2012.23 PCI of antibody-based drugs such as immunotoxins provides high selectivity against specific receptors overexpressed in cancers. Such molecules have been used as model drugs for the development of the PCI method because they are often sequestered in endolysosomal vesicles after uptake by receptor-mediated endocytosis.18-24 EpCAM is known to be internalized by receptor-mediated endocytosis,25 which makes it a good target for antibody-based drugs with an intracellular drug target, or for compounds that require internalization for drug activation. Thus, combining EpCAM-targeting therapeutics with the PCI technology, where laser-controlled activation of the drug is confined to the tumor, should be beneficial as this should minimize the possible side effects of such drugs on normal epithelial cells expressing EpCAM in distal organs.
The aim of this study was to investigate the anti-cancer therapeutic potential of combining the EpCAM-directed mAb 3–17I linked to the potent ribosome inactivating protein toxin saporin with the drug delivery method PCI. 3–17I binds strongly in all carcinoma and adenocarcinoma tissue samples (n > 100) investigated. This proof-of-principle study clearly demonstrate that 3–17I is EpCAM-specific because it selectively binds to known EpCAM-positive cancer cell lines and not an EpCAM-negative cancer cell line. 3–17I is also taken up and sequestered in acidic endocytic vesicles. PCI successfully enhances the efficacy and selectivity of the model 3–17I-saporin in pancreas, colon and breast cell lines, suggesting a general applicability of this drug delivery and targeting approach.
Results
Binding profile of 3–17I in vitro and in clinical samples
The human mAb 3–17I was discovered using Affitech’s human naïve phage display library, maturation techniques and proprietary CBAS technology.16
Flow cytometric analysis of binding of 3–17I IgG, and the therapeutic mAbs MT201 IgG and MOC31 IgG to Kato III cells was performed. 3–17I displayed a binding profile close to that of MOC31, while MT201 seemed to have a lower affinity than MOC31 and 3–17I for EpCAM on Kato III cells (Fig. 1). The negative control antibody (anti-GFP IgG) showed no binding to Kato III cells. Figure 1 is reproduced with permission from Ref. 16.
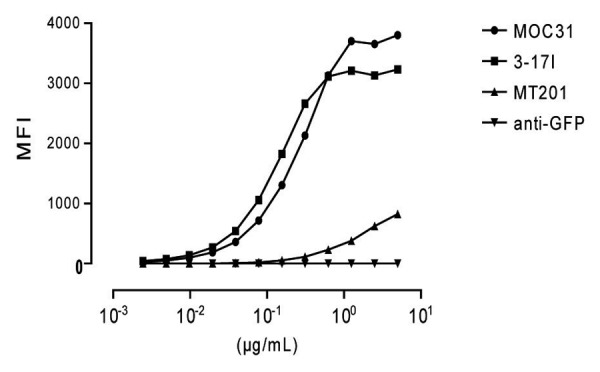
Figure 1. Flow cytometric analysis of 3–17I IgG, MOC31 IgG, and MT201 IgG binding to the EpCAM-positive Kato III cell line. Anti-green fluorescent protein (GFP) antibody is used as a negative control. An RPE-conjugated goat anti-human IgG antibody was used as detection antibody. MFI = Median Fluorescence Intensity. Reproduced from ref. 16 with permission from Affitech Research AS.
Immunohistochemistry studies of the reactivity of mAb 3–17I IgG2A was performed on a panel of normal human tissues. Although the antibody reacted with a number of epithelial tissues as expected, weak or no binding was observed in adrenal, esophagus, lung, skin, thymus, tonsil, and uterus, where the competition antibody MOC31 was more positive (summarized in Table S1B). Representative images are shown in Figure 2 (Images from tonsil tissue are not shown). In addition, no binding of the antibody was observed in cerebellum, cerebrum, heart, skeletal muscle, mesothelium, peripheral nerve, placenta, ovary, spleen, and bone marrow tissues (Table S1A). 3–17I showed a strong reaction (3+) to sloughed cells in one esophagus sample while MOC31 showed a moderate reaction (2+) to epithelial cells in two of three esophagus samples. No or very limited binding to healthy tissues was seen with MT201.

Figure 2. 3–17I shows less reaction in healthy tissue than MOC31. Images of immunohistochemistry studies of 3–17I and MOC31 (both IgG2A), and IgG2A isotype control, binding to healthy tissue samples. Figure shows the tissues listed in Table S1B, in which 3–17I reacts less than MOC31. All antibody concentrations were set to 1 µg/ml. Binding is shown as brown stain (DAB). Immunohistochemistry images are included with permission from Affitech Research AS.
In immunohistochemistry studies on cancer tissues the anti-EpCAM antibody 3–17I showed strong binding in 36 of 37 colon cancers (sample C3 contained no tumor cells and had to be excluded), 37/37 breast cancers, and in 36 of 37 lung cancers with epithelial origin (Fig. 3; lung tissue sample B3 was a sarcoma and thus had no epithelial origin, which explains the negative result, as seen in Table S2C). No or very limited reaction was seen with MT201. The limited reaction was interpreted as background staining and the same reaction was also found in the isotype control. MOC31 displayed similar binding as 3–17I in all cancer tissues investigated (Fig. 3, and Tables S2A-C). Immunohistochemistry images are included with permission from Affitech Research AS.

Figure 3. 3–17I IgG2A displays a similar reactivity as MOC31 IgG2A in breast, colon, and lung tumor tissue samples. Immunohistochemistry studies of 3–17I, MOC31, MT201 (all IgG2A), and IgG2A isotype control binding to tumor tissue samples of colon, breast, and lung origin. Figure shows representative images (from 36–37 samples per tumor type) from each of the three tumor tissues. Binding is shown as brown stain (DAB). Immunohistochemistry images are included with permission from Affitech Research AS.
