

Abstract
Multiple Myeloma (MM) is a common hematologic malignancy of plasma cells representing an excellent model of epigenomics dysregulation in human disease. Importantly, these findings, in addition to provide a better understanding of the underlying molecular changes leading to this malignance, furnish the basis for an innovative therapeutic approach. Histone deacetylase inhibitors (HDACIs), including Vorinostat and Panobinostat, represent a novel class of drugs targeting enzymes involved in epigenetic regulation of gene expression, which have been evaluated also for the treatment of multiple myeloma. Although the clinical role in this setting is evolving and their precise utility remains to be determined, to date that single-agent anti-MM activity is modest. More importantly, HDACIs appear to be synergistic both in vitro and in vivo when combined with other anti-MM agents, mainly proteasome inhibitors including bortezomib. The molecular basis underlying this synergism seems to be multifactorial and involves interference with protein degradation as well as the interaction of myeloma cells with microenvironment. Here we review molecular events underling antitumor effects of HDACIs and the most recent results of clinical trials in relapsed and refractory MM.
Keywords: Multiple myeloma, HDACIs, apoptosis, proteasome inhibitor, novel therapy
Introduction
Multiple myeloma (MM) is a malignant plasma-cell disorder characterized by expansion in the bone marrow microenvironment of secretory plasma cells with a low proliferative index and long life span (1) associated with serum and/or urine monoclonal (M) protein and end-organ damage including renal failure, anemia, hypercalcemia and bone lesions. (2)
Although MM represents 1–2% of cancer cases and 13% of hematologic diseases, (3) it accounts for approximately 20% of annual mortality worldwide in hematological malignancy (4) and remains difficult to treat. (5, 6) Over the past two decades, treatment for MM has improved with the use of autologous stem-cell transplantation and novel therapeutics options including Proteasome inhibitors (PI) and Immunomodulatory drugs (IMiDs), changing the treatment paradigm in myeloma and improving overall survival. (7) In younger patients, survival now may extend beyond 10 years and obtaining complete remission has become the goal of therapy with novel drug combinations. Despite these advances, acquired or intrinsic resistance to therapy leads to disease progression, and novel treatment strategies are urgently needed.
MM cells exhibit intrinsic genomic alterations that enable them to survive, proliferate, and avoid apoptotic cell death. Moreover, oncogenomic studies have also shown that signals from surrounding accessory cells and extracellular matrix (ECM) are essential for MM pathogenesis and contribute to MM cell growth, migration, and drug resistance (8–10). Novel therapies are able to induce cytotoxicity in MM cells even in the context of the BM milieu in preclinical models, and importantly, overcome conventional drug resistance in clinical trials.(11)
It is well recognized that cancer cells exhibit high level of epigenetic modifications, which modulate expression and regulation of genes, as well as cellular processes such as differentiation/development and tumorigenesis. (12) Therefore there is a strong preclinical rationale for testing histone deacetylase inhibitors (HDACIs) to revert aberrant epigenetic changes associated with cancer, thereby potentially reversing the malignant phenotype. (13) Indeed, epigenetically targeted agents affect gene expression by modulating histone deacetylase function are being tested broadly in clinical trials. (14) Importantly, these small-molecule drugs have a broad range of effects on cancer cells including cell cycle arrest, apoptosis, cell differentiation, autophagy, and anti-angiogenesis.(15) A landmark in HDACIs therapy is the FDA approval of vorinostat (Zolinza; Merck) and romidepsin (Istodax; Celgene) for treatment of cutaneous T cell lymphoma. (16–18) Recent studies have indicated that additional substrates and mechanism of action besides modulation of gene expression,(13) and remarkably hematologic malignancies appear to be more sensitive to HDACIs treatment than solid malignancies, including acute myeloid leukemia (AML), myelodysplastic syndrome (MDS) and multiple myeloma. Specifically, in vitro data show the antitumor activity of HDACIs against MM cells resistant to conventional or novel anti-MM drugs, suggesting that these agents can overcome the protective effect of the bone marrow microenvironment. Based on these preclinical results, the combination treatments of HDACIs with a variety of anti-MM drugs have been evaluated in clinical trials (see below) with promising. Here, we will discuss current knowledge in the area of anti-tumor activity of HDACIs, and focus on the most important preclinical studies providing the basis for use of HDACIs in the clinical management of MM.
Histone Modification and HDAC Inhibition
The regulation of gene expression can be occurred by post-translational modification of proteins binding DNA (chromatin), without altering the genetic sequence. (15) The nucleosome, the basic structure of chromatin, consists of a protein envelope (histone) packaging DNA. (19) Histone proteins compact a huge amount of DNA into a conformation easily contained inside cells. Indeed Histone tails can be modified by numerous post-translational modifications: methylation, acetylation, phosphorylation, ubiquitination, and SUMOlation, as well as addition of poly (ADP-ribose) moieties. The combination of specific histone modifications is dynamic, and provides for modulation of gene expression by promoting accessibility of the DNA for transcriptional activation. (20, 21) One of the most widely studied histone modification in myeloma, and in tumor cells more generally, is regulation of acetylation of specific histone lysine residues, by two enzymes with opposite activities: histone acetylases (HATs) and histone deacetylases (HDACs). These enzymes catalyze acetylation and deacetylation, respectively, regulating chromatin conformation and consequently gene expression by three different mechanisms. First, histone hypoacetylation increases the charge density on the N-terminal of core histones, leading to condensed chromatin structure and reduced accessibility of transcriptional machinery to DNA, thereby resulting in transcriptional repression. A second mechanism causing transcription blockade is deacetylation of specific DNA sequences binding transcription factors. Hypo- or acetylation of specific DNA sequences can promote either increase or decrease of DNA-transcription factor binding, enhancing or repressing the transcription.(12) Finally, HDACs affect number of cytoplasmic proteins, including tubulin and HSP90, with distinct functional sequelae. (22)
HDACs are expressed in almost all eukaryotic cells, playing a pivotal role in cellular mechanisms such as proliferation, differentiation and homeostasis. Based on their homology to yeast HDACs, the 18 human HDACs identified are classified into 4 different families: Class I includes the human HDACs 1,2,3 and 8; class IIa is the human HDACs 4,5,7 and 9; class IIb has HDACs 6 and 10 and class IV is HDAC 11. Class III encompasses the seven sirtuins, NAD+-dependent HDACs related to the yeast Sir2 proteins. (23) Overall all HDACs, except class III, contain highly conserved enzymatic domains and are zinc (Zn2+)-dependent.
Hypoacetylation of histones, due to overexpression of HDACs, is frequently observed in different types of human cancer (gastric, pancreatic, colorectal, prostate and hepatocellular), suggesting their role in both cell survival and proliferation. It has been also shown that HDACs expression affects the prognosis of several types of human cancers. (24) Accordingly, several HDACIs have been synthesized to test their antitumor effects, with the nonselective-HDACIs vorinostat (SAHA) and the cyclic peptide romidepsin (FK228), representing the first to be approved by FDA in cancer treatment. (17, 18)
HDACIs differ in their chemical structure as well as selectivity towards different HDAC enzymes. Based on their structure, these chemicals are subdivided into short-chain fatty acids, hydroxamic acids, cyclic tetrapeptides, aliphatic acids, and benzamides. Additionally, according to their specificity for HDACs, HDACIs are classified: nonselective-deacetylase inhibitors (vorinostat, panobinostat, belinostat, resminostat and trichostatin A); class I and IIa inhibitors (butyrate and valproic acid); class I selective inhibitor (romidepsin, etinostat and mocetinostat); and HDAC6 inhibitors (Tubacin and ACY-1215).
Mechanisms of action of HDACIs
HDACIs have great potential as anti-cancer agents. Classically HDACs act as repressors of gene expression, tethered to sequence-specific transcription factors; however more interacting processes have been observed.
A recent high-throughput screening in cancer cell lines revealed 3600 lysine acetylation sites on 1750 proteins, associated with various intracellular functions: cell growth, chromatin remodeling, DNA replication and repair, cytoskeletal reorganization, autophagy, angiogenesis, and protein chaperone activity.(25) Importantly, acetylation changes as well as a gene expression profile in response to the deacetylase inhibitors are individuated. (26) However, mechanisms of action of HDACIs remain under active investigation. (Figure 1)
Figure 1.
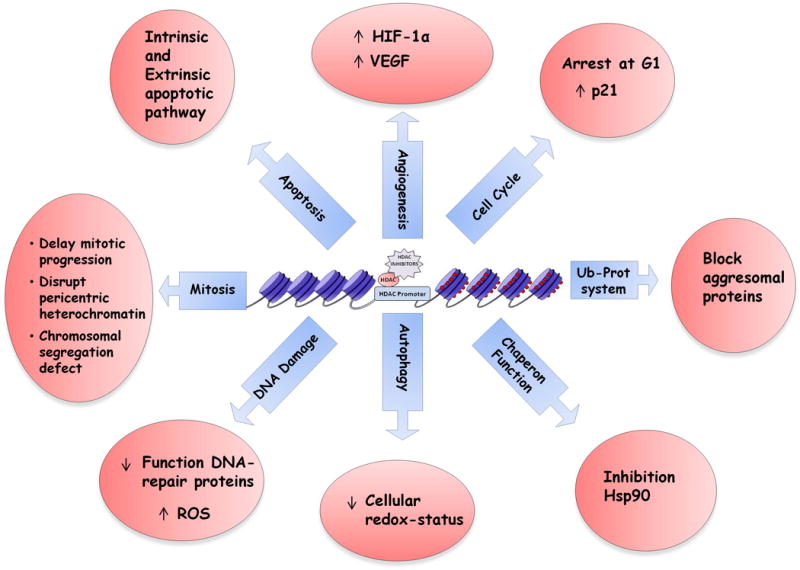
HDAC Inhibitors have great potential as anti-cancer agents, but their exact mechanisms of action are not completely established. Acetylation of histone induces gene expression by altering chromatin activity; acetylation of non-histone proteins affects a variety of physiological pathways including cell growth, chromatin remodeling, DNA replication and repair, cytoskeletal reorganization, autophagy, angiogenesis, and protein chaperone activity.
