

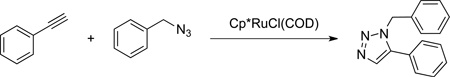
1. Procedure
1-Benzyl-5-phenyl-1H-1,2,3-triazole
Benzyl azide (10.0 g, 0.075 mol) (Note 1) is placed in a 500-mL, three-necked round-bottomed flask, equipped with an 8.0 × 30 mm, octagon-shaped Teflon coated-magnetic stirring bar and a rubber septum. The reaction vessel is purged with argon. 150 mL of DCE (Note 2) followed by phenylacetylene (Note 3) (8.06 g, 0.0789 mol, 8.66 mL) is added to the flask, and the reaction is placed in a 45 °C oil bath. After five min, a solution of chloro(1,5-cyclooctadiene)(pentamethylcyclopentadiene)ruthenium (285 mg, 0.752 mmol) (Note 4) in 3 mL of DCE is added to the reaction vessel via syringe. The reaction is monitored by GC-MS or 1H NMR (Notes 5 and 6) and is found to be completed within 30 min, over which period the orange solution becomes dark brown. The reaction mixture is cooled to room temperature, silica gel (35 g) (Note 7) is added, and the solvent is removed by rotary evaporation (35 °C, 40 mmHg). The resulting light brown powder is placed in a 5 cm diameter column and flushed with ethyl acetate (2 × 200 mL). The dark brown solution is concentrated again by rotary evaporation to give a dark brown solid. The solid material is transferred to a 250-mL one-necked round-bottomed flask, 200 mL of hexanes are added and the heterogeneous solution is triturated (Note 8) for 15 h. The mixture is filtered over a Büchner funnel, rinsed 4 times with 25 mL of hexanes and dried in vacuo (rt, 18 mmHg) to afford 15.9–16.2 g (90–92%) of a beige powder as the titled compound (Note 9).
Safety and Waste Disposal Information
All hazardous materials should be handled and disposed of in accordance with “Prudent Practices in the Laboratory”; National Academies Press; Washington, DC, 2011.
3. Discussion
Ruthenium-catalyzed azide-alkyne cycloaddition (RuAAC)2 is a sister process of a widely utilized CuAAC reaction.3 In contrast to the copper catalysis, which requires the formation of σ-copper(I) acetylides (when terminal alkynes are used), ruthenium catalysts activate alkynes via π-interactions, increasing their nucleophilicity and thereby promoting the addition of the alkyne’s most nucleophilic carbon to the electrophilic N3 terminus of the azide. The currently accepted mechanism of the reaction is shown in Scheme 1.4 In reactions with terminal alkynes, RuAAC produces 1,5-disubstituted 1,2,3-triazoles (instead of the 1,4-regiosiomers that are the exclusive products of CuAAC). Ruthenium catalysts are also active in the cycloaddition of internal alkynes with organic azides to provide fully substituted 1,2,3-triazoles.2,4,6 Regioselectivity is often high for unsymmetrically substituted internal alkynes and is generally influenced by the electronic properties of the substituents, their steric demands, and the ability to engage in hydrogen bonding.2,4–7 Thus, hydrogen bond donors (alcohols and amines) in the propargylic position of the alkyne invariably end up at the C-5 position of the product triazole.4 This directing effect can be explained by the formation of a strong H-bond between the chloride ligand on the ruthenium and the H-bond donor group. The effect of the electronic properties of the alkyne on regioselectivity is exemplified by reactions of ynones and propiolic esters, which usually result in regioselective formation of substituted triazoles with those groups at the C-4 position of the heterocycle.
Scheme 1.

Key intermediates in the RuAAC catalytic cycle.
Among the many ruthenium catalysts that have been examined, only four complexes containing the [Cp*RuCl] fragment proved viable for this reaction: Cp*RuCl(PPh3)2, [Cp*RuCl]4,5 Cp*RuCl(COD) and Cp*RuCl(NBD). For a mechanistic study as well as reaction scope and limitations, see references 4,6.
