

Abstract
Understanding cancer cell signal transduction is a promising lead for uncovering therapeutic targets and building treatment-specific markers for epithelial ovarian cancer. To brodaly assay the many known transmembrane receptor systems, previous studies have employed gene expression data measured on high-throughput microarrays. Starting with the knowledge of validated ligand-receptor pairs (LRPs), these studies postulate that correlation of the two genes implies functional autocrine signaling. It is our goal to consider the additional weight of evidence that prognosis (progression-free survival) can bring to prioritize ovarian cancer specific signaling mechanism. We survey three large studies of epithelial ovarian cancers, with gene expression measurements and clinical information, by modeling survival times both categorically (long/short survival) and continuously. We use differential correlation and proportional hazards regression to identify sets of LRPs that are both prognostic and correlated. Of 475 candidate LRPs, 77 show reproducible evidence of correlation; 55 show differential correlation. Survival models identify 16 LRPs with reproduced, significant interactions. Only two pairs show both interactions and correlation (PDGFA PDGFRA and COL1A1
PDGFRA and COL1A1 CD44) suggesting that the majority of prognostically useful LRPs act without positive feedback. We further assess the connectivity of receptors using a Gaussian graphical model finding one large graph and a number of smaller disconnected networks. These LRPs can be organized into mutually exclusive signaling clusters suggesting different mechanisms apply to different patients. We conclude that a mix of autocrine and endocrine LRPs influence prognosis in ovarian cancer, there exists a heterogenous mix of signaling themes across patients, and we point to a number of novel applications of existing targeted therapies which may benefit ovarian cancer.
CD44) suggesting that the majority of prognostically useful LRPs act without positive feedback. We further assess the connectivity of receptors using a Gaussian graphical model finding one large graph and a number of smaller disconnected networks. These LRPs can be organized into mutually exclusive signaling clusters suggesting different mechanisms apply to different patients. We conclude that a mix of autocrine and endocrine LRPs influence prognosis in ovarian cancer, there exists a heterogenous mix of signaling themes across patients, and we point to a number of novel applications of existing targeted therapies which may benefit ovarian cancer.
Background
Signal transduction contains cluses to how abberation in cancer cells may lead to uncontrolled growth and division. It has been hypothesized that evidence of functional signal transduction can be found by examining the mRNA expression-based correlation structure of known ligand-receptor pairs (LRPs) [1]. If correlation is found between a pair, one infers that they form a positive-feedback loop, an autocrine signaling relationship.
A survey in 2006 examined autocrine signaling pairs in epithelial ovarian cancer (EOC) [2] and confirmed their findings by immunohistochemical staining. Importantly, this study continued to develop the idea that differential signaling (a change in autocrine status) might be associated with prognosis.
Statistical differential correlation or differential co-expression (DC) techniques have advanced significantly in recent years allowing for the consideration of multivariate associations beyond treating LRPs one at a time [3]. For example, a Gaussian graphical model (GGM) studies the precision matrix (the inverse of the correlation) to infer signaling and statistical work has developed techniques for proper false discovery control [4]. Recently, it has been proposed to directly estimate the difference in precision matrices to study differential signaling [5].
Combining new correlation techniques with the foreknowledge of candidate signaling pairs derived from protein folding models and confirmatory biochemical experiments [6], we conjectured that a multivariate survey may yield better functional understanding of clinically relevant signaling with strong translational potential. We focus on the Cancer Genome Atlas (TCGA) study of ovarian cancer [7] as a discovery set paired with two large, independent studies for validation [8], [9]. Our plan is an update of the study conducted by Castellano et al. (n = 522) in that each of the new datasets totals over 500 patients and comprise a more clinically relevant set – a higher percentage of high-grade EOC and long follow up periods – each conducted as single study instead of several small studies.
We augment the autocrine signaling hypothesis with similar correlation-based models of cancer-relevant signal transduction. Zandi and colleagues reviewed a number of modes of deleterious signaling in the EGFR family of transmembrane receptors [10], relevant to expression data are: the overexpression of the ligand, the overexpression of the receptor, and receptor-receptor crosstalk. In addition to DC analysis, we consider whether the interaction term in a survival regression model can detect an association directly. Finally, we consider whether prognostic LRPs vary over time and heterogenously across patients.