Effect on cell cycle
The cell cycle perturbation observed after HDACIs treatment, represents one the most important functional sequelae with therapeutic implications in cancer. Almost all HDACIs induce cell-cycle arrest at G1, due to increase of histone acetylation and upregulation of cyclin-dependent kinase inhibitor CDKN1A (p21WAF/CIP1) (27, 28) followed by cell death. Indeed an inhibition/depletion of p21WAF/CIP1 increases the cytotoxic effect of HDACIs treatment in a synergistic fashion. Moreover these compounds induce downregulation of several cyclins, causing a modulation of Rb-E2F1 pathway activity and subsequent cell death. (29) Remarkably, G2/M cell-cycle arrest triggered by HDACIs both in normal as well as in transformed cells results in a selective anti-tumor effect since the loss of G2 checkpoint function, typically observed in tumor cells, renders these cells unable to rescue as normal cells. (30–32)
Induction of apoptosis
HDAC inhibitors have demonstrated anticancer efficacy across a range of malignancies, most impressively in the hematological cancers. However, it remains unclear whether the induction of apoptosis or cell-priming to other pro-death stimuli represents the main mechanism of their antitumor effect. Significantly, induction of apoptotic cell-death triggered by HDACIs treatment involves intrinsic (mitochondrial) as well as extrinsic (death receptor) pathways. (33) In such a scenario, an upregulation of cell death-receptors and -ligands (ligands FasL, TRAIL and DR5), commonly observed after HDACIs treatment, explains the synergistic effect with TRAIL observed in pancreatic cancer cells. (34) Importantly, this cell death-receptors modulation is not observed in normal cells, suggesting selectivity of treatment by HDACIs against malignant cells.
The mitochondrial pathway also mediates induction of apoptosis by HDACIs. Specifically, inhibition of HDACs upregulates expression of proapoptotic Bcl-2 family proteins (Bax, Bak, Bim) (29, 35) and downregulates antiapoptotic proteins (Bcl-2, Bcl-xl, MCL1 and XIAP). (36, 37) Overall, these events trigger increased mitochondrial membrane permeability and cytosolic release of proteins such as cytochrome C and Smac followed by intrinsic apoptotic pathway activation. (27, 38) Consistent with this data, activation of caspases 8, 9 and 3 is responsible of the anti-MM effect of vorinostat. (30, 31) Moreover, downregulation of antiapoptotic proteins Bcl2 and BCL-XL is associated with anti MM effect of romidepsin. (32)
DNA damage and oxidative stress
The modulation of DNA-damage response plays an important role in the biological effects of HDACIs in cancer cells. Treatment of leukemia cells with these drugs induces the appearance of cellular markers such as phosphorylation of γH2AX nuclear foci and ATM, suggesting DNA double strands break (DSBs).(39) Indeed HDACIs can enhance their antitumor activity when combined with ionizing radiation and DNA damaging agents. (40) Several cellular mechanisms are involved in generating and stabilizing DNA DSBs during treatment with HDACIs. In particular, these compounds prevent deacetylation and disrupt the function of DNA-repair proteins such as Ku70, thereby inhibiting Bax-mediated apoptosis. Likewise HDACIs may act by a transcriptional mechanism reducing DNA repair proteins such as RAD51, RAD50, DNA-PKcs, BRCA1 and BRCA2. (41, 42) Importantly, inhibition of HDACs does not induce DNA DSBs alone, but the hyper-acetylation of chromatin resulting after this treatment makes DNA more sensitive to radiation, drugs, and reactive oxygens species. (43) Finally, recent studies suggest a deregulation of both homologous recombination as well as non-homologous end-joining DSB repair, triggered by HDACIs. The synergistic effect observed in the treatment of lymphomas and MM cells combining HDACIs and proteasome inhibitors may be, at least in part, due to perturbation in DNA DSB repair mechanisms. (44)
The production of reactive oxygen species (ROS) observed after HDAC inhibition also plays a crucial role in the cytotoxic effect of these compounds. An increase of endogenous antioxidant protein in tumor cells exposed to HDACIs is frequently observed (45); conversely, the selective anti tumor effect of HDACIs is abolished after pretreatment with the antioxidant N-acetylcysteine. Consistent with these observations, treatment of U937 leukemic cells with vorinostat leads to nuclear localization of the transcriptor factor erythroid 2-like 2 (Nrf2), followed by an upregulation of several antioxidant genes such as GST (glutathione s-transferase), GSR (glutathione reductase) and SOD2 (superoxide dismutase 1 and 2). Furthermore, the use of phenylethyl isothiocyanate, a natural compound capable of depleting cellular glutathione, increases cytotoxicity of vorinostat in leukemia cells by inhibiting the cytoprotective antioxidant response. Collectively, these data suggest that ROS generation in HDACIs treated cells enhances their cytotoxic effect. (46)
Effects on mitosis
HDACIs have emerged as key players in mitosis induction acting similarly to vinca alkaloids or taxane by overcoming the spindle assembly checkpoint. Therefore HDAC inhibition delays mitotic progression through prometaphase due to disruption of pericentric hetrochromatin and consequent chromosomal segregation defects.(47) The premature exit from mitosis or mitotic slippage observed during HDACIs treatment is due to the failure of accumulation of chromosomal passenger protein BubRi, hBub1 Mad2, CENP-F and CENP-E at the centromere. (48) Additionally, HDACIs can modulate mitotic kinases such as Aurora A and B, thereby impairing phosphorylation and stimulating proteasomal degradation, respectively. (49) Overall, the aberrant mitosis and chromosomal disruption are further molecular events participating in tumor cell death observed with HDACIs treatment.
Disruption chaperone function
It is noteworthy that HDACIs treatment also results in inhibition of heat-shock protein 90 (Hsp90). Hsp90 is a cellular chaperone required for assembly and stability of clients proteins involved in intracellular signaling including Akt, Raf, Her2/neu, ERK, pS6 and NF-kB (50). In addition, Hsp90 chaperone prevents client protein degradation by the proteasome. In this context, HDAC6 is activated by acetylation of Hsp90 and its dependent pathways; conversely, specific inhibition or depletion of HDAC6 results in hyperacetylation and decreased activity of Hsp90 function, with consequent polyubiquitination and proteosomal degradation of its clients protein. Therefore, the selective HDAC6 inhibitor tubacin increases its cytotoxic effect when combined with bortezomib in multiple myeloma cells. (51) Since HDACIs resistant cells are frequently characterized by overexpression of HDAC1, 2 and 4 but decreased expression of HDAC6 and consequent hyper-acetylation of Hsp90, inhibition of chaperone protein would be a useful approach for the treatment of resistant tumor cells to HDACIs.
Effects on the ubiquitin-proteasome system and the misfolded protein response
The misfolded protein response (MPR) is a cellular stress response linked to endoplasmic reticulum (ER) and involving the chaperone Hsp90.(52) It is a set of cellular events triggered by toxicity, resulting from unfolded proteins accumulating and representing a protective cellular mechanism. Indeed, apoptosis will occur if the cellular stress is prolonged or the MPR system is not able to restore normal cellular function. The unfolded proteins are ubiquitinilated to be eliminated by proteasome; thus when proteasome function is compromised, substrate proteins are not degradated leading to consequent cellular accumulation of cytotoxic aggregate named aggresomes.(53) Ubiquitinated misfolded proteins bind to HDAC6 on the one hand and to the tubulin and dynein complex on the other to be shuttled to the lysosome for destruction. (54) Therefore, the aggresome represents a salvage pathway for misfolded proteins in the context of proteasome blockade (55), and HDAC6 plays a pivotal role in aggresome formation(56).
Targeting aggresome with HDACIs therefore represents a useful therapeutic approach for cancer. (55, 57, 58) A novel oral HDAC6 selective inhibitor ACY-1215 recently showed ability to potently block aggresomal protein degradation with anti-MM activity, either alone or in combination with bortezomib. (59) It is now under evaluation in preclinical studies, in phase I /II clinical trials in MM.
Anti-angiogenic effects
Several malignant tumors including MM, breast, lung and prostate carcinomas are considered to be angiogenesis-dependent. This biological tumor feature is often due to hypoxia secondary to tumor growth or to increase oncogenic signaling. Both mechanisms result in increased hypoxia-inducible factor-1 alpha (HIF-1α) and a transcriptional target vascular endothelial growth factor (VEGF). (60) Consistent with this notion, much attention has recently been focused on the effect of HDACIs treatment on tumor angiogenesis. (61, 62)
The post translational modifications by acetylation and deacetylation represent critical events in HIF-1α signaling activity. Importantly, in conditions of normoxia Hif-1α is inactivated by ubiquitination and proteasomal degradation, whereas in hypoxia conditions, in malignant cells, there are higher levels of Hif-1α triggering angiogenesis. HDAC function under hypoxic and normoxic conditions are able to regulate expression of several genes including HIF-1α. This regulation occurs by direct and indirect mechanisms. Under hypoxic conditions, malignant cells exhibit increased expression of HDAC1, HDAC2 and HDAC3, resulting in over-expression of HIF-1α and VEGF mediated by p53, pVHL, proteasome and Hsp90. (63–65) Furthermore, HDAC7 under hypoxic conditions translocates from the cytoplasm to the nucleus to bind HIF-1α and increase its transcriptional activity. (66) Therefore the inhibition of HDACs activity results also in anti-angiogenic effect.