3–17I efficiently induces ADCC and CDC compared with MT201
Antibody-dependent cell cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC) assays were performed to compare the ability of 3–17I and MT201 (IgG1 isotype) to induce ADCC and CDC in vitro in the presence of human PBMCs that will target cells bound by the antibody. The ability of 3–17I to induce ADCC was analyzed using the three different breast cancer cell lines MDA-MB-453, MDA-MB-231, and BT-474, which cover a range of more than 100-fold difference in surface density of EpCAM.26 3–17I induced a higher cytotoxic response in ADCC than MT201 in MDA-MB-453, MDA-MB-231, and BT-474 (Fig. 4A-C, respectively). MT201 did not induce a cytotoxic response in MDA-MB-231(Fig. 4B). 3–17I induced CDC on the human gastric carcinoma cell line Kato III and breast carcinoma cell line MT-3 in the presence of human PBMCs. At a concentration of 1 ng/ml, 3–17I induces more than 80% cytotoxicity (CDC) in both Kato III and MT-3 cells (Fig. 4D and E, respectively). In comparison, MT201 does not induce a cytotoxic response at this antibody concentration. In summary, Figure 4 shows that 3–17I is a more potent inducer of ADCC and CDC than MT201 in selected human carcinoma cell lines. Figure 4 is reproduced with permission from Ref. 16.
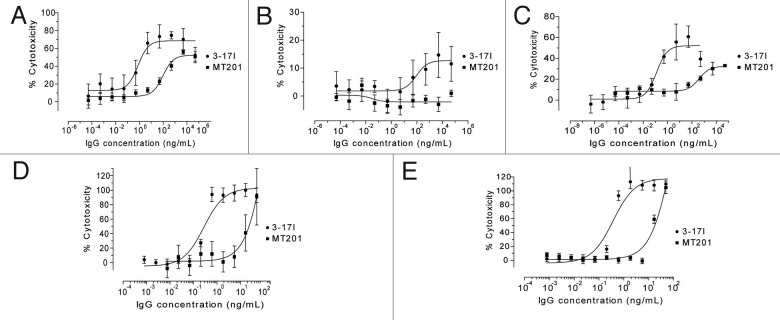
Figure 4. 3–17I induces ADCC- and CDC. Comparison of ADCC induced by 3–171 IgG and MT201 IgG in (A) MDA-MB-453, (B) MDA-MB-231, and (C) BT-474 cells, in the presence of human PBMCs, and comparison of CDC induced by 3–171 IgG and MT201 IgG in (D) KATO III and (E) MT-3 cells in the presence of human serum. The data presented is percentage lysis relative to control. Reproduced from Ref. 16 with permission from Affitech Research AS.
Selective binding and intracellular sequestration of 3–17I
The 3–17I antibody was biotinylated, and flow cytometry was used to confirm successful biotinylation and binding of the biotinylated 3–17I antibody to the EpCAM-positive cell lines MCF7, WiDr, and BxPC-3 cells and lack of binding to the EpCAM-negative cell line COLO320DM (Fig. S1). These cell lines were further used in the PCI-based drug (3–171-saporin) delivery study. To investigate whether the 3–17I antibody was taken up into the cells, we studied the uptake of 3–17I by confocal and fluorescence microscopy. Strep-Cy3 was used to label the biotinylated 3–17I mAb (named 3–17I-Cy3). Images were taken after 18 h of incubation followed by four hours of incubation in medium without the antibody present (chase), to mimic the PCI-protocol. 3–17I-Cy3 did bind to and was selectively taken up into in the EpCAM-expressing cell lines MCF7, WiDr, and BxPC-3 (Fig. 5A, E and I), whereas EpCAM negative cells (COLO320DM) did not show any binding nor uptake of 3–17I-Cy3 (Fig. 5M). To determine the potential localization of 3–17I in endolysosomal vesicles, Lysotracker® Green (LTG) was included (Fig. 5B, F, J and N). Indeed, 3–17I-Cy3 and LTG colocalized to various degrees (BxPC-3 > MCF-7 > WiDr) in all EpCAM-positive cell lines (Fig. 5C, G and K).

Figure 5.Cellular uptake and endolysosomal sequestration of 3–17I in EpCAM-positive cell lines. Live cell confocal fluorescence microscopy was performed after 18 h incubation with 4.7 nM 317-I-Cy3 (red), mimicking the PCI protocol (see Photochemical internalization and viability assay in the Materials and Methods section). The cells were incubated with 1 µM LysoTracker Green (green), which stains acidic vesicles, for 30 min prior to microscopy in MCF7 (A-D); WiDr (E-H); BxPC-3 (I-L); and COLO320DM (M-P) cells.
Efficient, selective and light controlled targeting of EpCAM-positive cancer cells after PCI of 3–17I-saporin
Next, the biotinylated 3–17I antibody was linked to Strep-saporin, creating the immunotoxin 3–17I-saporin as a model immunoconjugate. The effect of photochemical internalization of 3–17I-saporin on cell growth and viability was tested on the four cell lines, using light doses ranging from 0 to 4 J/cm2 (illumination times from 0 to 300 s). An overview of treatment groups is shown in Table 1. Exposure of the cells to 3–17I (mAb alone) or to 3–17I-saporin without the presence of the photosensitizer TPCS2a did not have any significant cytotoxic effects (Fig. 6) neither with nor without illumination. In contrast, in all three EpCAM-positive cell lines a strong cytotoxic response after PCI of 3–17I-saporin was seen, increasing with the light dose (Fig. 6A-C). The effect on viability of PCI of 3–17I-saporin was significantly (P < 0.05) higher than the effect of PCI of saporin in MCF7 and BxPC-3 (Fig. 6A and B). In all EpCAM-expressing cell lines we tested, the cytotoxic effect of PCI of 3–17I-saporin was much higher than that of photochemical treatment alone (equivalent to PDT). In the EpCAM-negative cell line COLO320DM (Fig. 6D) no difference in cytotoxicity was observed between the different treatment strategies at light doses up to 240 s. At the highest light dose (300 s. = 4 J/cm2), the cytotoxic response of PCI of 3–17I-saporin and PCI of saporin was still equal, however better than photochemical treatment alone.