Methods
Ligand-Receptor Pair Database
We identified interacting pairs from the Database of Ligand-Receptor Partners (DLRP, http://dip.doe-mbi.ucla.edu/ [1]), matching 162 ligands, 131 receptors and 419 paired interactions on the Affymetrix array. We augmented this set with KEGG pathways [6] hsa04060 (Cytokines/Chemokines) hsa04512 (Cell adhesion molecules) and hsa04514 (ECM interactions). In total, there are 475 pairs (200 ligands, 166 receptors) for consideration after verifying the ligand/receptor functions (Table S1).
Expression and Clinical Data
Correlation and survival analysis depend on 503 TCGA samples [7] (Table S2) and 500 validation samples collected from two studies by Tothill and colleagues [8] and Yoshihara and colleagues [9]. In each study, measurements are made on tumor tissue excised during surgery prior to chemotherapy. Expression is measured on Affymetrix's HT-HG-U133A and HG-U133 Plus 2.0 arrays (GPL570, GPL571) for the TCGA and Tothill (GSE9899) studies as well as an Agilent platform (GPL56480). Notably the TCGA study uses 3 independent platforms, we opted to analyze just the U133A assay as it was most complete at the time of data acquisition. We follow the standard RMA [11] algorithm and further reduce data to gene level measurements by the brightest-spot rule. Standard gene names are then matched across platforms.
While TCGA samples are selected for advanced serous EOC, Tothill's study contains a representative number of endometriod subtypes; cases with low maligant potential are omitted. Yoshihara's dataset are strictly high-grade, high-stage ovarian cancers. Further, the rate of optimal debulking surgeries is high in the Yoshihara data set (40% vs. 23% p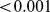 , Table 1). Beyond this difference, these patients all undergo debulking surgery and adjuvant platinum-based chemotherapy as standard treatment.
, Table 1). Beyond this difference, these patients all undergo debulking surgery and adjuvant platinum-based chemotherapy as standard treatment.
Table 1. Clinical differences between discovery and validation studies.
| Studies | |||||
| Discovery | Validation | ||||
| TCGA | Tothill | Yoshihara | |||
| N | 503 | 240 | 260 | ||
| Age | Mean | 59.8 | 60.2 | NA |
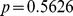
|
| Stage | I, II | 24 | 9 | 0 | |
| III, IV | 463 | 215 | 260 |
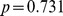 *
*
|
|
| Grade | 1 | 4 | 5 | 0 | |
| 2,3 | 473 | 218 | 260 |
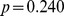 ** ** |
|
| Cytoreduction | Optimal | 102 | 53 | 103 | |
| Suboptimal | 348 | 140 | 157 |
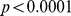 +
+
|
|
| Follow Up | |||||
| OS | Median | 30 | 28 | 41.5 | |
| PFS | Median | 13.2 | 14 | 19 | |
*excludes Yoshihara data, p-value = .001597 when including Yoshihara data.
definition of optimal/suboptimal not clearly defined in Yoshihara data.
We have focused on progression-free survival (PFS) instead of overall survival as post-recurrence treatments are varied and will confound the genetic signals apparent in pre-treatment tissue. Below, we will stratify patients based on PFS to 18 months, the typical median time to progression for advanced EOC [12]. This is a clinically meaningful endpoint as patients who progress after this time will typically be called “platinum sensitive” and will receive a second regimen of platinum (and taxane) chemotherapy. Standard treatment for patients who recur before this interval is uncertain.
Models and Hypotheses
We summarize the various LRP models to be tested. Each of these models will have its own specialized analysis. Throughout the paper, we will refer to a specific LRP as Ligand Receptor to emphasize the pairing.
Receptor to emphasize the pairing.
Model 1. Correlation of ligand-receptor pair expression implies autocrine signaling. [1]
Model 2. Correlation in short PFS subset and lack of or inverse correlation in long PFS subset implies prognostic autocrine signaling. [2]
Model 3. Significant statistical interactions crossed by correlated sets should imply functionally relevant signaling.