Finally HDACIs treatment induces alteration of numerous pro- and anti-angiogenic genes (angiopoietin, TIE2, eNOS, p53, pVHL and thrombospondin 1), further suggesting an attractive role of HDACs as novel targets in cancers that rely on angiogenesis. (61, 62, 67)
Induction of autophagy
A number of anticancer therapies, including HDAC inhibitors, have been observed to induce autophagy in human cancer cell lines. (68) Autophagy is a conserved process of normal cell turnover by regulating degradation of its components, and is characterized by the formation of autophagosomes, which are double-membrane cytoplasmic vescicles engulfing intracellular material including protein, lipids, as well as organelles such as mitochondria and endoplasmic reticulum. Subsequently autophagosomes fuse with lysosomes, and their contents are degradated by lysosomal enzymes.(69, 70) This self-cannibalization is a highly-conserved response to metabolic stress, in which cellular components are degraded for the maintenance of homeostasis.(71) Intriguingly, the waste removal function of autophagy appears as to be double-edged sword, since it can either lead to cell survival or death.(72) A recent report shows that chloroquine, a chemical inhibitor of autophagy, significantly increased viability of leukemia cells treated with HDACIs. (73) Although the clinical relevance of autophagy induced by HDACIs is not totally defined, the above data suggest an intriguing role of autophagy in the treatment with HDACIs. These results also suggesting a rationale for targeting tumor cells harboring defects in apoptosis mechanisms.
Antimyeloma activity of HDAC Inhibitors
Histone deacetylase represents a hopeful clinical target for development of novel anti-MM therapeutic options. Indeed several compounds have already been tested in preclinical MM models, revealing promising antimyeloma activity. In many of these preclinical studies, inhibition of HDAC resulted in addictive or synergistic effects when combined with novel or classical anti-MM drugs, providing the rationale for derived clinical studies. Importantly, early-phase clinical trials of class I and II have reveal modest single agent activity in patients with advanced MM, but encouraging clinical response rates have been reported with HDACIs combination treatment when combined with proteasome inhibitors, lenalidomide and dexamethasone. A common pattern of toxicity, observed with most of these compounds, is fatigue, nausea, vomiting and diarrhea.
VORINOSTAT
Vorinostat (SAHA) is a potent HDACI with a hydroxamic acid moiety targeting class I and II HDACs, causing their reversible inhibition (74) As above mentioned, it was the first epigenetic agent approved for the treatment of malignant disease (cutaneous T-cell lymphoma). This nonselective-HDACI showed an in vitro broad anti-proliferation effect against a variety of transformed cells, including myeloma cells, with an IC50 values ranging from 0.5 to 10μM (75, 76). Various antiproliferative and/or proapototic sequelae are observed in vorinostat-treated MM cells, including a decrease in transcript levels for growth factors and/or their surface receptors, caspase inhibitors, proteasome subunits and DNA synthesis/repair enzymes. These anti-MM effects of HDACIs provided the framework for an initial phase-I trial for 13 heavily pre-treated relapsed myeloma patients. vorinostat monotherapy was administered orally with an escalating dosing schedule to a maximum of 200 mg twice daily for 14d of a 3 weeks cycle. The most important side effects reported were fatigue, diarrhea, and nausea and were mostly <or=2 grade. Among 10 evaluable patients, 1 had a minimal response and 9 had stable disease. Overall these data showed modest single agent activity of vorinostat for treatment of relapsed/refractory MM patients.(77)
Several in vitro studies showed a synergistic cytotoxic effect when this HDACI was added to bortezomib, by disruption of aggresome function and induction of ER stress. Accordingly subsequent clinical studies have focused on combination regimens including vorinostat and the proteasome inhibitor bortezomib for the treatment of advanced MM patients. (54, 78) The first study of this kind was a phase I trial involving 23 patients with relapsed MM (median number of previous regimens was 7, including Bortezomib). The dose-limiting toxicity (DLT) of vorinostat was seen in 2 patients at 500mg/die vorinostat (prolonged QT interval and fatigue), with a maximum tolerated dose (MTD) of 400 mg /die SAHA (days 4–11), and 1.3 mg/m2 bortezomib (days 1, 4, 8, and 11 of a 21-day cycles). The most common toxicities were hematologic, gastrointestinal, and fatigue. The events were mild-to-moderate in severity, and were similar to those observed in previous trials with vorinostat monotherapy (16, 77, 79). After 2 cycles of therapy, response rates in 21 patients (pts) evaluable for response were reported as very good partial response (VGPR) in 2 pts and partial response (PR) in 7 pts, resulting in an ORR of 42%. Importantly stable disease (SD) was observed in 10 pts and progressive disease (PD) in 2 pts. (80) Overall, these preliminary data demonstrated that the combination of vorinostat and bortezomib, is well tolerated, paving the way for the two recently concluded phase II and Phase III clinical trials VANTAGE088 and VANTAGE095 in the relapsed MM setting.
In the large phase III international, multicenter, randomized VANTAGE088 trial, 637 MM patients who had progressive disease after one to three prior anti-MM treatments, but were still Bortezomib sensitive, were randomized to receive 21-day cycles of either vorinostat (oral 400mg/die) in combination with bortezomib (1.3 mg/m2 intravenously; days 1, 4, 8, and 11) or placebo on days 1 to 14. A median of 7 cycles (mean: 7.6 cycles; range 1–30 cycles) was administrated. The combination of bortezomib plus vorinostat increased the primary end point of progression-free survival from 6.83 months to 7.63 months (hazard ratio, 0.774; P= .01), a 25-days advantage. In addition, median overall survival was not significantly different between the bortezomib/vorinostat and control arms. However, using the European Bone and Marrow Transplantation Group (EBMT) criteria, the combination of vorinostat plus bortezomib significantly improved the overall response (56% vs. 41%) and clinical benefit rates (71% vs. 53%) compared with bortezomib alone (both P < .0001). (81)
More encouraging results were presented recently at American Society of Hematology (ASH) meeting about multicenter, open-label phase IIB study (VANTAGE095) in Bortezomib relapsed/refractory population. 143 patients whose disease was described as refractory to available therapies including bortezomib were enrolled to receive the same combination regimen that was used in VANTAGE088. The patients received vorinostat 400 mg orally daily for 14 days in combination with bortezomib (1.3 mg/m2 intravenously; days 1, 4, 8, and 11). This treatment continued up to disease progression, unacceptable toxicities, or patient withdrawal from the study. The response rate was complete response (CR) in 1%, VGPR in 4%, PR in 12%, minimal response 14% and SD in 47%. Applying the EBMT criteria, the median duration of response was 7.0 months and the overall response rate 11%. Indeed, the 2-year overall survival rate was 32%, with a median overall survival of 11.1 months. The most common treatment-emergent adverse events included thrombocytopenia (70%), nausea (57%), diarrhea (54%), and anemia (52 %). Serious adverse events (SAE), reported in 65% of patients, resulted in treatment discontinuations in only 11%. (82) Overall, these data show that the combination of vorinostat and bortezomib is a suitable therapeutic option for heavily pretreated and refractory MM patients. (Table1) However, the research of other novel HDACIs as well as proteasome inhibitors will help elucidate which treatment combinations and dosing regimens are optimal in the context of varied MM patient characteristics. Finally, more clinical trials are currently underway aimed to establish whether the combination of vorinostat and novel or standard anti-MM drugs are superior to monotherapy. (Table 2)
Table 1.
Clinical studies of Vorinostat (SAHA) in relapsed patients with MM
| Phase | Regimen | Schedule | Response | Toxic Effectsgrade ≥2 occurring in >10% of patients | References |
|---|---|---|---|---|---|
| I | SAHA | SAHA Escalating dose up to 250mg orally bd × 5d/w or 14d/3w | 10% MR; 90% SD |
Anorexia 38% Deydration 31% Nausea 31% Lymphopenia 31% Weight decrease 23% Fatigue 38% |
Richardson et al, 2008 |
| I | SAHA + BORTEZOMIB | SAHA 400 mg/d × 8d + BORTEZOMIB 1.3 mg/m2 days 1, 4, 8, 11 (21-day cycles) | 9% VGPR 33% PR 43% OR |
Anemia 12% Thrombocytopenia 14% Peripheral Neuropathy 10% |
Badros et al, 2009 |
| IIB (VANTAGE095) | SAHA + BORTEZOMIB | SAHA 400 mg od, day 1–14; BORTEZOMIB 1.3 mg/m2 iv, Day 1, 4, 8, 11 (21-day cycle) | 1%CR 4%VGPR 12%PR 14%MD 47% SD |
Anemia 38% Thrombocytopenia 68% Neutropenia 32% Diarrhea 17% |
Siegel et al, ASH 2011 |
| III (VANTAGE088) | SAHA + BORTEZOMIB | SAHA 400 mg od, day 1–14; BORTEZOMIB 1.3 mg/m2 iv, Day 1, 4, 8, 11 (21-day cycle) | 8%CR 20%VGPR 28%PR 15%MR 25% SD |
Anemia 17% Thrombocytopenia 45% Neutropenia 28% Diarrhea 17% Fatigue 17% |
Dimopoulos et al, ASH 2011 |
Bd, twice daily; od, once daily; CR, complete response; VGPR, very good partial response; PR, partial response; MR, minimal response; SD, stable disease; IV, intravenous;
National Cancer Institute Common Terminology Criteria for Adverse Events
Table 2.