Table 1. Overview of treatment groups.
| Sample name | Drug | TPCS2a | Light exposure |
|---|---|---|---|
| NT | No | No | No |
| 3–17I | 3–17I | No | Yes |
| PCT | No | Yes | Yes |
| PCI saporin | Strep-saporin | Yes | Yes |
| PCI IT | Strep-saporin + 3–17I | Yes | Yes |
| IT | Strep-saporin + 3–17I | No | Yes |
For viability assays, light exposure ranged from 0 to 300 s. For colony formation assay, light exposure was 0 or 180 s. Abbreviations: PCT, photochemical therapy; IT, immunotoxin; sap, saporin; NT, no treatment.

Figure 6. PCI of 3–17I-saporin attenuates cellular viability in three different EpCAM-positive cancer cell lines. Viability (MTS) assay of (A) MCF7; (B) BxPC-3; (C) WiDr; and (D) the EpCAM-negative cell line COLO320DM after treatment with PCI of 3–17I-saporin; PCT; PCI of saporin; IT (3–17I-saporin); and 3–17I alone, all relative to no treatment. The MTS assays were performed 96 h after light treatment, following the PCI protocol in section 2.13. Error bars represent standard deviation. Statistically significant difference between PCI 3–17I-saporin vs. PCI saporin (P < 0.05) is indicated by *.
PCI of 3–17I-saporin strongly attenuates both proliferation and colony forming ability of EpCAM-positive MCF7 cells
It has been shown that local control of chest wall recurrence following mastectomy can be obtained with PDT.27,28 We have previously demonstrated that PCI of both bleomycin and targeted toxins are superior to photochemical treatment alone. Thus, PCI may have a future role in the treatment of local recurrences of breast cancer. MCF7 cells show low sensitivity to PDT compared with the other cell lines, however, a significantly higher cytotoxic response to PCI of 3–17I compared with that of PCI of saporin. Therefore, we decided to further investigate the effect of PCI of 3–17I-saporin in the MCF7 cells with regards to cell viability, proliferation, and clonal growth. In these experiments a higher concentration to (4.7 nM) of immunotoxin was employed. MCF7 cells show a significant cytotoxic response of PCI of 4.7 nM 3–17I-saporin, compared with PCI of 4.7 nM saporin, already at 120 s light treatment. The effect of photochemical treatment alone, 3–17I, and 3–17I-saporin alone, remained subtoxic even at 300 s light exposure (Fig. 7A).

Figure 7. Sub-toxic treatment with PCI of 3–17I-saporin inhibits cellular proliferation. (A) Viability (MTS) assay of MCF7 96 h after treatment with PCI of 3–17I-saporin; PDT; PCI of saporin; IT (3–17I-saporin); and 3–17I alone, all relative to no treatment. The MTS assays were performed 96 h after light treatment, following the PCI protocol (see Photochemical internalization and viability assay in the Materials and Methods section) (B) The proliferative capacity of MCF7 cells was measured by cell confluence using live cell imaging 0- 96 h post 180 s of light exposure, following the same treatment regime as described above. (C) Representative cell confluency images of PCI of 3–17I-saporin and control groups PCT and PCI of saporin at time 0 and 96 h post treatment. (D) Images of colony growth assay after treatment with PCT; PCI of 3–17I-saporin; and PCI of saporin, with or without 180 s light treatment. (E) Colony growth assay show colony forming ability per 1000 cells (%) relative to NT/no light exposure. Graph shows the average of two individual experiments. Error bars represent standard deviation. Statistically significant difference between PCI 3–17I-saporin vs. PCI saporin (P < 0.05) is indicated by *.
To measure cell proliferation following the different treatment strategies, the degree of confluence was measured from directly after 180 s light exposure and for the next 96 h using an IncuCyte live cell imaging system, as described under proliferation assay in the methods section. Proliferation of the MCF7 cells treated with PCI of 3–17I-saporin was impaired as compared with proliferation for cells treated with PDT or PCI of saporin alone (Fig. 7B). Live cell images taken by the Incucyte reveal strongly reduced cell density the MCF7 cells treated with PCI of 3–17I-saporin compared with cells treated PDT, PCI of saporin alone, or no treatment (Fig. 7C). Next we investigated the effect of PCI of 3–17I-saporin on MCF7 breast carcinoma cells with regards to clonal growth ability. Our results show that the number of colonies formed per 1000 cells plated was strongly reduced (~80%) following treatment as compared with the untreated cells. In comparison, with PCI of saporin alone the number of colonies was reduced only by about 40% showing that PCI of the immunotoxin gave a substantially better cytotoxic effect than PCI of the toxin (Fig. 7D and E).
Discussion
The low cure rates by many traditional cancer treatment regimens used today are not due to lack of drug efficiency per se, but mostly due to limited specificity, often resulting in dose-limiting toxicity and severe side effects. Antibody-based drugs have the potential to provide very high selectivity, but a major limitation of treatment is limited penetration of the molecules into the malignant tissue and intracellular entrapment in endocytic vesicles.29 In addition, the treatment usually involves many injections that may cause induction of neutralizing antibodies.30 In many cases, the antigen may be expressed also on non-target cells that may reside in vital organs of the body, perhaps leading to off-target toxicities. By utilizing PCI in combination with targeted immunotoxins, most of these obstacles can be prevented. PCI treatment gives primarily a localized effect due to the activation of photosensitizer and subsequently delivery of the drug to the cytosol of targeted cancer cells only in the illuminated tissue.31 As previously demonstrated, only one treatment may be required (i.e., one injection of targeting drug and one following light exposure),32 thus the patient could avoid multiple injections and development of treatment resistance.
Although EpCAM is an attractive cancer target, to our knowledge no therapeutic antibody targeting EpCAM alone has been approved for clinical use. There is a clear need for the discovery of new anti-EpCAM antibodies with improved or alternative properties to the existing clinical trial candidates. In this study, we describe results utilizing the novel human mAb 3–17I16 that specifically targets EpCAM-positive cells in vitro and in clinical samples, and shows promising potential as a drug carrier in combination with the drug delivery technology PCI.