The RuAAC reaction is compatible with a range of aprotic solvents including 1,2-dichloroethane, acetone, toluene, dioxane, dichloromethane, dimethylformamide, and chloroform, and it is important that the solution be completely homogeneous. Water, ethyl acetate, methanol, isopropyl alcohol, hexanes, and diethyl ether were detrimental to the catalysis. The procedures provided in this work call for moderate temperatures of 50 °C; however, for some substrates room temperature is sufficient to achieve high conversions with low catalyst loading. In general, ruthenium complexes containing labile ligands, such as cyclooctadiane, or the “ligand-free” tetramer [Cp*RuCl]4 require lower temperature to achieve full conversion, as opposed to the bis(triphenylphosphine) complexes where it is advisable to perform the reactions at temperatures exceeding 60 °C.
At this stage of development, RuAAC is not as robust as the CuAAC with respect to functional group tolerance and reaction conditions compatibility, primarily due to potential catalyst deactivation, as shown in Scheme 2. For example, organic azides react with 1 to generate ruthenium tetraazadiene complex 2. These complexes are exceedingly stable, can be isolated by column chromatography and are often easily identified by the presence of a green spot on TLC (or a green band on a silica gel column). Thus, the catalyst should not be mixed with the azide in the absence of alkyne.
Scheme 2.

Catalyst deactivation pathways in CuAAC.
Ruthenium-catalyzed reactions of alkynes are numerous and have evolved into an extraordinary number of synthetically useful transformations during the last decades. Among them is the cyclotrimerization of alkynes to generate benzene rings (4),10 which was shown to proceed through a ruthenacyclopentatriene 3 intermediate.10c There is no direct evidence that ruthenacycle 3 results in deactivation of the catalyst or is even formed during standard RuAAC reactions. However, an isolated ruthenacycle similar to 3 was catalytically incompetent and also did not react with azides, even at the elevated temperature.4
Cyclobutadiene ruthenium complex 5 (characterized by X-ray crystallography) was recently isolated from a dimerization reaction of propargylic alcohols.11 It is a very non-polar, stable to column chromatography compound and is reminiscent of ruthenium species prepared by Kirchner et. al.12 As with 2 and 3, complex 5 does not react with azides.
RuAAC reaction is quite sensitive to atmospheric oxygen and must be run under an inert atmosphere. It is known that Cp*RuCl(PPh3)2 reacts with dioxygen to give Cp*RuCl(O2)(PPh3), where the dioxygen ligand is reportedly not tightly bound and can be replaced by a phosphite ligand, Cp*RuCl(L)(PPh3).13 However, when employing the Cp*RuCl(COD) catalyst the effects of molecular oxygen can be drastic in the RuAAC reaction. Figure 1 shows the rate profile of the reaction of benzyl azide with phenylacetylene (0.1 M in DCE with 0.004 M Cp*RuCl(COD), at 45 °C) using reaction heat flow calorimetry. The area under the curve is directly correlated with the overall conversion. As seen in this example, the maximum rate and overall yield is significantly higher when the reaction is performed under argon. The conversion was quantitative under argon and only 20% under air.
The RuAAC process significantly expands the reach of catalytic dipolar cycloaddition reactions. Its applications are beginning to appear14 and should continue to grow as the reaction matures and new and improved catalysts are developed.
Figure 1. Effects of atmospheric oxygen on RuAAC.

Calorimetry trace of the reaction of benzyl azide and phenyl acetylene (0.1 M in DCE) Several conditions were xamined for both reactions under 4 mol % catalyst loading (0.004 M in DCE) including: degassed DCE under argon, degassed DCE under atmospheric oxygen, untreated DCE under atmospheric oxygen, and untreated DCE under argon. These results illustrate the necessity ofnplacing the RuAAC reaction under an inert atmosphere.
Biographies
Valery V. Fokin received his undergraduate education at the University of Nizhny Novgorod, Russia, and his Ph.D. degree at the University of Southern California under the tutelage of Prof. Nicos A. Petasis. After a postdoctoral stint with Prof. K. Barry Sharpless at The Scripps Research Institute in La Jolla, California, he joined the Scripps faculty, where he is currently Associate Professor in the Department of Chemistry. His research is focused on new reactivity of organometallic compounds and catalysis, and on applying them to the studies of macromolecular and biological phenomena. His research group is working on the development of new catalytic reactions, asymmetric synthesis, studies of organic and organometallic mechanisms, medicinal chemistry, and smart polymeric materials.