Ongoing Vorinostat clinical trials in relapsed myeloma
| PHASE | ClinicalTrials.gov Identifier: | Experimental Treatment | |
|---|---|---|---|
| II | Bortezomib and Vorinostat in Treating Patients With Multiple Myeloma Who Have Undergone Autologous Stem Cell Transplant | NCT00839956 | Bortezomib IV days 2 and 5; Vorinostat orally OD days 1–14. (28 days cycle) |
| I | Vorinostat and Lenalidomide After Autologous Stem Cell Transplant in Treating Patients With Multiple Myeloma | NCT00729118 | Lenalidomide days 1–21 (28-day cycle); Vorinostat orally beginning at dose level 1 starting day +90 ±6 days after HSCT days 1 and 15–21 (28-day cycle) |
| II | Study of Vorinostat With Doxil and Bortezomib for Patients With Relapsed/Refractory Multiple Myeloma | NCT01492881 | Vorinostat Oral, 400mg 4–11; Bortezomib SC, 1.3mg/m2, Days 1, 4, 8, 11 ; Pegylated Liposomal Doxorubicin IV, 30mg/m2 day 4 |
| I | A Study of the Combination Vorinostat With Lenalidomide, Bortezomib and Dexamethasone for Patients With Newly Diagnosed Multiple Myeloma | NCT01038388 | Lenalidomide 25mg daily days 1–14; Bortezomib 1.3mg/m2 daily days 1, 4, 8, 11; Dexamethasone 20 mg daily days 1, 2, 4, 5, 8, 9, 11, 12; Vorinostat escalating dose up to 300mg daily days 1–14 |
| I/II | Vorinostat, Lenalidomide and Dexamethasone in Multiple Myeloma Refractory to Previous Lenalidomide Containing Regimens | NCT01502085 | Vorinostat: 400 mg orally days 1–7 and 15–21 Lenalidomide: 25 mg oral days 1–21; Dexamethasone: 40mg orally days 1, 8, 15 and 22 for patients aged less than 75 years, 20mg for those aged 75 years |
| II | Vorinostat Plus Lenalidomide and Dexamethasone or Lenalidomide Plus Dexamethasone in Multiple Myeloma Patients Who Experience Biochemical Relapse During Lenalidomide Maintenance Therapy | NCT01501370 | Vorinostat orally 400 mg/day, days 1–7 and 15–21 (28-day cycle); Lenalidomide orally 25 mg/day 21 days; Dexamethasone orally 40 mg day 1,8, 15, 22 (28-day cycle) |
| I/II | Vorinostat in Combination With Bortezomib, Doxorubicin and Dexamethasone in Patients With Refractory or Relapsed Multiple Myeloma | NCT01394354 | Vorinostat escalating dose up to 300 mg/d orally, day 1–4, 8–11, 15–18; Bortezomib IV 1.3mg/m2 days 1,8,15; Doxorubicin 18mg/m2 IV days 1 and 8; Dexamethasone 40mg oral. days 1,8,15,22 1st treatment cycle, 20mg oral days 1,8,15,22 2–6 treatment cycles; (28-day cycle). |
| I/II | Study of Vorinostat Plus Melphalan and Prednisone in Advanced, Refractory Multiple Myeloma Patients | NCT00857324 | Melphalan 0.18 mg/Kg for 4 days; Prednisone 1.5 mg/Kg for 4 days; Vorinostat escalating dose up to 400 mg/d oral., (28 days cycle) |
| I/II | A Study of Carfilzomib, Lenalidomide, Vorinostat, and Dexamethasone in Relapsed and/or Refractory Multiple Myeloma | NCT01297764 | Escalation Schema Cohort Carfilzomib 15–20 mg/m2, Lenalidomide 15–25 mg, Vorinostat 300–400 mg, Dexamethasone 40mg; |
OD, once daily; IV, intravenous; SC, subcutaneous;
PANOBINOSTAT
Panobinostat (LBH589) is a cinnamic hydroxamic acid analog that has potent activity as HDACI and has been shown, to have, a 10-fold higher inhibitory activity in vitro against Class I, II and IV HDACs than vorinostat. The inhibitory activity and cytotoxic effect have been demonstrated in a broad range of hematologic and solid tumor cell lines including cutaneous T cell lymphoma, acute myeloid leukemia, chronic myeloid leukemia, Hodgkin lymphoma, colon, breast, pancreas and prostate. (83, 84) Potent in vitro antimyeloma activity as a single agent as well as in combination with bortezomib (54) has been shown, providing the rationale for derived clinical protocols. Encouraging results were obtained in a phase II, single arm and multicenter study enrolling 38 heavily pretreated and refractory MM patients. The dosing schedule was 20 mg/day of oral Panobinostat given on a Monday/Wednesday/Friday (MWF) In general, the treatment was well tolerated, with mild or moderate level of nausea and fatigue in half of the patients. Grade 3/4 cytopenia, with neutropenia (32%), thrombocytopenia (26%) and anemia (16%) was observed. The responses obtained were 1 VGPR, 1 MR and 3 SD for longer than 3 months.(85) Based on these data, panobinostat was tested in combination with Bortezomib in a phase I study involving 62 relapsed or relapsed and refractory MM patients, with a predictable and manageable safety profile as well as promising activity in this setting. (86) These observations are being evaluated further in phase II and III international clinical trials, (87, 88) combining panobinostat with bortezomib and dexamethasone in patients with relapsed or refractory MM. The Phase III Study PANobinostat ORAl in Multiple myelomA (PANORAMA) 1 is an international, randomized, double-blind, phase III study of panobinostat (or placebo) + bortezomib + dexamethasone. Patients with bortezomib-refractory MM are excluded from this trial, comprising 2 treatment phases. Treatment phase 1 consists of eight 3-week cycles of panobinostat (oral 20 mg) or placebo administered thrice weekly and bortezomib (intravenous 1.3 mg/m2) administered twice weekly, each for 2 of 3 weeks. Dexamethasone (oral 20 mg) is administered on the days of and after bortezomib dosing. If clinical benefit is observed, patients proceed to treatment phase 2, which consists of four 6-week cycles with a modified (once-weekly) bortezomib schedule. Preliminary safety blinded data from 273 enrolled patients demonstrated no novel or unpredicted side effects, with diarrhea (36%) thrombocytopenia (41%), anemia (24%), fatigue (24%) and neutropenia (12%) representing the most commonly observed side effects.
PANORAMA2 is a single-arm, open-label, phase II study of panobinostat + bortezomib + dexamethasone in bortezomib-refractory patients, to assess if the combination with HDAC inhibitor can re-sensitize patients to a bortezomib containing therapeutic regimen. Patients with relapsed and bortezomib-refractory MM are treated in 2 phases. Treatment phase 1 consists of 8 three-week cycles of oral panobinostat (20 mg days 1, 3, 5, 8, 10, 12) + intravenous bortezomib (1.3 mg/m2 days 1, 4, 8, 11) + oral dexamethasone (20 mg on day of and after bortezomib). Patients demonstrating clinical benefit (stable disease) can proceed to treatment phase 2, consisting of 4 six-week cycles of panobinostat (20 mg TIW 2 weeks on 1 week off, and repeat) + bortezomib (1.3 mg/m2 days 1, 8, 22, 29) + dexamethasone (20 mg on day of and after bortezomib). The most common adverse events include diarrhea (41%) thrombocytopenia (38%), anemia (24%), fatigue (52%) and nausea (38%). A preliminary analysis (ASH 2011) showed a PR in 9 pts and MR in 9 pts. Collectively the preliminary results of PANORAMA1 and PANORAMA2 therefore show that the combination of panobinostat and bortezomib is a promising treatment option for relapsed MM patients, including bortezomib-resistant MM. (Table 3)
Table 3.