Our results show that 3–17I binds less to normal human tissues than MOC31, although it displays a similar binding profile to cancer tissues of different origins. Previously published results16 indicate that both MOC31 and MT201 bind to different epitopes on human EpCAM than 3–17I. As stated in the antibody patent, 16 3–17I was determined to have a binding affinity (KD) close to that of MOC31, while the KD of MT201 is described as much lower. The fact that 3–17I has a similar affinity as MOC31, but a different binding epitope on EpCAM could be a reason for the beneficial binding profile of 3–17I compared with MOC31 on normal tissue. The low binding of MT201 seen in the immunohistochemistry study is in line with its much lower binding affinity to EpCAM.
The reduced binding to healthy tissue compared with that of MOC31, coupled with the precisely localized activation afforded by PCI technology (ref), indicate that 3–17I–conjugate may be a candidate with exquisite selectivity for diagnostic or therapeutic uses or as a basis for engineering enhanced anti-EpCAM molecules, such as bispecific antibodies, immunotoxins, or ADCs.
The main aim of this study was to evaluate the cytotoxic effect of PCI of the EpCAM-targeting antibody 3–17I linked to saporin, creating the per se immunotoxin 3–17I-saporin, in different cancer cell lines. The PCI technology allows for light-controlled release of intracellularly sequestered 3–17I-saporin from endolysosomes into the cytosol. Colocalization of the clinically tested photosensitizer TPCS2a and the immunoconjugate to be delivered in endolysosomal compartments is a prerequisite for the PCI technology. Our confocal and fluorescence microscopy results clearly demonstrate cellular uptake of the endolysosomal targeting photosensitizer TPCS2a and 3–17I-Cy3. Cy3 labeled 3–17I was colocalized to a high degree with Lysotracker green in the MCF7 cell line, partially in the BxPC-3 cells and to a lesser degree in the WiDr cells. Together with previous evidence of colocalization of TPCS2a and Lysotracker green,33 and the strong cytotoxic response obtained after PCI of 3–17I –saporin, our data suggest co-localization of TPCS2a and 3–7I-saporin in endolysosomal vesicles. The present report indicate that PCI of 3–17I can significantly reduce target cell viability, proliferation and colony forming ability in all tested EpCAM-positive cells, indicating a potential for in vivo light-controlled activation of 3–17I-drug conjugates. As in other PCI-based studies, we demonstrate that employment of the PCI technology, is superior to using single treatments, such as mAb-treatment alone or photochemical treatment alone. Since PCI of 3–17I-saporin gives higher cytotoxic responses than PCI of saporin in EpCAM-positive cell lines and not in an EpCAM-negative cell line, we conclude that our targeting strategy is specific. It should also be very well suited for tumor-specific therapy in vivo due to: (1) the preferential accumulation of photosensitizers in tumor tissues; (2) the mAb-based targeting of the immunoconjugate; and (3) the light-controlled activation of the immunoconjugate. Thus, we hypothesize that our PCI-based targeting of EpCAM will provide an excellent spatio-temporal controlled activation of therapy in EpCAM-positive tumor tissues and spare distant EpCAM-positive normal tissues that are not subject to light exposure.
A critical requirement for the efficiency of the treatment is that an immunoconjugate is able to pass through the intra-body barriers and reach its target cells. Thus, the 3–17I-saporin immunotoxin described here (theoretically > 700 kDa) is probably too large for in vivo use and has only been intended as a model conjugate to establish an in vitro proof-of-concept. Future efforts would include reducing the size of the immunotoxin, by a chemical conjugation between the mAb and a toxin/drug or by making a recombinant fusion toxin and performing in vivo PCI.
In conclusion, our results show that PCI-based drug delivery using the human and novel EpCAM-targeting mAb 3–17I linked to saporin strongly and selectively reduces cellular viability, proliferative capacity, and colony forming ability in selected EpCAM-expressing breast carcinoma, pancreatic adenocarcinoma, and colon adenocarcinoma cell lines. The promising binding profile of 3–17I in patient samples suggest a potential clinical application for detection and diagnosis of EpCAM-positive cancers. PCI-controlled cytosolic delivery of 3–17I-based drug conjugates has a therapeutic potential, and it would be of high interest to further explore this strategy in vivo.
Materials and Methods
Cells and culturing
The adenocarcinoma cell lines BxPC-3 (pancreatic), MCF7 (breast), and WiDr and COLO320DM (both colorectal) were all obtained from ATCC, and cultured at 37 °C in a humidified atmosphere of 5% CO2. BxPC-3, WiDr, and COLO320DM were cultured in RPMI medium (R8758, Sigma-Aldrich) containing 10% FBS (16000–044, Invitrogen) and penicillin/streptomycin (17–603E, BioWhittaker). MCF7 was cultured in EMEM medium (M4655, Sigma-Aldrich) containing 10% FBS and penicillin/streptomycin. All cell lines used were mycoplasma negative.
Cell lines used in ADCC studies
The MDA-MB-453, MDA-MB-231, BT-474 cell lines (all breast mammary gland) were obtained from the American Type Culture Collection (ATCC), and cultured at 37 °C in a humidified atmosphere of 5% CO2. The MDA-MB-453 and BT-474 cells were maintained in RPMI-1640 culture medium and the MDA-MB-231 cells were maintained in Leibovitz’s culture medium (L1518, Sigma-Aldrich). All media were supplemented with 10% FBS and penicillin/streptomycin. All cell media and supplements were obtained from PAA.
Cell lines used in binding profile and CDC studies
KATO III (gastric carcinoma) and MT-3 (breast carcinoma) cell lines were obtained from the American Type Culture Collection (ATCC) and DSMZ, respectively, and cultured at 37 °C in a humidified atmosphere of 5% CO2. The KATO III and MT-3 cells were maintained in RPMI-1640 culture medium with the addition of FBS at 20% and 10%, respectively. All media were supplemented with Penicillin/Streptomycin. All cell media and supplements were obtained from PAA.