Model 4. Receptor-Receptor correlation implies receptor cross-talk or heterodimerization. [10]
Correlation Models
We compute Pearson's correlation between the expression of ligand and receptor genes. Significance tests are based on the statistic 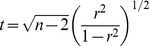 that follows a t-distribution with n-2 degrees of freedom [13]. Note that correlation is invariant to scaling and centering. To account for the effect of outliers, we performed a sensitivity analysis using Spearman's rank correlation (the effect of a small number of outliers will be attenuated). Our results did not change substantially.
that follows a t-distribution with n-2 degrees of freedom [13]. Note that correlation is invariant to scaling and centering. To account for the effect of outliers, we performed a sensitivity analysis using Spearman's rank correlation (the effect of a small number of outliers will be attenuated). Our results did not change substantially.
Differential correlation is based on stratifying patients into long PFS intervals (PFS  18 months) and short PFS intervals. Patients censored prior to 18 months are excluded from analysis, because these patients have insufficient followup to positively identify them into a long or short response; that is, patients who are known to recur before 18 months are assigned to the short interval group. There are 227 long PFS intervals and 186 short in the TCGA study. For each LRP, we computed correlation in each set separately (Bonferroni
18 months) and short PFS intervals. Patients censored prior to 18 months are excluded from analysis, because these patients have insufficient followup to positively identify them into a long or short response; that is, patients who are known to recur before 18 months are assigned to the short interval group. There are 227 long PFS intervals and 186 short in the TCGA study. For each LRP, we computed correlation in each set separately (Bonferroni 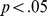 ) and classified pairs as “both” if long and short PFS sets have significant differences. We imagine that the long PFS subset represents nominal function, so that if a pair is correlated in the long PFS set and not in the short PFS set, we say the effect is a “loss” of function. Conversely, if a set is correlated in the short PFS set and not in the long PFS set we say the effect is a “gain” of deleterious function.
) and classified pairs as “both” if long and short PFS sets have significant differences. We imagine that the long PFS subset represents nominal function, so that if a pair is correlated in the long PFS set and not in the short PFS set, we say the effect is a “loss” of function. Conversely, if a set is correlated in the short PFS set and not in the long PFS set we say the effect is a “gain” of deleterious function.
Survival regression models
For each LRP, we regressed PFS on a set of candidate Cox proportional hazards regression models. In these models, the Ligand and Receptor may act additively or may interact. We selected the model maximizing Akaike's Information Criterion for each pair, classifying them as simply additive or synergistic if the interaction term was significant. We considered both scaled and centered data and expression rescaled to quantiles so that the hazard ratios have a more consistent interpretation. Again, the quantile scale reduces the potential effect of outliers.
Partial correlation for receptor crosstalk models
We estimated the Gaussian graphical model (GGM) via the GeneNet algorithm with edge posterior probability 0.90 [4], again stratifying patients into long and short PFS subsets. Edges that appear in both sets are classified as functional but not specific to prognosis, edges specific to the short survivors are termed “gain of function” and the edges specific to the long survivors are termed “loss of function.” We harmonized the estimated graphs by combining them with significant LRPs selected through the DC analysis. The graphical layout was done by hand aiming to emphasize clarity and size of the contiguous networks.
Heterogeneity and predictive models
Our analysis so far assumes homogeneity in time and across patients. We attempt to relax the first by noting that the DC analysis requires us to pick a timepoint to define long/short PFS. We vary this threshold across 1 to 60 months and record the change in DC test statistics. This sensitivity analysis highlights the range of time for which a particular signaling pair is relevant for prognosis.
To test homogeneity in signaling pairs across patients, we select only the LRPs from the DC analysis and run a supervised clustering algorithm to detect subgroups [14]. This algorithm clusters patients based on having similar survival models. We assigned patients to their maximum a posteriori class and computed an importance score: the absolute value of the expression level of the interaction multiplied by the cluster specific regression effect for each patient. Large scores reflect the determination that signaling in this LRP has a large influence on a particular patient's prognosis; small values imply little influence.