Clinical studies of Panobinostat (LBH589) in relapsed MM
| Phase | Regimen | Schedule | Response | Toxic Effects grade >2 occurring in >10% of patients | References |
|---|---|---|---|---|---|
| II | LBH589 | LBH589 20mg/d | 2% VGPR 2% MR 8% SD |
Anemia 16% Thrombocytopenia 26% Neutropenia 32% Fatigue/Nausea 50% |
Wolf et al, 2008 |
| Ib | LBH589 + BORTEZOMIB | LBH589 Escalating dose up to 20mg orally/tw; BORTEZOMIB 1.3 mg/m2 IV days 1, 4, 8, 11 (21-day cycles) |
76% ≥MR | Anemia 21% Thrombocytopenia 75% Neutropenia 53% Asthenia 16% |
San-Miguel et al, ASCO 2011 |
| III (PANORAMA1) | LBH589 + BORTEZOMIB + DEXAMETHASONE | LBH589 20 mg tw; BORTEZOMIB 1.3 mg/m2 IV, day 1, 4, 8, 11; DEX 20MG day 1,2, 4,5, 8,9, 11,12 (21-day cycle) | ongoing | ongoing | San-Miguel et al, ASCO 2011 |
| II (PANORAMA2) | LBH589 + BORTEZOMIB + DEXAMETHASONE | LBH589 20 mg tw; BORTEZOMIB 1.3 mg/m2 IV, day 1, 4, 8, 11; DEX 20MG day 1,2, 4,5, 8,9, 11,12 (21-day cycle) | Phase 1: 4% nCR 13% PR 13% MR Phase 2: ongoing |
Anemia 10% Thrombocytopenia 38% Neutropenia 12% Pneumonia 10% Peripheral Neuropathy 17% |
Richardson et al, ASH 2011 |
Tw, thrice weekly; CR, complete response; nCR, near complete response, VGPR, very good partial response; PR, partial response; MR, minimal response; SD, stable disease; IV, intravenous;
National Cancer Institute Common Terminology Criteria for Adverse Events
Finally, panobinostat is also undergoing evaluation in at least 8 different clinical trials in combination with bortezomib, dexamethasone, melphalan, lenalidomide, everolimus and carfilzomib to determine both efficacy and safety. (89, 90)
ROMIDEPSIN
Romidepsin (FR901228 or FK228) is a depsipeptide derived from the bacterium c. violaceum with mainly class I inhibitory activity. It was approved by the Food and Drug Administration in 2009 for relapsed cutaneous T cell lymphoma as a single-agent. Preclinical data has demonstrated activity against several MM cell lines, as well as primary patient myeloma cells. (32) Based on these preclinical data and the minimal toxicity of this compound, a phase II study evaluated its activity in heavily pretreated patients with MM who were refractory to multiple therapies, often including stem cell transplantation, thalidomide, and bortezomib. Patients received romidepsin at a dose of 13 mg/m2 as a 4-hour infusion on days 1, 8, and 15 of a 28-days cycle. All patients were treated for at least 4 weeks. Although no objective response was achieved, approximately 30% of patients exhibited stabilization of M-protein and resolution of hypercalcemia or improvement in bone pain. (91) The most common adverse events included grade 1 to 2 nausea in 54% of pts, grade 2 fatigue in 31% pts and grade 2 taste alteration in 8% of pts. Of the 27 cycles administered, 22 were given at full dose, and 5 doses were given at a reduced dose in 5 pts. Electrocardiographic changes were common but clinically insignificant, including asymptomatic and reversible QT interval prolongation, ST segment depression, and T wave inversion. Importantly, all the above side effects were previously described. (92) In summary, the treatment of MM patients with romidepsin as single agent induces biological but only modest clinical effects. However, romidepsin exhibits synergistic cytotoxicity when combined with bortezomib (93).
Encouraging results have been reported in a phase 2 trial of romidepsin in combination with dexamethasone and bortezomib. Twenty-five patients were enrolled, and all were assessable for toxicity. The MTD identified was bortezomib 1.3 mg/m2 (days 1, 4, 8, and 11), dexamethasone 20 mg (days 1, 2, 4, 5, 8, 9, 11, and 12), and romidepsin 10 mg/m2 (days 1, 8, and 15) every 28 days. The incidence of grade 3 neutropenia and anemia were acceptable and similar to those reported in previous phase 2 or 3 trials using bortezomib and dexamethasone. Non-hematologic toxicities at the MTD were all manageable with standard approaches. 72% of patients exhibited an OR > minor response with 8% complete remissions and 52% partial responses (including 28% with VGPR). The median time to progression was 7.2 months, and the median OS was > 36 months. (94) (Table 4)
Table 4.
Clinical studies of Romidepsin (FR901228) in relapsed MM
| Phase | Regimen | Schedule | Response | Toxic Effects grade >2 occurring in >10% of patients | References |
|---|---|---|---|---|---|
| II | FR901228 | FR901228 13 mg/m2 (4-hour infusion) days 1, 8, 15 (28-days cycle) | 30% stabilization of M protein | Thrombocytopenia 23% Fatigue 31% |
Niesvizky et al, 2011 |
| II | FR901228 + BORTEZOMIB + DEXAMETHASONE | FR901228 Escalating dose 8–14 mg/m2 on days 1, 8, and 15; BORTEZOMIB 1.3 mg/m2 IV days 1, 4, 8, 11; DEX 20MG day 1,2, 4,5, 8,9, 11,12 (28-day cycle) | 8% CR 52% PR 3% MR |
Anemia 36% Thrombocytopenia 64% Neutropenia 36% Fatigue 20% |
Harrison et al, 2011 |
CR, complete response; PR, partial response; MR, minimal response; IV, intravenous;
National Cancer Institute Common Terminology Criteria for Adverse Events
OTHER HDAC INHIBITORS IN MM CLINICAL TRIALS
The experience with Vorinostat and Panobinostat in MM has fueled interest in the development of other HDAC inhibitors for the treatment of MM. Many other HDACIs show promising in vitro effects and are currently being taken forward into clinical trials. The most encouraging results have been obtained with the following:
ACY-1215 it is the only small molecule developed to target HDAC6 optimized for oral delivery. Recent preclinical data have demonstrated its ability to inhibit MM cell growth, osteoclastogenesis and to enhance osteoblastogenesis, alone and in combination with bortezomib.(59) A Phase 1/2, open-labeled, multicenter study, to evaluate the side effects and determine the best dose of oral ACY-1215 as monotherapy, and in combination with bortezomib and dexamethasone, in patients with relapsed or refractory MM is ongoing.
Belinostat (PXD101) it is hydroxamic acid class of HDACIs with a non-selective-HDAC inhibitor effect. It has demonstrated activity in lymphoma and leukemia patients and in certain types of solid tumors (26) A Phase II study enrolled 24 MM relapsed and refractory patients, at a dose of 1g/m2 per day IV for 5 days of a 21 days cycle, in monotherapy as well as combination with high dose of dexamethasone. This treatment was tolerated, with minimal side effects obtaining 1 MR and 5 SD. (95)
ITF2357 (Givinostat) it is an orally active HDACI which has been given to 19 patients with relapsed or progressive MM to determine the maximum tolerated dose in phase-II, multiple-dose clinical trial. When given at a dose of 100 mg twice daily, alone or combined with dexamethasone, it proved tolerable but showed only a modest clinical benefit in advanced MM. Five patients achieved stable disease, five had disease progression, and nine had died of progressive MM. Three patients experienced grade 3–4 gastro-intestinal toxicity, and three had transient electrocardiographic abnormalities. All patient experimented thrombocytopenia, grade 3–4. (96)
FUTURE PERSPECTIVES
Although promising, the precise role of HDACIs in the therapy of MM remains to be defined, as well as the specific mechanisms by which anti-MM activity is mediated. Overall, the use of HDACIs as single agents in MM patients results in modest activity. However, the combination with other anti-MM drugs, mainly bortezomib, results in marked clinical response. One of the most convincing mechanisms underlying this synergism is interference with protein degradation by blockade of both proteasome and aggresome inhibition or deregulation of the transcriptional regulator KLF9. (97) A large number of clinical trials have been completed and are ongoing using a wide variety of HDACIs in combination with novel or conventional anti-MM agents. Although there have been some promising results in patients with advanced disease, it is likely that the full potential of this such a therapy will only be obtained once rational combination therapies and more tolerated and selective HDACIs are developed. Moreover impaired understanding of the in vivo mechanisms of action of HDAC inhibitor-induced tumor cell death will identify biomarkers associated with clinical response. Overall this approach will better define the potential of HDACIs for the management of MM and other hematologic malignances.
Acknowledgments
This study is supported by National Institutes of Health (grants RO-1 CA-50947, RO-1 CA-73878), DF/HCC SPORE in Multiple Myeloma (PO-50100707); American Italian Cancer Foundation (M.C.), Fondazione Italiana per la Ricerca sul Cancro (M.C.) and International Multiple Myeloma Foundation (A.C.). K.C.A. is an American Cancer Society Council Research Professor.