James Oakdale received a B.Sc. in chemistry in 2009 from the University of California, Davis, where he pursued undergraduate research in the laboratory of Prof. Mark Kurth. He is currently pursuing a Ph.D. as an NSF graduate research fellow at The Scripps Research Institute under the advisement of Prof. Valery V. Fokin. His research efforts include the development and mechanistic investigation of ruthenium catalyzed reactions and the design and manipulation of functional materials.
Appendix
Chemical Abstracts Nomenclature; (Registry Number)
Benzyl Azide: Azidomethyl benzene; Triazotoluene; (622-79-7)
Chloro(1,5-cyclooctadiene)(pentamethylcyclopentadienyl)ruthenium(II): (92390-26-6)
Phenylacetylene: Ethynyl benzene; Acetylene benzene; (536-74-3)
Footnotes
The submitters purchased benzyl azide, pract. (94%) from Frinton Laboratories, Inc., and used after flushing the compound through a short column of silica gel with diethyl ether. The submitters noted that three times as much catalyst was needed to complete the reaction without purification. The checkers purchased benzyl azide (97%) from Wako Pure Chemical Industries, Ltd. and used as received.
1,2-Dichloroethane was purchased from Fisher Scientific (certified ACS, submitters) or Kanto Chemical Co., Inc. (>99.5%, Conforms to ACS, checkers), and degassed prior to use by bubbling a stream of nitrogen (submitters) or argon (checkers) through the solvent for 1 hour. No precautions were taken to dry the solvent.
Phenylacetylene (98%) was purchased from Acros Organics and used as received.
Chloro(1,5-cyclooctadiene)(pentamethylcyclopentadienyl)ruthenium( II) (98%) was purchased from Stream Chemicals Inc. (submitters) or Aldrich Chemical Company, Inc. (checkers) and used as received.
The submitters monitor the reaction by GC-MS. Gas chromatography was performed using a Hewlett Packard 5890A Gas Chromatograph. GCMS data was recorded on an Agilent 7890A GC system with an Agilent 5975C Inert MSD system operating in the electron impact (EI+) mode. Column; Agilent 19091S-433: 325 °C: 30 m × 320 µm × 0.25 µm, H-5MS 5% phenyl methyl silox. Split Ratio 20:1. Front Inlet; 200 °C. Front Detector FID; 300 °C. Oven program; 50 °C for 2.25 min then 60 °C/min to 300 °C for 4 min. Retention times are as follows; phenylacetylene (2.9 min), benzyl azide (4.03 min) and 1-benzyl-5-phenyl-1H-1,2,3-triazole (6.88 min).
The checkers monitor the reaction by 1H NMR. Chemical shifts of benzyl proton are used for monitoring. Chemical shifts are as follows; benzyl azide (4.35 ppm) and 1-benzyl-5-phenyl-1H-1,2,3-triazole (5.55 ppm)
The submitters purchased silica gel 60 from EMD Chemicals Inc.; particle size 40–63 µm. The checkers purchased silica gel (acidic) from Kanto Chemical Co., Inc., particle size 40–100 µm.
Three stirring bars (8.0 × 30 mm, octagon-shaped) are added to the hexane solution and the mixture is vigorously stirred in order to grind the solid into a fine powder. The submitters note that it is critical that the material be completely solid prior to the addition of hexanes.
The properties of 1-benzyl-5-phenyl-1H-1,2,3-triazole are as follows: 1H NMR (CDCl3, 400 MHz) δ: 5.55 (s, 2 H), 7.06–7.10 (m, 2 H), 7.24–7.32 (m, 5 H), 7.38–7.47 (m, 3 H), 7.75 (s, 1 H); 13C NMR (CDCl3, 100 MHz) δ: 51.7, 126.9, 127.1, 128.1, 128.8, 128.8, 128.9, 129.4, 133.2, 135.6, 138.1; IR (thin film) 1483, 1455, 1435, 1242, 1210, 1126 cm−1; mp 76–78 °C; HRMS (ESI) m/z calcd. for C15H13N3Na ([M+Na]+) 258.1007; found 258.1006; Anal. calcd. for C15H13N3: C, 76.57; H, 5.57; N, 17.86. found: C, 76.57; H, 5.72; N, 17.65.