Results
Global correlation and active autocrine signaling
Following Graeber and Eisenberg's hypothesis, we tested for significant correlation between 475 ligand-receptor pairs and found 96 significant pairs in the TCGA discovery set (Bonferroni 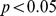 ). Of these, 77 are reproduced in our validation set with significant correlation and concordant direction of effect (Table S3). Using Spearman's rank correlation to attenuate the effect of outliers, 70 of the 77 are robust to extreme values. Three pairs have significant negative correlation: INHBA
). Of these, 77 are reproduced in our validation set with significant correlation and concordant direction of effect (Table S3). Using Spearman's rank correlation to attenuate the effect of outliers, 70 of the 77 are robust to extreme values. Three pairs have significant negative correlation: INHBA ACVR1B, INHBA
ACVR1B, INHBA ACVR2B and JAM2
ACVR2B and JAM2 F11R. We note that the magnitude of the selected correlations is remarkably strong (median
F11R. We note that the magnitude of the selected correlations is remarkably strong (median 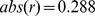 with 13/77, 16% greater than 0.50) which is consistent with previous studies [2]. A list of the full list of significant pairs, their estimated correlation and significance tests in both data sets is included in supplemental material.
with 13/77, 16% greater than 0.50) which is consistent with previous studies [2]. A list of the full list of significant pairs, their estimated correlation and significance tests in both data sets is included in supplemental material.
For reference, we considered the distribution of sample correlations after permuting the relationship between ligand and receptor. Pairs that were individually significant (p<0.05) had an average correlation of 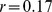 given the true pairings versus and average
given the true pairings versus and average 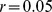 for pairs with significant associations given random associations (t-test p = 4.4e-11). If we randomly select 475 transcript pairs from any measured gene, the average effect drops to
for pairs with significant associations given random associations (t-test p = 4.4e-11). If we randomly select 475 transcript pairs from any measured gene, the average effect drops to 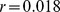 (t-test p = 2.8e-16). This implies that the average direction and magnitude of the observed LRPs are unlikely to be coincidental.
(t-test p = 2.8e-16). This implies that the average direction and magnitude of the observed LRPs are unlikely to be coincidental.
Ligand-receptor correlation stratified by long/short prognosis
Stratifying patients by long and short PFS, we identify 63 pairs with significant correlation in one or both subgroups. Of these 55 are found to have a significant gain or loss in the validation set. We note in Figure 1B, that significant correlations are uniformly positive consistent with a positive feedback loop. A pair can have positive or non-significant (n/s) correlation in the (long; short) PFS strata: we classify them as (positive; n/s), a loss of function; (n/s, positive), a gain of function; (positive, positive) functional but not specific to survival.
Figure 1. Correlation among ligand-receptor pairs.

(A) Correlation in ligand-receptor pairs (LRPs) in the discovery and validation datasets is largely concordant. (B) Stratified by prognosis, there is evidence of differential correlation. (C) The intersection of correlation and survival regression model interactions suggests that some LRPs are prognostic but not necessarily correlated (not autocrine).
Using the discovery cohort, the selected set of pairs comprises 39 receptors and 56 ligands (Table S4). The most frequently represented receptors are FGFR2, FGFR3, and EPHA5, each with at least 5 connections. While most of the remaining ligands are specific to one receptor (34/39, 87%), CCL13, CCL7, CCL8, FGF9, IL15 each form pairs with 3 receptors. With reference to the findings by Castellano and colleagues [2], we find that in at least one dataset, ENFB2 EPHA4 had a gain of function, LIF
EPHA4 had a gain of function, LIF IL6ST had loss of function and IL15
IL6ST had loss of function and IL15 IL2RG had non-specific function. The rest of the pairs had no significant change in correlation in either dataset.
IL2RG had non-specific function. The rest of the pairs had no significant change in correlation in either dataset.
This set is largely concordant with the analysis using all patients together. All pairs labelled with correlation in both sets were previously selected (34 of 63). Three pairs identified by stratified analysis had no significant marginal correlation (JAG2 NOTCH3, NGF
NOTCH3, NGF NGFR, and INHBC
NGFR, and INHBC ACVR2B); these pairs would have been missed without stratification. A significant subset of previously identified pairs were not significant in the stratified analysis (36/96, 38%), likely due to a loss of power due to splitting the sample, implying they are likely lower confidence results.