ABBREVIATIONS
- MM
Multiple myeloma
- M
monoclonal
- PI
proteasome inhibitor
- IMiDs
immunomodulatory drugs
- ECM
extracellular matrix
- BM
bone marrow
- HDACIs
histone deacetylase inhibitors
- AML
acute myeloid leukemia
- MDS
myelodisplastic syndrome
- BMSC
bone marrow stromal cells
- HATs
histone acetylases
- DSBs
double strands break
- Hsp90
heat shock protein 90
- MPR
misfolded protein response
- HIF-1α
hypoxia inducible factor-1 alpha
- VEGF
vascular endothelial growth factor
- ER
endoplasmic reticulum
- DLT
dose-limiting toxicity
- MTD
maximum tolerated dose
- VGPR
very good partial response
- pts
patients
- PR
partial response
- ORR
overall response rate
- SD
stable disease
- PD
progressive disease
- EBMT
European bone and marrow transplantation
- CR
complete response
- SAE
serious adverse event
- MR
minimal response
- OR
overall response
- OS
overall survival
- BD
twice daily
- OD
once daily
- IV
intravenous
- SC
subcutaneous
- Tw
thrice weekly
- nCR
near complete response
References
- 1.Hallek M, Bergsagel PL, Anderson KC. Multiple myeloma: increasing evidence for a multistep transformation process. Blood. 1998 Jan 1;91(1):3–21. [PMC free article] [PubMed] [Google Scholar]
- 2.Kyle RA, Rajkumar SV. Multiple myeloma. N Engl J Med. 2004 Oct 28;351(18):1860–73. doi: 10.1056/NEJMra041875. [DOI] [PubMed] [Google Scholar]
- 3.Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. 2011 Mar 17;364(11):1046–60. doi: 10.1056/NEJMra1011442. [DOI] [PubMed] [Google Scholar]
- 4.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008 Mar-Apr;58(2):71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 5.Barlogie B. Advances in therapy of multiple myeloma: lessons from acute leukemia. Clin Cancer Res. 1997 Dec;3(12 Pt 2):2605–13. [PubMed] [Google Scholar]
- 6.Durie BG, Salmon SE. A clinical staging system for multiple myeloma. Correlation of measured myeloma cell mass with presenting clinical features, response to treatment, and survival. Cancer. 1975 Sep;36(3):842–54. doi: 10.1002/1097-0142(197509)36:3<842::aid-cncr2820360303>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 7.Kristinsson SY, Landgren O, Dickman PW, Derolf AR, Bjorkholm M. Patterns of survival in multiple myeloma: a population-based study of patients diagnosed in Sweden from 1973 to 2003. J Clin Oncol. 2007 May 20;25(15):1993–9. doi: 10.1200/JCO.2006.09.0100. [DOI] [PubMed] [Google Scholar]
- 8.Chauhan D, Uchiyama H, Akbarali Y, Urashima M, Yamamoto K, Libermann TA, et al. Multiple myeloma cell adhesion-induced interleukin-6 expression in bone marrow stromal cells involves activation of NF-kappa B. Blood. 1996 Feb 1;87(3):1104–12. [PubMed] [Google Scholar]
- 9.Chauhan D, Catley L, Li G, Podar K, Hideshima T, Velankar M, et al. A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from Bortezomib. Cancer Cell. 2005 Nov;8(5):407–19. doi: 10.1016/j.ccr.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 10.Moreaux J, Legouffe E, Jourdan E, Quittet P, Reme T, Lugagne C, et al. BAFF and APRIL protect myeloma cells from apoptosis induced by interleukin 6 deprivation and dexamethasone. Blood. 2004 Apr 15;103(8):3148–57. doi: 10.1182/blood-2003-06-1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anderson KC. The 39th David A. Karnofsky Lecture: Bench-to-Bedside Translation of Targeted Therapies in Multiple Myeloma. J Clin Oncol. 2012 Feb 1;30(4):445–52. doi: 10.1200/JCO.2011.37.8919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Glozak MA, Seto E. Histone deacetylases and cancer. Oncogene. 2007 Aug 13;26(37):5420–32. doi: 10.1038/sj.onc.1210610. [DOI] [PubMed] [Google Scholar]
- 13.Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006 Jan;6(1):38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- 14.Richon VM, Emiliani S, Verdin E, Webb Y, Breslow R, Rifkind RA, et al. A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases. Proc Natl Acad Sci U S A. 1998 Mar 17;95(6):3003–7. doi: 10.1073/pnas.95.6.3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Inche AG, La Thangue NB. Chromatin control and cancer-drug discovery: realizing the promise. Drug Discov Today. 2006 Feb;11(3–4):97–109. doi: 10.1016/S1359-6446(05)03691-3. [DOI] [PubMed] [Google Scholar]
- 16.Duvic M, Talpur R, Ni X, Zhang C, Hazarika P, Kelly C, et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL) Blood. 2007 Jan 1;109(1):31–9. doi: 10.1182/blood-2006-06-025999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist. 2007 Oct;12(10):1247–52. doi: 10.1634/theoncologist.12-10-1247. [DOI] [PubMed] [Google Scholar]
- 18.StatBite: FDA oncology drug product approvals in 2009. J Natl Cancer Inst. 2010 Feb 24;102(4):219. doi: 10.1093/jnci/djq030. [DOI] [PubMed] [Google Scholar]
- 19.Kouzarides T. Chromatin modifications and their function. Cell. 2007 Feb 23;128(4):693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 20.Felsenfeld G, Groudine M. Controlling the double helix. Nature. 2003 Jan 23;421(6921):448–53. doi: 10.1038/nature01411. [DOI] [PubMed] [Google Scholar]
- 21.Rice JC, Allis CD. Code of silence. Nature. 2001 Nov 15;414(6861):258–61. doi: 10.1038/35104721. [DOI] [PubMed] [Google Scholar]
- 22.Fiskus W, Rao R, Fernandez P, Herger B, Yang Y, Chen J, et al. Molecular and biologic characterization and drug sensitivity of pan-histone deacetylase inhibitor-resistant acute myeloid leukemia cells. Blood. 2008 Oct 1;112(7):2896–905. doi: 10.1182/blood-2007-10-116319. [DOI] [PubMed] [Google Scholar]
- 23.Bruzzone S, Parenti MD, Grozio A, Bauer I, Del Rio A, Nencioni A. Rejuvenating sirtuins: the rise of a new family of cancer drug targets. Current Pharmaceutical Design. 2012 doi: 10.2174/138161213804581954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Osada H, Tatematsu Y, Saito H, Yatabe Y, Mitsudomi T, Takahashi T. Reduced expression of class II histone deacetylase genes is associated with poor prognosis in lung cancer patients. Int J Cancer. 2004 Oct 20;112(1):26–32. doi: 10.1002/ijc.20395. [DOI] [PubMed] [Google Scholar]
- 25.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009 Aug 14;325(5942):834–40. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 26.Stimson L, La Thangue NB. Biomarkers for predicting clinical responses to HDAC inhibitors. Cancer Lett. 2009 Aug 8;280(2):177–83. doi: 10.1016/j.canlet.2009.03.016. [DOI] [PubMed] [Google Scholar]
- 27.Rosato RR, Almenara JA, Grant S. The histone deacetylase inhibitor MS-275 promotes differentiation or apoptosis in human leukemia cells through a process regulated by generation of reactive oxygen species and induction of p21CIP1/WAF1 1. Cancer Res. 2003 Jul 1;63(13):3637–45. [PubMed] [Google Scholar]
- 28.Nebbioso A, Clarke N, Voltz E, Germain E, Ambrosino C, Bontempo P, et al. Tumor-selective action of HDAC inhibitors involves TRAIL induction in acute myeloid leukemia cells. Nat Med. 2005 Jan;11(1):77–84. doi: 10.1038/nm1161. [DOI] [PubMed] [Google Scholar]
- 29.Zhao Y, Tan J, Zhuang L, Jiang X, Liu ET, Yu Q. Inhibitors of histone deacetylases target the Rb-E2F1 pathway for apoptosis induction through activation of proapoptotic protein Bim. Proc Natl Acad Sci U S A. 2005 Nov 1;102(44):16090–5. doi: 10.1073/pnas.0505585102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Catley L, Weisberg E, Tai YT, Atadja P, Remiszewski S, Hideshima T, et al. NVP-LAQ824 is a potent novel histone deacetylase inhibitor with significant activity against multiple myeloma. Blood. 2003 Oct 1;102(7):2615–22. doi: 10.1182/blood-2003-01-0233. [DOI] [PubMed] [Google Scholar]
- 31.Mitsiades N, Mitsiades CS, Richardson PG, McMullan C, Poulaki V, Fanourakis G, et al. Molecular sequelae of histone deacetylase inhibition in human malignant B cells. Blood. 2003 May 15;101(10):4055–62. doi: 10.1182/blood-2002-11-3514. [DOI] [PubMed] [Google Scholar]
- 32.Khan SB, Maududi T, Barton K, Ayers J, Alkan S. Analysis of histone deacetylase inhibitor, depsipeptide (FR901228), effect on multiple myeloma. Br J Haematol. 2004 Apr;125(2):156–61. doi: 10.1111/j.1365-2141.2004.04882.x. [DOI] [PubMed] [Google Scholar]
- 33.Dickinson M, Johnstone RW, Prince HM. Histone deacetylase inhibitors: potential targets responsible for their anti-cancer effect. Invest New Drugs. 2010 Dec;28(Suppl 1):S3–20. doi: 10.1007/s10637-010-9596-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Frew AJ, Lindemann RK, Martin BP, Clarke CJ, Sharkey J, Anthony DA, et al. Combination therapy of established cancer using a histone deacetylase inhibitor and a TRAIL receptor agonist. Proc Natl Acad Sci U S A. 2008 Aug 12;105(32):11317–22. doi: 10.1073/pnas.0801868105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang XD, Gillespie SK, Borrow JM, Hersey P. The histone deacetylase inhibitor suberic bishydroxamate regulates the expression of multiple apoptotic mediators and induces mitochondria-dependent apoptosis of melanoma cells. Mol Cancer Ther. 2004 Apr;3(4):425–35. [PubMed] [Google Scholar]