References
- 1.The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037. fokin@scripps.edu. This work was supported by the National Science Foundation (CHE-0848982).
- 2.Zhang L, Chen X, Xue P, Sun HHU, Williams ID, Sharpless KB, Fokin VV, Jia G. J. Am. Chem. Soc. 2005;127:15998–15999. doi: 10.1021/ja054114s. [DOI] [PubMed] [Google Scholar]
- 3.(a) Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew. Chem. Int. Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]; (b) Tornøe CW, Christensen C, Meldal M. J. Org. Chem. 2002;67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 4.Boren BC, Narayan S, Rasmussen LK, Zhang L, Zhao H, Liu Z, Jia G, Fokin VV. J. Am. Chem. Soc. 2008;130:8923–8930. doi: 10.1021/ja0749993. [DOI] [PubMed] [Google Scholar]
- 5.Rasmussen LK, Boren BC, Fokin VV. Org. Lett. 2007;9:5337–5339. doi: 10.1021/ol701912s. [DOI] [PubMed] [Google Scholar]
- 6.Majireck MM, Weinreb SM. J. Org. Chem. 2006;71:8680–8683. doi: 10.1021/jo061688m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oppilliart S, Mousseau G, Zhang L, Jia G, Thuery P, Rousseau B, Cintrat JC. Tetrahedron. 2007;63:8094. [Google Scholar]
- 10.For examples see Yamamoto Y, Kinpara K, Saigoku T, Takagishi H, Okuda S, Nishiyama H, Itoh K. J. Am. Chem. Soc. 2005;127:605–613. doi: 10.1021/ja045694g. Yamamoto Y, Arakawa T, Ogawa RJ, Itoh K. J. Am. Chem. Soc. 2003;125:12143–12160. doi: 10.1021/ja0358697. Kirchner K, Calhorda MJ, Schmid R, Veiros LF. J. Am. Chem. Soc. 2003;125:11721–11729. doi: 10.1021/ja035137e.
- 11.Oakdale JS, Fokin VV. Unpublished Results. [Google Scholar]
- 12.Paih JP, Dérien S, Demerseman B, Bruneau C, Dixneuf PH, Toupet L, Dazinger G, Kirchner K. Chem. Eur. J. 2005;11:1312–1324. doi: 10.1002/chem.200400899. [DOI] [PubMed] [Google Scholar]
- 13.Trost BM. Acc. Chem. Res. 2002;35:695–705. doi: 10.1021/ar010068z. [DOI] [PubMed] [Google Scholar]
- 14.(a) Poecke SV, Negri A, Gago F, Van Daele I, Solaroli N, Karlsson A, Balzarini J, Van Calenbergh S. J. Med. Chem. 2010;53:2902–2912. doi: 10.1021/jm901532h. [DOI] [PubMed] [Google Scholar]; (b) Horne WS, Olsen CA, Beierle JM, Montero A, Ghadiri MR. Angew. Chem. Int. Ed. 2009;48:4718–4724. doi: 10.1002/anie.200805900. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chemama M, Fonvielle M, Arthur M, Valéry JM, Etheve-Quelquejeu M. Chem. Eur. J. 2009;15:1929–1938. doi: 10.1002/chem.200801563. [DOI] [PubMed] [Google Scholar]; (d) Hirose T, Sunazuka T, Sugawara A, Endo A, Iguchi K, Yamamoto T, Ui H, Shiomi K, Watanabe T, Sharpless KB, Omura S. J. Antibiot. 2009;62:277–282. doi: 10.1038/ja.2009.28. [DOI] [PubMed] [Google Scholar]; (e) Nulwala H, Takizawa K, Odukale A, Khan A, Thibault RJ, Taft BR, Lipshutz BH, Hawker CJ. Macromolecules. 2009;42:6068–6074. [Google Scholar]; (f) Kelly AR, Wei J, Kesavan S, Marié JC, Windmon N, Young DW, Marcaurelle LA. Org. Lett. 2009;11:2257–2260. doi: 10.1021/ol900562u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Tam A, Arnold U, Soellner MB, Raines RT. J. Am. Chem. Soc. 2007;129:12670–12671. doi: 10.1021/ja075865s. [DOI] [PMC free article] [PubMed] [Google Scholar]