ACVR2B); these pairs would have been missed without stratification. A significant subset of previously identified pairs were not significant in the stratified analysis (36/96, 38%), likely due to a loss of power due to splitting the sample, implying they are likely lower confidence results.
Ligand-receptor interaction in regression models
No single ligand or single receptor was associated with PFS without considering its paired relationship (Cox regression, Bonferroni-adjusted likelihood ratio test). We subsequently identify significant paired ligands and receptors which supports the hypothesis that signal transduction is associated with survival versus simple overexpression of the ligand or receptor.
Regressing on the quantiles of expression, 29 pairs have selected interaction models (Table S5). Of these, 16 have interaction or additive models in the validation cohort. Four have consistent directions of effects across datasets; three of these are in the ephrin family and one is FGF1-FGFR2. If we consider scaled and centered expression data (Table S6), 9 pairs are again selected and validated: PDGFA PDGFRA, COL1A1
PDGFRA, COL1A1 CD44, IFNA14
CD44, IFNA14 IFNAR2, JAG2
IFNAR2, JAG2 NOTCH2, EFNA3
NOTCH2, EFNA3 EPHA1, EFNB3
EPHA1, EFNB3 EPHA4, FGF1
EPHA4, FGF1 FGFR2, VEGFA
FGFR2, VEGFA NRP1, and EREG
NRP1, and EREG ERBB4.
ERBB4.
When cross-tabulated by the correlation analysis, most pairs with significant interaction regression models do not have marginal correlation between the ligand and receptor. Thus, we infer the existence of a class of prognostic signaling pairs that operate without an autocrine feedback loop and we hypothesize that these ligand-receptor pairs act on prognosis in a typical endocrine fashion.
Graphical model of receptor crosstalk
We considered whether there is evidence of receptor heterodimerization and crosstalk functioning as a mechanism of oncogenic signaling. Continuing to assume that correlation implies signaling cooperativity, we used Gaussian graphical modeling techniques to estimate the graphical edges between receptors. We discovered 29 edges (posterior probability 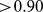 ) between the 39 receptors with significant differential associations from the stratified analysis. These edges are again classified into Loss/Gain/Non-specific function based on whether they are significant in the long PFS strata, the short PFS strata or both; the majority are losses (22/29, 76%) suggesting a breakdown in signaling coincides with poorer outcomes.
) between the 39 receptors with significant differential associations from the stratified analysis. These edges are again classified into Loss/Gain/Non-specific function based on whether they are significant in the long PFS strata, the short PFS strata or both; the majority are losses (22/29, 76%) suggesting a breakdown in signaling coincides with poorer outcomes.
Figure 2 connects the active ligand-receptor pairs to one another via cooperating receptors. We see that there is one large network of ephrins, the fibroblast growth factor (FGF), and Notch signaling that, as noted, tends to lose its coherence in the short PFS strata. There exists a small CCR chemokine network that is strongly interconnected (13/18 possible edges) indicating both their promiscuity and potential for a multivariate risk phenotype. Other small graphs include a PDGFR family and VEGF/PGF growth factor family. The remaining receptors operate independently.
Figure 2. Multivariate correlation graph.

Connecting all of the validated LRPs to one another via estimated receptor cross-talk shows a mix of small independent graphs and a large single signaling graph. Grey edges are not specific to prognosis; blue edges are gained in poor prognosis patients.
Heterogeneity in prognostic signaling
To study heterogeneity in signaling across patients, we applied a supervised clustering algorithm to identify prognostic, multivariate sets of LRPs. It identifies four subsets of patients defined by the relationship of signaling pairs and PFS visualized in Figure 3. Patients are arranged by cluster (columns) and LRPs are in rows where LRPs with significant univariate associations with PFS are highlighted.
Figure 3. Heterogeneity analysis by patient and in time.

(A) Patient heterogeneity implies four prognosis clusterse driven by different LRPs. (B) Each of these clusters has a distinct prognosis. (C) Over time, there are significant changes in the prognostic association and specific LRPs at about 9 and 27 months, close to the second treatment times for platinum resistant and sensitive patients.