- 36.Xu W, Ngo L, Perez G, Dokmanovic M, Marks PA. Intrinsic apoptotic and thioredoxin pathways in human prostate cancer cell response to histone deacetylase inhibitor. Proc Natl Acad Sci U S A. 2006 Oct 17;103(42):15540–5. doi: 10.1073/pnas.0607518103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rosato RR, Maggio SC, Almenara JA, Payne SG, Atadja P, Spiegel S, et al. The histone deacetylase inhibitor LAQ824 induces human leukemia cell death through a process involving XIAP down-regulation, oxidative injury, and the acid sphingomyelinase-dependent generation of ceramide. Mol Pharmacol. 2006 Jan;69(1):216–25. doi: 10.1124/mol.105.017145. [DOI] [PubMed] [Google Scholar]
- 38.Ruefli AA, Ausserlechner MJ, Bernhard D, Sutton VR, Tainton KM, Kofler R, et al. The histone deacetylase inhibitor and chemotherapeutic agent suberoylanilide hydroxamic acid (SAHA) induces a cell-death pathway characterized by cleavage of Bid and production of reactive oxygen species. Proc Natl Acad Sci U S A. 2001 Sep 11;98(19):10833–8. doi: 10.1073/pnas.191208598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gaymes TJ, Padua RA, Pla M, Orr S, Omidvar N, Chomienne C, et al. Histone deacetylase inhibitors (HDI) cause DNA damage in leukemia cells: a mechanism for leukemia-specific HDI-dependent apoptosis? Mol Cancer Res. 2006 Aug;4(8):563–73. doi: 10.1158/1541-7786.MCR-06-0111. [DOI] [PubMed] [Google Scholar]
- 40.Sanchez-Gonzalez B, Yang H, Bueso-Ramos C, Hoshino K, Quintas-Cardama A, Richon VM, et al. Antileukemia activity of the combination of an anthracycline with a histone deacetylase inhibitor. Blood. 2006 Aug 15;108(4):1174–82. doi: 10.1182/blood-2005-09-008086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rosato RR, Almenara JA, Maggio SC, Coe S, Atadja P, Dent P, et al. Role of histone deacetylase inhibitor-induced reactive oxygen species and DNA damage in LAQ-824/fludarabine antileukemic interactions. Mol Cancer Ther. 2008 Oct;7(10):3285–97. doi: 10.1158/1535-7163.MCT-08-0385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen CS, Wang YC, Yang HC, Huang PH, Kulp SK, Yang CC, et al. Histone deacetylase inhibitors sensitize prostate cancer cells to agents that produce DNA double-strand breaks by targeting Ku70 acetylation. Cancer Res. 2007 Jun 1;67(11):5318–27. doi: 10.1158/0008-5472.CAN-06-3996. [DOI] [PubMed] [Google Scholar]
- 43.Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003 Jan 30;421(6922):499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 44.Feng R, Oton A, Mapara MY, Anderson G, Belani C, Lentzsch S. The histone deacetylase inhibitor, PXD101, potentiates bortezomib-induced anti-multiple myeloma effect by induction of oxidative stress and DNA damage. Br J Haematol. 2007 Nov;139(3):385–97. doi: 10.1111/j.1365-2141.2007.06772.x. [DOI] [PubMed] [Google Scholar]
- 45.Ungerstedt JS, Sowa Y, Xu WS, Shao Y, Dokmanovic M, Perez G, et al. Role of thioredoxin in the response of normal and transformed cells to histone deacetylase inhibitors. Proc Natl Acad Sci U S A. 2005 Jan 18;102(3):673–8. doi: 10.1073/pnas.0408732102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Quintas-Cardama A, Santos FP, Garcia-Manero G. Histone deacetylase inhibitors for the treatment of myelodysplastic syndrome and acute myeloid leukemia. Leukemia. 2011 Feb;25(2):226–35. doi: 10.1038/leu.2010.276. [DOI] [PubMed] [Google Scholar]
- 47.Taddei A, Maison C, Roche D, Almouzni G. Reversible disruption of pericentric heterochromatin and centromere function by inhibiting deacetylases. Nat Cell Biol. 2001 Feb;3(2):114–20. doi: 10.1038/35055010. [DOI] [PubMed] [Google Scholar]
- 48.Stevens FE, Beamish H, Warrener R, Gabrielli B. Histone deacetylase inhibitors induce mitotic slippage. Oncogene. 2008 Feb 28;27(10):1345–54. doi: 10.1038/sj.onc.1210779. [DOI] [PubMed] [Google Scholar]
- 49.Li Y, Kao GD, Garcia BA, Shabanowitz J, Hunt DF, Qin J, et al. A novel histone deacetylase pathway regulates mitosis by modulating Aurora B kinase activity. Genes Dev. 2006 Sep 15;20(18):2566–79. doi: 10.1101/gad.1455006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pearl LH, Prodromou C. Structure and mechanism of the Hsp90 molecular chaperone machinery. Annu Rev Biochem. 2006;75:271–94. doi: 10.1146/annurev.biochem.75.103004.142738. [DOI] [PubMed] [Google Scholar]
- 51.Hideshima T, Bradner JE, Wong J, Chauhan D, Richardson P, Schreiber SL, et al. Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc Natl Acad Sci U S A. 2005 Jun 14;102(24):8567–72. doi: 10.1073/pnas.0503221102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li X, Zhang K, Li Z. Unfolded protein response in cancer: the physician’s perspective. J Hematol Oncol. 2011;4:8. doi: 10.1186/1756-8722-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bellavista EAF, Parenti MD, Martucci M, Santoro A, Salvioli S, Capri M, Baruzzi A, Del Rio A, Franceschi C, Mishto M. Immunoproteasome in cancer and neuropathologies: a new therapeutic target? Current Pharmaceutical Design. 2012 [PubMed] [Google Scholar]
- 54.Catley L, Weisberg E, Kiziltepe T, Tai YT, Hideshima T, Neri P, et al. Aggresome induction by proteasome inhibitor bortezomib and alpha-tubulin hyperacetylation by tubulin deacetylase (TDAC) inhibitor LBH589 are synergistic in myeloma cells. Blood. 2006 Nov 15;108(10):3441–9. doi: 10.1182/blood-2006-04-016055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hideshima T, Richardson PG, Anderson KC. Mechanism of action of proteasome inhibitors and deacetylase inhibitors and the biological basis of synergy in multiple myeloma. Mol Cancer Ther. 2011 Nov;10(11):2034–42. doi: 10.1158/1535-7163.MCT-11-0433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Simms-Waldrip T, Rodriguez-Gonzalez A, Lin T, Ikeda AK, Fu C, Sakamoto KM. The aggresome pathway as a target for therapy in hematologic malignancies. Mol Genet Metab. 2008 Jul;94(3):283–6. doi: 10.1016/j.ymgme.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Namdar M, Perez G, Ngo L, Marks PA. Selective inhibition of histone deacetylase 6 (HDAC6) induces DNA damage and sensitizes transformed cells to anticancer agents. Proc Natl Acad Sci U S A. 2010 Nov 16;107(46):20003–8. doi: 10.1073/pnas.1013754107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kozikowski AP, Tapadar S, Luchini DN, Kim KH, Billadeau DD. Use of the nitrile oxide cycloaddition (NOC) reaction for molecular probe generation: a new class of enzyme selective histone deacetylase inhibitors (HDACIs) showing picomolar activity at HDAC6. J Med Chem. 2008 Aug 14;51(15):4370–3. doi: 10.1021/jm8002894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Santo L, Hideshima T, Kung AL, Tseng JC, Tamang D, Yang M, et al. Preclinical activity, pharmacodynamic and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood. 2012 Jan 19; doi: 10.1182/blood-2011-10-387365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kerbel RS. Tumor angiogenesis. N Engl J Med. 2008 May 8;358(19):2039–49. doi: 10.1056/NEJMra0706596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Qian DZ, Wang X, Kachhap SK, Kato Y, Wei Y, Zhang L, et al. The histone deacetylase inhibitor NVP-LAQ824 inhibits angiogenesis and has a greater antitumor effect in combination with the vascular endothelial growth factor receptor tyrosine kinase inhibitor PTK787/ZK222584. Cancer Res. 2004 Sep 15;64(18):6626–34. doi: 10.1158/0008-5472.CAN-04-0540. [DOI] [PubMed] [Google Scholar]
- 62.Ellis L, Hammers H, Pili R. Targeting tumor angiogenesis with histone deacetylase inhibitors. Cancer Lett. 2009 Aug 8;280(2):145–53. doi: 10.1016/j.canlet.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim MS, Kwon HJ, Lee YM, Baek JH, Jang JE, Lee SW, et al. Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nat Med. 2001 Apr;7(4):437–43. doi: 10.1038/86507. [DOI] [PubMed] [Google Scholar]
- 64.Fath DM, Kong X, Liang D, Lin Z, Chou A, Jiang Y, et al. Histone deacetylase inhibitors repress the transactivation potential of hypoxia-inducible factors independently of direct acetylation of HIF-alpha. J Biol Chem. 2006 May 12;281(19):13612–9. doi: 10.1074/jbc.M600456200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kim SH, Jeong JW, Park JA, Lee JW, Seo JH, Jung BK, et al. Regulation of the HIF-1alpha stability by histone deacetylases. Oncol Rep. 2007 Mar;17(3):647–51. [PubMed] [Google Scholar]
- 66.Kato H, Tamamizu-Kato S, Shibasaki F. Histone deacetylase 7 associates with hypoxia-inducible factor 1alpha and increases transcriptional activity. J Biol Chem. 2004 Oct 1;279(40):41966–74. doi: 10.1074/jbc.M406320200. [DOI] [PubMed] [Google Scholar]