Drugs and chemicals
The photosensitizer meso-tetraphenyl chlorin disulfonate (TPCS2a/Amphinex®) was provided by PCI Biotech (Lysaker, Norway). TPCS2a was diluted in 3% polysorbate 80, 2.8% mannitol and 50 nM Tris (pH 8.5) to a final concentration of 0. 35 mg/ml. The biotinylated anti-EpCAM from Affitech Research AS was used to target EpCAM in the PCI experiments, and streptavidin-saporin (Strep-ZAP from Advanced Targeting Systems, #IT-27) was used as the toxin. Lysotracker® Green (pH 5.2) (L7526, Invitrogen) was used as a marker of acidic vesicles in the live confocal microscopy studies. Streptavidin-Cy3 (016- 160- 084, Jackson Immunoresearch Laboratories) was used as the fluorescent dye, bound to biotinylated 3–17I, both in the live confocal microscopy studies and in the flow cytometry studies.
Discovery and synthesis of 3–17I
The mAb 3–17I was discovered, patented and is owned by Affitech Research.16
For isolation of antibodies against EpCAM, phage display selection was performed using a scFv antibody library derived from the naïve human IgM/IgD repertoire34 as a first step. Initial candidates obtained from that selection were then optimized for improved binding characteristics and screened in a soluble scFv format. The best candidate, 3–17I, was expressed as a human IgG1 variant, or as a chimeric form, with the human variable regions being paired with murine IgG2a and kappa constant domains. Detailed descriptions of methods related to the discovery of the antibody were previously reported.16
For binding profile studies and for ADCC and CDC experiments, a human IgG1 variant of the antibody was used. For IHC studies and for all experiments related to photochemical internalization, a chimeric IgG2A antibody was used.
Binding profile studies
For flow cytometry, Kato III cells were harvested from the culture flasks, washed 2 times with PBS, re-suspended in PBS with 0.2% BSA and 0.09% NaN3 and finally distributed at 1 × 105 cells per well into V-shaped 96-well plates (Greiner Bio-One). Cells were centrifuged at 400 × g for 5 min and then incubated at 4 °C for 45 min with 50 µL of each different antibody dilution (all dilutions made in PBS with 0.2% BSA and 0.09% NaN3). After washing in PBS with 0.2% BSA and 0.09% NaN3, the cells were stained with 10 µg/mL of RPE-conjugated goat anti-human IgG (204009, AbD Serotec) for 30 min at 4 °C. The stained cells were washed, re-suspended in 200 µL PBS with 0.2% BSA and 0.09% NaN3 and transferred to a U-shaped 96-well plate (Corning) for analysis on EasyCyte flow cytometer (Guava Technologies, Hayward, CA, USA). An anti-green fluorescent protein (GFP) antibody was used as a negative control.
Immunohistological investigation of mouse anti-human EpCAM antibodies
The antibodies used in the immunohistological study were provided by Affitech Research AS and were designated as follows: MOC31B (mouse IgG2A), MT201 (mouse IgG2A), 3–17I (mouse IgG2A) and an IgG2A isotype control. The study was performed at MicroMorph Histology Services. In two validation experiments, optimal protocol and concentrations of the antibodies were determined. For this work, colon cancer (n = 4) and spleen (n = 1) from MicroMorph tissue bank were used. For assessment of normal tissue cross-reactivity, tissues from BioChain were purchased (prod. # BT6234701). Tumor tissues were also purchased from BioChain: colon cancer, n = 37 (T6235090), breast cancer, n = 37 (T6235086) and lung cancer, n = 37 (T6235152–5). All tissue from BioChain arrived as cryo-sectioned, acetone-fixed frozen tissue micro array slides. In two validation experiments, the antibodies were tested at concentrations from 0.05 µg/ml to 16 µg/ml (16, 8, 4, 2, 1, 0.5, 0.1, and 0.05 µg/ml). Experiments were performed as follows: the cryo-sectioning was air-dried at room temperature overnight, and fixed in acetone for 10 min before they were blocked in 5% human normal serum (Jackson ImmunoResearch, 009–000–121) for 30 min. Sections were then stained with primary antibodies in 2% human normal serum for 1 h, before they were washed 4X in PBS. Subsequently, sections were stained with Envision mouse (Dako, K4001) in 2% human normal serum for 30 min. Lastly the sections were washed 2X in PBS and 2X in Tris, before they were stained with DAB stain for 5 min. Images were taken using HTX imaging. Staining intensity was judged as; negative (0), weak reaction (1+), moderate reaction (2+), or strong reaction (3+).
ADCC assay
MDA-MB-453, MDA-MB-231, and BT-474 cells were harvested by trypsin-EDTA (17–161E, Lonza), sedimented by centrifugation, and resuspended twice in RPMI-1640 culture medium. One ml containing 2.5x106 cells was mixed with calcein-AM (C-3099, Invitrogen) to a final concentration of 10 µM and then incubated at 37 °C for 30 min on a vertical rotating wheel (7 rpm). The cells were washed three times in RPMI-1640 with 10% FCS and the cell density was adjusted to 3 x 105 per ml. The peripheral blood mononuclear cells (PBMC) were prepared from blood of a single healthy volunteer by Ficoll–Hypaque gradient centrifugation. The isolated PBMC were washed in RPMI-1640 with 10% FCS and resuspended at 6 x 106 per ml. 50 µl of each target and effector cell suspension were added to the same wells in a 96–well microtiter plate providing a ratio of effector (E) to target (T) cells (E:T) of 20:1.
The antibody dilutions were added in a volume of 20 µl in quadruplicates for each concentration. The microtiter plate was then incubated for 4 h at 37 °C, and 20 µl 0.9% TritonX-100 (T8787, Sigma) was added to some of the wells after 3 h 45 min to achieve complete lysis of the target cells. 100 µl of the supernatant of each sample was then transferred to a black microtiter plate and the fluorescence (excitation: 488 nm, emission: 518 nm) was analyzed in a TECAN M200 plate reader (Mannedorf). The fluorescence intensity in the samples with no antibodies was subtracted from the intensity of all other samples. The percentage of lysis in samples with antibodies was estimated on the basis of fluorescence intensity in the samples with 100% cell lysis after treatment with Triton X-100. The dose–response curves were generated by nonlinear regression analysis using a three-parameter fit model of software Prism (GraphPad).