The first cluster of patients has no relation between signaling and PFS (Cox model 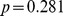 ) and has the worst prognosis (13.9 vs 18.0 months median, p = 1.8e-05) likely reflecting the fact that this cluster has the highest rate of patients with progressive disease after primary treatment. The second cluster's model has a mild prognostic effect (R2 = 0.254, p = 1.0e-06) with 6 significant pairs (PDGFA
) and has the worst prognosis (13.9 vs 18.0 months median, p = 1.8e-05) likely reflecting the fact that this cluster has the highest rate of patients with progressive disease after primary treatment. The second cluster's model has a mild prognostic effect (R2 = 0.254, p = 1.0e-06) with 6 significant pairs (PDGFA PDGFRA, COL1A
PDGFRA, COL1A CD44, COMP
CD44, COMP CD36, FGF9
CD36, FGF9 FGFR2, PGF
FGFR2, PGF FLT1, INHBA
FLT1, INHBA ACVR1). Notably, these interactions are differentially expressed between the first and second clusters implying they may be markers for identifying these patients. The third and fourth clusters are strong models (R2>0.600, p<1e-06 for both) with 7 and 10 significant, mutually exclusive LRPs. Patients in the third cluster have significantly shorter PFS (17.3 months versus 25.1, p = 0.00873). Both the longest and shortest PFS groups contain similar signaling pathways; notably the presence of ephrins and FGF differ between them. The models and their clinical associations are summarized in Table 2 with a summary of the drug target relevant signals.
ACVR1). Notably, these interactions are differentially expressed between the first and second clusters implying they may be markers for identifying these patients. The third and fourth clusters are strong models (R2>0.600, p<1e-06 for both) with 7 and 10 significant, mutually exclusive LRPs. Patients in the third cluster have significantly shorter PFS (17.3 months versus 25.1, p = 0.00873). Both the longest and shortest PFS groups contain similar signaling pathways; notably the presence of ephrins and FGF differ between them. The models and their clinical associations are summarized in Table 2 with a summary of the drug target relevant signals.
Table 2. Clinical associations for signaling clusters.
| Signaling Cluster | ||||||
| Plot Color | Black | Blue | Green | Red | ||
| n | 101 | 186 | 106 | 110 | ||
| Events | 100 | 95 | 93 | 73 | ||
| PFS Model fit | LRT | 0.281 | 0.001* | 0.001* | 0.001* | |
| R2 | 0.196 | 0.254 | 0.620 | 0.675 | ||
| Stage | % III/IV | 93% | 94% | 91% | 91% | p = 0.665 |
| Grade | % 3/4 | 86% | 85% | 80% | 88% | p = 0.420 |
| Clinical | Complete Response | 56 | 106 | 62 | 64 | |
| Response | Partial Response | 13 | 17 | 17 | 12 | |
| Stable Disease | 2 | 12 | 7 | 5 | ||
| Progressive Disease | 14 | 8 | 6 | 8 | ||
| Objective Response Rate | 68% | 66% | 75% | 69% | p = 0.522 | |
| Disease Control Rate | 70% | 73% | 81% | 74% | p = 0.292 | |
| OS | Median Months | 38.3 | 41.0 | 44.8 | 51.9 | p = 0.0875 |
| PFS | Median Months | 13.9 | 16.6 | 17.3 | 25.1 | p<0.001 |
| Notable Effects | VEGF | VEGF | Immune | VEGF | ||
| PDGF | PDGF | PDGF | ||||
| Ephrins | FGF | FGF | ||||
| Notch | TGFβ | Notch | ||||
*: p-value smaller than 0.001.
OS: Overall survival.
PFS: Progression-free survival.
LRT: Likelihood-ratio test.