- 67.Liu T, Kuljaca S, Tee A, Marshall GM. Histone deacetylase inhibitors: multifunctional anticancer agents. Cancer Treat Rev. 2006 May;32(3):157–65. doi: 10.1016/j.ctrv.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 68.Rikiishi H. Autophagic and apoptotic effects of HDAC inhibitors on cancer cells. J Biomed Biotechnol. 2011;2011:830260. doi: 10.1155/2011/830260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nature reviews Molecular cell biology. 2007 Nov;8(11):931–7. doi: 10.1038/nrm2245. [DOI] [PubMed] [Google Scholar]
- 70.Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008 Feb;4(2):151–75. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004 Apr;6(4):463–77. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 72.Levine B. Cell biology: autophagy and cancer. Nature. 2007 Apr 12;446(7137):745–7. doi: 10.1038/446745a. [DOI] [PubMed] [Google Scholar]
- 73.Wei Y, Kadia T, Tong W, Zhang M, Jia Y, Yang H, et al. The combination of a histone deacetylase inhibitor with the Bcl-2 homology domain-3 mimetic GX15-070 has synergistic antileukemia activity by activating both apoptosis and autophagy. Clin Cancer Res. 2010 Aug 1;16(15):3923–32. doi: 10.1158/1078-0432.CCR-10-0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Marks PA, Miller T, Richon VM. Histone deacetylases. Curr Opin Pharmacol. 2003 Aug;3(4):344–51. doi: 10.1016/s1471-4892(03)00084-5. [DOI] [PubMed] [Google Scholar]
- 75.Kelly WK, Marks PA. Drug insight: Histone deacetylase inhibitors--development of the new targeted anticancer agent suberoylanilide hydroxamic acid. Nat Clin Pract Oncol. 2005 Mar;2(3):150–7. doi: 10.1038/ncponc0106. [DOI] [PubMed] [Google Scholar]
- 76.Siegel D, Hussein M, Belani C, Robert F, Galanis E, Richon VM, et al. Vorinostat in solid and hematologic malignancies. J Hematol Oncol. 2009;2:31. doi: 10.1186/1756-8722-2-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Richardson P, Mitsiades C, Colson K, Reilly E, McBride L, Chiao J, et al. Phase I trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) in patients with advanced multiple myeloma. Leuk Lymphoma. 2008 Mar;49(3):502–7. doi: 10.1080/10428190701817258. [DOI] [PubMed] [Google Scholar]
- 78.Pei XY, Dai Y, Grant S. Synergistic induction of oxidative injury and apoptosis in human multiple myeloma cells by the proteasome inhibitor bortezomib and histone deacetylase inhibitors. Clin Cancer Res. 2004 Jun 1;10(11):3839–52. doi: 10.1158/1078-0432.CCR-03-0561. [DOI] [PubMed] [Google Scholar]
- 79.Garcia-Manero G, Yang H, Bueso-Ramos C, Ferrajoli A, Cortes J, Wierda WG, et al. Phase 1 study of the histone deacetylase inhibitor vorinostat (suberoylanilide hydroxamic acid [SAHA]) in patients with advanced leukemias and myelodysplastic syndromes. Blood. 2008 Feb 1;111(3):1060–6. doi: 10.1182/blood-2007-06-098061. [DOI] [PubMed] [Google Scholar]
- 80.Badros A, Burger AM, Philip S, Niesvizky R, Kolla SS, Goloubeva O, et al. Phase I study of vorinostat in combination with bortezomib for relapsed and refractory multiple myeloma. Clin Cancer Res. 2009 Aug 15;15(16):5250–7. doi: 10.1158/1078-0432.CCR-08-2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dimopoulos MAJS, Yoon S-S, Siegel DS, Lonial S, Hajek R, Facon T, Rosiñol L, Blacklock HA, Goldschmidt H, Hungria V, Spencer A, Palumbo A, Reece DE, Graef T, Houp J, Sun L, Eid JE, Anderson KC. Vantage 088: Vorinostat in Combination with Bortezomib in Patients with Relapsed/Refractory Multiple Myeloma: Results of a Global, Randomized Phase 3 Trial. ASH. 2011 [Google Scholar]
- 82.Siegel DSDM, Yoon S-S, Laubach JP, Kaufman JL, Goldschmidt H, Reece DE, Leleu X, Durrant S, Offner FC, Cavo M, Nagler A, Jagannath S, Graef T, Houp J, Sun L, Howe J, Wear SM, Anderson KC. Vantage 095: Vorinostat in Combination with Bortezomib in Salvage Multiple Myeloma Patients: Final Study Results of a Global Phase 2b Trial. ASH. 2011 [Google Scholar]
- 83.Ellis L, Pan Y, Smyth GK, George DJ, McCormack C, Williams-Truax R, et al. Histone deacetylase inhibitor panobinostat induces clinical responses with associated alterations in gene expression profiles in cutaneous T-cell lymphoma. Clin Cancer Res. 2008 Jul 15;14(14):4500–10. doi: 10.1158/1078-0432.CCR-07-4262. [DOI] [PubMed] [Google Scholar]
- 84.Ottmann OGSA, Prince HM, Bhalla KN, Fischer T, Liu A, Parker K, Jalaluddin M, Laird G, Woo M, Scott JW, DeAngelo DJ. Phase IA/II Study of Oral Panobinostat (LBH589), a Novel Pan- Deacetylase Inhibitor (DACi) Demonstrating Efficacy in Patients with Advanced Hematologic Malignancies. Blood (ASH Annual Meeting Abstracts) 2008;112 [Google Scholar]
- 85.Wolf LWSD, Matous J, Lonial S, Goldschmidt H, Schmitt S, Vij R, De Malgalhaes-Silverman M, Abonour R, Jalaluddin M, Li M, Hazell K, Bourquelot PM, Mateos MV, Anderson KC, Spencer A, Harousseau JL, Bladé J. A Phase II Study of Oral Panobinostat (LBH589) in Adult Patients with Advanced Refractory Multiple Myeloma) Blood ASH Annual Meeting Abstracts. 2008;112 [Google Scholar]
- 86.San-Miguel JRP, Sezer O, Guenther A, Siegel DS, Blade J, LeBlanc R, Sutherland HJ, Mateos M, Gramatzki M, Hazell KM, Bengoudifa B, Bourquelot PM, Anderson KC. A phase lb study of oral panobinostat and IV bortezomib in relapsed or relapsed and refractory multiple myeloma. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings. 2011;29(15) [Google Scholar]
- 87.San-Miguel JFdMHV, Yoon S-S, Wiktor-Jedrzejczak W, Elghandour A, Siritanaratkul N, Dimopoulos MA, Corradini P, Nakorn TN, Shelekhova T, Günther A, Yong K, Schlossman R, Wroclawska-Swacha M, Weber HJ, Bourquelot P, Hou J, Einsele H, Lee JH, Moreau P, Lonial S, Richardson PG. Update on a Phase III Study of Panobinostat with Bortezomib and Dexamethasone in Patients with Relapsed Multiple Myeloma: PANORAMA 1. Blood (ASH Annual Meeting Abstracts) 2011;118 [Google Scholar]
- 88.Richardson PGAM, Weber DM, Coutre SE, Lonial S, Gasparetto C, Warsi G, Ondovik M, Mukhopadhyay S, Snodgrass S, Schlossman R. Phase II Study of the Pan-Deacetylase Inhibitor Panobinostat in Combination with Bortezomib and Dexamethasone in Relapsed and Bortezomib-Refractory Multiple Myeloma (PANORAMA 2) Bood (ASH Annual Meeting Abstracts) 2011;118 [Google Scholar]
- 89.Kazamel TBJ, Yellin O, Boccia RV, Matous J, Dressler KA, Nassir Y, Rothstein K, Swift R. A phase I/II study of oral melphalan (MEL) combined with panobinostat (PAN) for patients with relapsed or refractory (R/R) multiple myeloma (MM) J Clin Oncol. 2011;29 [Google Scholar]
- 90.Offidani M, Polloni C, Cavallo F, Liberati AM, Ballanti S, Pulini S, et al. Phase II study of Melphalan, Thalidomide and Prednisone (MPT) combined with oral Panobinostat in patients with relapsed/refractory MM. Leuk Lymphoma. 2012 Feb 15; doi: 10.3109/10428194.2012.664844. [DOI] [PubMed] [Google Scholar]
- 91.Niesvizky R, Ely S, Mark T, Aggarwal S, Gabrilove JL, Wright JJ, et al. Phase 2 trial of the histone deacetylase inhibitor romidepsin for the treatment of refractory multiple myeloma. Cancer. 2011 Jan 15;117(2):336–42. doi: 10.1002/cncr.25584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Piekarz RL, Frye AR, Wright JJ, Steinberg SM, Liewehr DJ, Rosing DR, et al. Cardiac studies in patients treated with depsipeptide, FK228, in a phase II trial for T-cell lymphoma. Clin Cancer Res. 2006 Jun 15;12(12):3762–73. doi: 10.1158/1078-0432.CCR-05-2095. [DOI] [PubMed] [Google Scholar]
- 93.Sutheesophon K, Kobayashi Y, Takatoku MA, Ozawa K, Kano Y, Ishii H, et al. Histone deacetylase inhibitor depsipeptide (FK228) induces apoptosis in leukemic cells by facilitating mitochondrial translocation of Bax, which is enhanced by the proteasome inhibitor bortezomib. Acta Haematol. 2006;115(1–2):78–90. doi: 10.1159/000089471. [DOI] [PubMed] [Google Scholar]
- 94.Harrison SJ, Quach H, Link E, Seymour JF, Ritchie DS, Ruell S, et al. A high rate of durable responses with romidepsin, bortezomib, and dexamethasone in relapsed or refractory multiple myeloma. Blood. 2011 Dec 8;118(24):6274–83. doi: 10.1182/blood-2011-03-339879. [DOI] [PubMed] [Google Scholar]
- 95.Sullivan DSS, Schuster M, Berenson J, Gimsing P, Wislöff F, Waage A, Alsina M, Gerwien R, Clarke A, Moller K, Eng Ooi C. A Phase II Study of PXD 101 in Advanced Multiple Myeloma. Blood (ASH Annual Meeting Abstracts) 2006;108 [Google Scholar]
- 96.Galli M, Salmoiraghi S, Golay J, Gozzini A, Crippa C, Pescosta N, et al. A phase II multiple dose clinical trial of histone deacetylase inhibitor ITF2357 in patients with relapsed or progressive multiple myeloma. Ann Hematol. 2010 Feb;89(2):185–90. doi: 10.1007/s00277-009-0793-8. [DOI] [PubMed] [Google Scholar]
- 97.Mannava S, Zhuang D, Nair JR, Bansal R, Wawrzyniak JA, Zucker SN, et al. KLF9 is a novel transcriptional regulator of bortezomib- and LBH589-induced apoptosis in multiple myeloma cells. Blood. 2012 Feb 9;119(6):1450–8. doi: 10.1182/blood-2011-04-346676. [DOI] [PMC free article] [PubMed] [Google Scholar]