CDC assay
KATO III and MT-3 cells were sedimented by centrifugation and resuspended twice in RPMI-1640 culture medium. Five ml containing 12.5 x 106 cells were mixed with calcein-AM to a final concentration of 10 µM and then incubated at 37 °C for 30 min on a vertical rotating wheel (7 rpm). The cells were washed three times in RPMI-1640 with 10% heat inactivated fetal calf serum (hiFCS) (10082147, Invitrogen) and the cell density was adjusted to 4 x 106 /ml. Twenty-five µl of target cell suspension was mixed with 25 µl human serum and 50 µl antibody dilutions in RPMI-1640 with 10% hiFCS. The final antibody concentrations in the wells ranged from 0.8 ng/ml to 50 µg/ml. The assay was performed in quadruplicates. Twenty µl of 0.9% Triton X-100 was added to some wells to achieve complete lysis of the target cells and the plate was then incubated for one hour at 37 °C. 100 µl of RPMI-1640 with 10% hiFCS was added to the wells to increase the volume and the cells were then sedimented by centrifugation. 100 µl of the supernatant was transferred to a black microtiter plate and the fluorescence (excitation: 488 nm, emission: 518 nm) was analyzed using TECAN M200 plate reader. The fluorescence intensity in the samples with no antibodies was subtracted from the intensity of all other samples. The percentage of lysis in samples with antibodies was estimated on the basis of fluorescence intensity in the samples with 100% cell lysis after treatment with TritonX-100. The dose–response curves were generated by nonlinear regression analysis using a three-parameter fit model of software Prism (GraphPad)
Biotinylation of antibody
3–17I was biotinylated using a commercial biotinylation kit (21327, Sulfo-NHS-LC-Biotin, Pierce) according to manufacturer’s protocol. Biotinylation was confirmed in flow cytometry experiments, as described below, on BxPC-3 cells using strep-PE as detection antibody.
Flow cytometric analysis of EpCAM-expressing cancer cell lines
To investigate cell surface expression of EpCAM and to demonstrate binding of biotinylated 3–17I to the cell lines selected for PCI experiments, we performed flow cytometric analysis with biotinylated 3–17I in combination with strep-Cy3. Cell lines were detached using Accutase (L11–007, PAA), washed 1x in medium and 2x in PBS, before they were resuspended in PBS containing 3–17I (0.7 µg/ml) and strep-Cy3 (SA1004–4, Life Tech.) diluted 1:200, or strep-Cy3 (1:200) alone (as background control) and incubated at room temperature for 1 h. Cells were washed twice in PBS after antibody incubation. Acquisition of data was performed using an EasyCyte flow cytometer (Guava Technologies). A minimum of 5000 cells were acquired per sample and exclusion of non-viable cells and debris was based on lower forward scatter and side scatter properties. Data analysis was performed using FlowJo software (Treestar) or Flowing Software.
Confocal fluorescence microscopy
Cells were grown in their respective medium on coverslips overnight. Biotinylated 3–17I mAb was mixed to a ratio of 4:1 with streptavidin–Cy3 (the final concentration of 3–17I-Cy3 was 4.7 nM) and incubated for 18 h, before the cells were washed three times with their respective medium. One μM Lysotracker Green (pH 5.2) was added 30 min-1 h prior to live cell microscopy, marking acidic vesicles. The cells were then washed with cold PBS containing Ca2 + and Mg2 + prior to microscopy. The cells were examined with a Zeiss LSM 710 confocal microscope (Carl Zeiss MicroImaging GmbH) equipped with an Ar-Laser Multiline (458/488/514 nm), a DPSS-561 10 (561 nm), a Laser diode 405–30 CW (405 nm), and a HeNe laser (633 nm). For live cell imaging, the coverslips were placed in 35 mm glass bottom MatTek dishes (MatTek), and the objective used was a Zeiss C-Apochromat 40 X/1.20 Water Imm DIC III. Image processing was performed with basic software ZEN 2009 (Carl Zeiss), Photoshop CS4 (Adobe), and Imaris 7.1.1 (Bitplane AG).
Photochemical internalization and viability assay
Three thousand cells per well were seeded per well in 96-well plates in respective cell culture medium and allowed to attach overnight. The cells were co-incubated with 0.35 μg/ml photosensitizer TPCS2a and immunotoxin 3–17I-saporin for 18 h. The concentration of immunotoxin 3–17I–saporin used for MTS assay in Figure 6 was 0.4 nM for MCF7 and BxPC-3, and 4.7 nM for WiDr and COLO320DM. The concentration of immunotoxin 3–17I–saporin used for all experiments in Figure 7 was 4.7 nM. Biotinylated antibody and Strep-saporin was mixed 1:1, according to manufacturer’s instructions.
A number of control treatments, listed in Table S2, were also performed. Following treatment, the cells were washed twice with drug-free culture medium, before they were incubated in drug-free culture medium and chased for 4 h. Cells were subsequently exposed to light, as described below, from 0 to 300 s. The viability of the cells was determined by performing an MTS assay 96 h after treatment according to manufacturer’s protocol [G3582, CellTiter 96® AQueous Non-Radioactive Cell Proliferation Assay (MTS), Promega], and absorbance at 490 nm was measured using an SLT SPECTRA plate reader (Perkin Elmer). Wells without cells incubated with MTS solution were used for background subtraction.
Proliferation assay
Cellular proliferation rates were determined using the IncuCyte live cell imaging system (Essen Bioscience). More specifically, 3000 cells were plated in 96-well dishes and placed in the IncuCyte device. Real-time imaging was performed by taking phase contrast photographs automatically of the same area of the well every second hour, for the duration of the experiment.
Light source
Illuminations of the cells were performed by using Lumisource® (PCI Biotech). This light source consists of four standard light tubes (18 W/tube, Osram L 18/67), which emit blue light with a main peak at ~435 nm. The irradiance varies less than 10% across the illumination area (765 cm2) with an output of ~13.5 mW/cm2. The light box is air-cooled during light exposure, preventing cells from being exposed to hyperthermia and ensuring that the irradiance is stable.