Varying the cutpoint for long/short PFS, we considered whether the DC test statistic was sensitive to a particular time range for each LRP (Figure 3). While all pairs remain significantly DC throughout the range of followup, there are two critical periods where significance drops for several pairs implying they may be more relevant to early followup: at 8.75 months, CCL12 CCR7 and CCL8
CCR7 and CCL8 CCR2; and at 26.63 months, CCL13
CCR2; and at 26.63 months, CCL13 CCR2, CCL7
CCR2, CCL7 CCR2, and CCL7
CCR2, and CCL7 CCR5. Notably, these times seem to correspond to the median time to initiation of secondary therapies for platinum resistant and platinum sensitive cancers suggesting that these cytokines are related to therapy response.
CCR5. Notably, these times seem to correspond to the median time to initiation of secondary therapies for platinum resistant and platinum sensitive cancers suggesting that these cytokines are related to therapy response.
Discussion
We have extended autocrine signaling loop screening techniques to incorporate survival outcomes more comprehensively. We confirmed the association of ephrin family receptors [2] as well as NOTCH signaling [7] with ovarian cancer prognosis. We find a class of LRPs showing autocrine-type correlation but with no effect on survival; these would have been false-positive associations made in previous studies. Conversely, we find a class of LRPs with survival associations that have no significant correlation which implies their nominal function affects prognosis without any system feedback. In addition, we have shown that mutually exclusive sets of LRPs may function in different programs of both gain and loss of signaling function. The connection between these LRPs might be made via correlated changes between receptors and we have estimated the relevant wiring between them.
Drawn from a solely computational analysis, our conclusions are limited to transcriptome-type studies. Two recent concerns with these studies are run batch effects and the influence of tumor purity. We have used multiple large and independent datasets to reproduce candidate signals to alleviate some of these concerns. While a natural next step is to verify that these transcripts are indeed expressed in these tissues, the finding that they are reproducible using similar probes is promising.
In the context of chemotherapy, assessing the prognostic importance of signal transduction is an appealing avenue for biomarker development relevant for ovarian cancers: nearly 75% of cases eventually fail surgical control with primary platinum/taxane adjuvant chemotherapy [12] and common second-line treatments (gemicitabine [15], topotecan [16]) show differences in immune effect/response.
We have highlighted three particular receptor tyrosine kinases (VEGFR, PDGFR and FGFR) each of which is targeted individually or in combination by one of a number of inhibitors at various stages of approval [17]. Among these compounds are bevacizumab, targeting VEGF signaling [18]; sunitinib, targeting VEGF and PDGF; nintedanib targeting all three; and imatinib targeting just PDGFR. The prevalence of these three signaling interactions leads us to conjecture that different patients may respond to therapies targeting different combinations of receptors which might be inferred from expression data.
We conjecture that the prevalence of immune related LRPs in our validated sets and the per patient heterogeneity suggest the potential for an actionable biomarker to distinguish the utility of chemotherapy or immunotherapy treatment strategies. Further, the discovery of so many immune interactions is likely to be representative of the immunogenic nature of ovarian cancers [19] and of chemotherapy; it is a positive control confirmation for this correlation-based approach.
Supporting Information
List of ligand-receptor pairs.
(CSV)
List of TCGA cases used in study.
(CSV)
Ligand-receptor correlation in TCGA and validation studies.
(CSV)
Stratified correlation in TCGA and validation studies.
(CSV)
Regression model selection using quantile scaled data.
(CSV)
Regression model selection using scaled and centered data.
(CSV)
Acknowledgments
The results published here are in whole or part based upon data generated by The Cancer Genome Atlas pilot project established by the NCI and NHGRI. Information about TCGA and the investigators and institutions who constitute the TCGA research network can be found at http://cancergenome.nih.gov/.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. Data are available from the TCGA data browser (cancergenome.nih.gov) and the NCBI Gene Expression Omnibus (GEO:GSE9899, GEO:GSE32062).
Funding Statement
This work was supported by Roswell Park Cancer Institute, National Cancer Institute grant P30CA016056 (www.cancer.gov), a grant from the Roswell Park Alliance Foundation (KHE), and the RPCI-UPCI Ovarian SPORE (P50CA159981). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Graeber TG, Eisenberg D (2001) Bioinformatic identification of potential autocrine signaling loops in cancers from gene expression profiles. Nature Genetics 29: 295–300. [DOI] [PubMed] [Google Scholar]
- 2. Castellano G, Reid JF, Alberti P, Carcangiu ML, Tomassetti A, et al. (2006) New potential ligandreceptor signaling loops in ovarian cancer identified in multiple gene expression studies. Cancer Research 66: 10709–10719. [DOI] [PubMed] [Google Scholar]
- 3.Shedden K, Taylor J (2005) Differential correlation detects complex associations between gene expression and clinical outcomes in lung adenocarcinomas. In: Methods of Microarray Data Analysis, Springer. pp. 121–131.
- 4. Schäfer J, Strimmer K (2005) An empirical bayes approach to inferring large-scale gene association networks. Bioinformatics 21: 754–764. [DOI] [PubMed] [Google Scholar]
- 5.Zhao SD, Cai TT, Li H (2014) Direct estimation of differential networks. Biometrika in press. [DOI] [PMC free article] [PubMed]
- 6. Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M (2012) KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Research 40: D109–D114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. The Cancer Genome Atlas Research Network (2011) Integrated genomic analyses of ovarian carcinoma. Nature 474: 609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tothill RW, Tinker AV, George J, Brown R, Fox SB, et al. (2008) Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clinical Cancer Research 14: 5198–5208. [DOI] [PubMed] [Google Scholar]
- 9. Yoshihara K, Tsunoda T, Shigemizu D, Fujiwara H, Hatae M, et al. (2012) High-risk ovarian cancer based on 126-gene expression signature is uniquely characterized by downregulation of antigen presentation pathway. Clinical Cancer Research 18: 1374–1385. [DOI] [PubMed] [Google Scholar]
- 10. Zandi R, Larsen AB, Andersen P, Stockhausen MT, Poulsen HS (2007) Mechanisms for oncogenic activation of the epidermal growth factor receptor. Cellular signalling 19: 2013–2023. [DOI] [PubMed] [Google Scholar]
- 11. Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, et al. (2003) Summaries of affymetrix genechip probe level data. Nucleic acids research 31: e15–e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vaughan S, Coward JI, Bast RC, Berchuck A, Berek JS, et al. (2011) Rethinking ovarian cancer: recommendations for improving outcomes. Nature Reviews Cancer 11: 719–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Best DJ, Roberts DE (1975) Algorithm AS89: The upper tail probabilities of Spearman's ρ . Applied Statistics 24: 377–379. [Google Scholar]
- 14. Eng KH, Hanlon BM (2014) Discrete mixture regression models for heterogenous time-to-event data: Cox assisted clustering. Bioinformatics 30: 1690–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nowak AK, Robinson BW, Lake RA (2002) Gemcitabine exerts a selective effect on the humoral immune response implications for combination chemo-immunotherapy. Cancer research 62: 2353–2358. [PubMed] [Google Scholar]
- 16. Trojandt S, Knies D, Pektor S, Ritz S, Mailänder V, et al. (2013) The chemotherapeutic agent topotecan differentially modulates the phenotype and function of dendritic cells. Cancer Immunology, Immunotherapy 62: 1315–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yap TA, Carden CP, Kaye SB (2009) Beyond chemotherapy: targeted therapies in ovarian cancer. Nature Reviews Cancer 9: 167–181. [DOI] [PubMed] [Google Scholar]
- 18. Burger RA, Brady MF, Bookman MA, Fleming GF, Monk BJ, et al. (2011) Incorporation of bevacizumab in the primary treatment of ovarian cancer. New England Journal of Medicine 365: 2473–2483. [DOI] [PubMed] [Google Scholar]
- 19. Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, et al. (2003) Intratumoral t cells, recurrence, and survival in epithelial ovarian cancer. New England Journal of Medicine 348: 203–213. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of ligand-receptor pairs.
(CSV)
List of TCGA cases used in study.
(CSV)
Ligand-receptor correlation in TCGA and validation studies.
(CSV)
Stratified correlation in TCGA and validation studies.
(CSV)
Regression model selection using quantile scaled data.
(CSV)
Regression model selection using scaled and centered data.
(CSV)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. Data are available from the TCGA data browser (cancergenome.nih.gov) and the NCBI Gene Expression Omnibus (GEO:GSE9899, GEO:GSE32062).