Colony forming assay
One thousand single cells were plated in 6-well plates and allowed to adhere overnight.Treatment was performed as described under the section Photochemical internalization and viability assay. Plates were left in the incubator and medium was changed twice a week for two weeks. Colonies were washed once with 0.9% mg/ml NaCl, fixed in 96% ethanol for ten minutes, and stained with a saturated solution of methylene blue (Sigma) for ten minutes. Subsequently, cells were washed in H2O and then dried before manual colony counting. Only colonies with a minimum of 50 cells were counted.
Statistical analysis
To assess whether the means of the different treatments results were significantly different we used the two-sided Student’s t test by Sigma plot 2001. A minimum significance level of P < 0.05 was used for all statistical tests.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
Thanks to members of the Krauss lab for providing helpful comments on the manuscript. We are grateful to Lavinia Cicortas Gunnarsson, Didrik Paus, Jenny Karlsson, Remko Griep, and Sergej Kiprijanov for their contributions in the antibody discovery process.
Thanks to MicroMorph AB and the Confocal Microscopy Core Facility at the Norwegian Radium Hospital.
Thanks to Affitech Research AS for permission to reproduce figures from the antibody patent.
This study was supported by a grant from the Norwegian Research Council (SFI-CAST), and Affitech Research AS.
Glossary
Abbreviations:
- ADCC
antibody-dependent cell-mediated cytotoxicity
- CDC
complement-dependent cytotoxicity
- ADC
antibody-drug conjugate
- PCI
photochemical internalization
- PDT
photodynamic therapy
- PCT
photochemical therapy
- Sap
Saporin
- TPCS2a
tetraphenyl chlorin disulfonate/ Amphinex®
- mAb
monoclonal antibody
- PBMCs
peripheral blood mononuclear cells
- MTS
(3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium)
References
- 1.Baeuerle PA, Gires O. EpCAM (CD326) finding its role in cancer. Br J Cancer. 2007;96:417–23. doi: 10.1038/sj.bjc.6603494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Trzpis M, McLaughlin PM, de Leij LM, Harmsen MC. Epithelial cell adhesion molecule: more than a carcinoma marker and adhesion molecule. Am J Pathol. 2007;171:386–95. doi: 10.2353/ajpath.2007.070152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balzar M, Winter MJ, de Boer CJ, Litvinov SV. The biology of the 17-1A antigen (Ep-CAM) J Mol Med (Berl) 1999;77:699–712. doi: 10.1007/s001099900038. [DOI] [PubMed] [Google Scholar]
- 4.Litvinov SV, Velders MP, Bakker HA, Fleuren GJ, Warnaar SO. Ep-CAM: a human epithelial antigen is a homophilic cell-cell adhesion molecule. J Cell Biol. 1994;125:437–46. doi: 10.1083/jcb.125.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spizzo G, Fong D, Wurm M, Ensinger C, Obrist P, Hofer C, Mazzoleni G, Gastl G, Went P. EpCAM expression in primary tumour tissues and metastases: an immunohistochemical analysis. J Clin Pathol. 2011;64:415–20. doi: 10.1136/jcp.2011.090274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Went PT, Lugli A, Meier S, Bundi M, Mirlacher M, Sauter G, Dirnhofer S. Frequent EpCam protein expression in human carcinomas. Hum Pathol. 2004;35:122–8. doi: 10.1016/j.humpath.2003.08.026. [DOI] [PubMed] [Google Scholar]
- 7.Schmelzer E, Reid LM. EpCAM expression in normal, non-pathological tissues. Front Biosci. 2008;13:3096–100. doi: 10.2741/2911. [DOI] [PubMed] [Google Scholar]
- 8.Varga M, Obrist P, Schneeberger S, Mühlmann G, Felgel-Farnholz C, Fong D, Zitt M, Brunhuber T, Schäfer G, Gastl G, et al. Overexpression of epithelial cell adhesion molecule antigen in gallbladder carcinoma is an independent marker for poor survival. Clin Cancer Res. 2004;10:3131–6. doi: 10.1158/1078-0432.CCR-03-0528. [DOI] [PubMed] [Google Scholar]
- 9.Gastl G, Spizzo G, Obrist P, Dünser M, Mikuz G. Ep-CAM overexpression in breast cancer as a predictor of survival. Lancet. 2000;356:1981–2. doi: 10.1016/S0140-6736(00)03312-2. [DOI] [PubMed] [Google Scholar]
- 10.Dalerba P, Dylla SJ, Park IK, Liu R, Wang X, Cho RW, Hoey T, Gurney A, Huang EH, Simeone DM, et al. Phenotypic characterization of human colorectal cancer stem cells. Proc Natl Acad Sci U S A. 2007;104:10158–63. doi: 10.1073/pnas.0703478104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, Wicha M, Clarke MF, Simeone DM. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67:1030–7. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- 12.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–8. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Münz M, Murr A, Kvesic M, Rau D, Mangold S, Pflanz S, Lumsden J, Volkland J, Fagerberg J, Riethmüller G, et al. Side-by-side analysis of five clinically tested anti-EpCAM monoclonal antibodies. Cancer Cell Int. 2010;10:44. doi: 10.1186/1475-2867-10-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moldenhauer G, Salnikov AV, Lüttgau S, Herr I, Anderl J, Faulstich H. Therapeutic potential of amanitin-conjugated anti-epithelial cell adhesion molecule monoclonal antibody against pancreatic carcinoma. J Natl Cancer Inst. 2012;104:622–34. doi: 10.1093/jnci/djs140. [DOI] [PubMed] [Google Scholar]
- 15.Simon M, Stefan N, Plückthun A, Zangemeister-Wittke U. Epithelial cell adhesion molecule-targeted drug delivery for cancer therapy. Expert Opin Drug Deliv. 2013;10:451–68. doi: 10.1517/17425247.2013.759938. [DOI] [PubMed] [Google Scholar]
- 16.Cicortas Gunnarsson LD, Paus D, Karlsson JM, Griep RA, Kiprijanov SM, inventors. Affitech Research AS, assignee Anti EpCAM antibodies. United States patent US008637017B2. January 28, 2014.
- 17.Agostinis P, Berg K, Cengel KA, Foster TH, Girotti AW, Gollnick SO, Hahn SM, Hamblin MR, Juzeniene A, Kessel D, et al. Photodynamic therapy of cancer: an update. CA Cancer J Clin. 2011;61:250–81. doi: 10.3322/caac.20114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Selbo PK, Weyergang A, Høgset A, Norum OJ, Berstad MB, Vikdal M, Berg K. Photochemical internalization provides time- and space-controlled endolysosomal escape of therapeutic molecules. J Control Release. 2010;148:2–12. doi: 10.1016/j.jconrel.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 19.Selbo PK, Sivam G, Fodstad O, Sandvig K, Berg K. In vivo documentation of photochemical internalization, a novel approach to site specific cancer therapy. Int J Cancer. 2001;92:761–6. doi: 10.1002/1097-0215(20010601)92:5<761::AID-IJC1238>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 20.Berg K, Dietze A, Kaalhus O, Høgset A. Site-specific drug delivery by photochemical internalization enhances the antitumor effect of bleomycin. Clin Cancer Res. 2005;11:8476–85. doi: 10.1158/1078-0432.CCR-05-1245. [DOI] [PubMed] [Google Scholar]
- 21.Berg K, Nordstrand S, Selbo PK, Tran DT, Angell-Petersen E, Høgset A. Disulfonated tetraphenyl chlorin (TPCS2a), a novel photosensitizer developed for clinical utilization of photochemical internalization. Photochem Photobiol Sci. 2011;10:1637–51. doi: 10.1039/c1pp05128h. [DOI] [PubMed] [Google Scholar]
- 22.Phase I, Dose-escalating Study to Evaluate Safety and Tolerance of Amphinex Based Photochemical Internalisation (PCI) of Bleomycin in Patients With Local Recurrence or Advanced/Metastatic, Cutaneous or Sub-cutaneous Malignancies. ClinicalTrials.gov identifier: NCT00993512 2011.
- 23.An Open-label, Single Arm, Multi-centre Phase II Study to Evaluate Safety and Efficacy of PC-A11 in Patients With Recurrent Non-metastatic Head and Neck Squamous Cell Carcinoma Unsuitable for Surgery and Radiotherapy. ClinicalTrials.gov Identifier:NCT01606566 2012.
- 24.Weyergang A, Selbo PK, Berstad ME, Bostad M, Berg K. Photochemical internalization of tumor-targeted protein toxins. Lasers Surg Med. 2011;43:721–33. doi: 10.1002/lsm.21084. [DOI] [PubMed] [Google Scholar]
- 25.Selbo PK, Sivam G, Fodstad O, Sandvig K, Berg K. Photochemical internalisation increases the cytotoxic effect of the immunotoxin MOC31-gelonin. Int J Cancer. 2000;87:853–9. doi: 10.1002/1097-0215(20000915)87:6<853::AID-IJC15>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 26.Prang N, Preithner S, Brischwein K, Göster P, Wöppel A, Müller J, Steiger C, Peters M, Baeuerle PA, da Silva AJ. Cellular and complement-dependent cytotoxicity of Ep-CAM-specific monoclonal antibody MT201 against breast cancer cell lines. Br J Cancer. 2005;92:342–9. doi: 10.1038/sj.bjc.6602310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schuh M, Nseyo UO, Potter WR, Dao TL, Dougherty TJ. Photodynamic therapy for palliation of locally recurrent breast carcinoma. J Clin Oncol. 1987;5:1766–70. doi: 10.1200/JCO.1987.5.11.1766. [DOI] [PubMed] [Google Scholar]
- 28.Allison R, Mang T, Hewson G, Snider W, Dougherty D. Photodynamic therapy for chest wall progression from breast carcinoma is an underutilized treatment modality. Cancer. 2001;91:1–8. doi: 10.1002/1097-0142(20010101)91:1<1::AID-CNCR1>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 29.Lammers T, Kiessling F, Hennink WE, Storm G. Drug targeting to tumors: principles, pitfalls and (pre-) clinical progress. J Control Release. 2012;161:175–87. doi: 10.1016/j.jconrel.2011.09.063. [DOI] [PubMed] [Google Scholar]
- 30.Pastan I, Kreitman RJ. Immunotoxins for targeted cancer therapy. Adv Drug Deliv Rev. 1998;31:53–88. doi: 10.1016/S0169-409X(97)00094-X. [DOI] [PubMed] [Google Scholar]
- 31.Berg K, Weyergang A, Prasmickaite L, Bonsted A, Høgset A, Strand MT, Wagner E, Selbo PK. Photochemical internalization (PCI): a technology for drug delivery. Methods Mol Biol. 2010;635:133–45. doi: 10.1007/978-1-60761-697-9_10. [DOI] [PubMed] [Google Scholar]
- 32.Selbo PK, Rosenblum MG, Cheung LH, Zhang W, Berg K. Multi-modality therapeutics with potent anti-tumor effects: photochemical internalization enhances delivery of the fusion toxin scFvMEL/rGel. PLoS One. 2009;4:e6691. doi: 10.1371/journal.pone.0006691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bostad M, Berg K, Høgset A, Skarpen E, Stenmark H, Selbo PK. Photochemical internalization (PCI) of immunotoxins targeting CD133 is specific and highly potent at femtomolar levels in cells with cancer stem cell properties. J Control Release. 2013;168:317–26. doi: 10.1016/j.jconrel.2013.03.023. [DOI] [PubMed] [Google Scholar]
- 34.Løset GA, Løbersli I, Kavlie A, Stacy JE, Borgen T, Kausmally L, Hvattum E, Simonsen B, Hovda MB, Brekke OH. Construction, evaluation and refinement of a large human antibody phage library based on the IgD and IgM variable gene repertoire. J Immunol Methods. 2005;299:47–62. doi: 10.1016/j.jim.2005.01.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.