

Abstract
The platelet-derived growth factor (PDGF) proteins are potent stimulators of cell proliferation/transformation and play a major role in cell-cell communication. For over two decades, PDGFs were thought to exist as three dimeric polypeptides (the homodimers AA and BB and the heterodimer AB). Recently, however, the PDGF C and D chains were discovered in a BLAST search of the expressed sequence tag databases. The PDGF CC and DD dimers have a unique two-domain structure with an NH2-terminal CUB (compliment subcomponents C1r/C1s, Uegf, and Bmp1) domain and a COOH-terminal PDGF/vascular endothelial growth factor domain. Whereas secreted PDGF AA, BB, and AB readily activate their cell surface receptors, it was suggested that extracellular proteolytic removal of the CUB domain is required for the PDGF/vascular endothelial growth factor domain of PDGF CC and DD to activate PDGF receptors. In the present study, we examined the processing of latent PDGF D into its active form and the effects of PDGF D expression on prostate cancer progression. We show that LNCaP cells auto-activate latent PDGF DD into the active PDGF domain, which can induce phosphorylation of the β-PDGF receptor and stimulates LNCaP cell proliferation in an autocrine manner. Additionally, LNCaP-PDGF D-conditioned medium induces migration of the prostate fibroblast cell line 1532-FTX, indicating LNCaP-processed PDGF DD is active in a paracrine manner as well. In a severe combined immunodeficient mouse model, PDGF DD expression accelerates early onset of prostate tumor growth and drastically enhances prostate carcinoma cell interaction with surrounding stromal cells. These demonstrate a potential oncogenic activity of PDGF DD in the development and/or progression of prostate cancer.
INTRODUCTION
Primary prostate carcinomas typically grow slowly and often do not progress to clinically relevant disease (1). The main cause for mortality from prostate cancer is metastasis, a complex process involving cancer cell invasion through stromal components and colonization at the distant organs (2, 3). Although it has become clear that paracrine signaling between the tumor cells and the surrounding mesenchymal cells plays a critical role for tumor progression, the molecular mechanisms of these interactions are much less understood.
The platelet-derived growth factor (PDGF) protein family is a potent stimulator of cell proliferation, chemotaxis, and transformation and is known to play a major role in cell-cell communication for normal development and also during pathogenesis (reviewed in Refs. 4 and 5). PDGF isoforms exert their biological functions through activation of two structurally related cell surface receptor tyrosine kinases [α-PDGF receptor (PDGFR) and β-PDGFR (4, 6)]. Immunohistochemical analysis showed that ~80% of prostate tumor tissues express PDGFRs at both primary and metastasized sites (7). Interestingly, β-PDGFR staining is more prominent in endothelial cells when they are exposed to prostate tumor cells that express PDGF (8), suggesting significance of PDGF signaling for prostate cancer progression in a paracrine manner. Consistently, PDGFR inhibitor STI571 (Gleevec) in combination with paclitaxel was shown to substantially reduce prostate cancer bone metastasis in a mouse model (8).
For more than two decades, PDGFs were thought to exist in the form of three dimeric polypeptides (the homodimers PDGF AA and BB and the heterodimer PDGF AB). The PDGF A chain can activate α-PDGFR only, whereas the PDGF B chain activates both α- and β-PDGFR (4, 6). Recently, however, the PDGF C and D chains were discovered in a BLAST search of the expressed sequence tag databases at the National Center for Biotechnology Information (9–11). The PDGF C and D chains have a unique two-domain structure with an NH2-terminal CUB (compliment subcomponents C1r/C1s, Uegf, and Bmp1) domain and a COOH-terminal PDGF/vascular endothelial growth factor domain. Whereas secreted PDGF AA, BB, and AB can readily activate their cell surface receptors, it was suggested that extracellular proteolytic removal of the CUB domain is required for the growth factor domain of PDGF CC and DD dimers to preferentially activate αα-PDGFR and ββ-PDGFR, respectively (9–11).
Whereas the PDGF A- and B-mediated signal transduction pathways leading to specific cellular processes have been extensively studied, little is known about the functions of the new family members, PDGF C and D. Increased expression of PDGF D has been found in ovarian, lung, renal, and glioma-derived cell lines as well as in clinical tumor samples, suggesting a correlation between increased PDGF D expression and human cancer (12, 13). Because β-PDGFR is thought to be critical for prostate cancer progression, and PDGF D is a specific ligand for β-PDGFR, we examined the effects of PDGF D expression in prostate cancer cell growth and its interactions with stromal cells. Whereas the previous studies suggested that the processing of latent PDGF DD into its active form requires proteases in serum (9, 10), we show in this study that human prostate carcinoma LNCaP processes latent PDGF DD into the active form under serum-free conditions, resulting in promotion of LNCaP growth. Additionally, PDGF DD produced by LNCaP cells induces cell motility of the prostate fibroblast cell line TFX-1532, indicating paracrine signaling of PDGF DD for the interactions between carcinoma and mesenchymal cells. We also show that PDGF DD expression greatly enhances prostate carcinoma cell interaction with the surrounding stromal layers in a severe combined immunodeficient (SCID) mouse model, demonstrating a potential oncogenic activity of PDGF D in human prostate cancer progression.
MATERIALS AND METHODS
Production of Recombinant PDGF D (rPDGF D) Protein
A two-step reverse transcription-PCR approach was taken to clone PDGF D into a vaccinia expression vector. Total RNA isolated from the prostate cell line DU145 using Trizol reagent was used in a cDNA synthesis reaction using SuperScript RTII (Invitrogen, Carlsbad, CA). The resultant cDNA was used in a first PCR reaction that yielded a 2107-bp product containing the 1113-bp open reading frame of PDGF D (forward, 5′-gga-gcgacgctgtctcta-3′; reverse, 5′-tttccagt-tacgtcgatg-3′). This PCR product was gel purified and used as a template in a second PCR reaction designed to add the restriction enzyme sites AflIII and BclI at the 5′ and 3′ ends, respectively, of the PDGF D open reading frame, as well as the addition of a 6×HIS epitope tag at the carboxyl end of the PDGF D protein (forward, 5′-ccacatgtcgcaccggctcatctt-3; reverse, 5′-catgcctgat-caaatatggtgatggtgatgatgtcgaggtggtcttga-3′; the 6×HIS tag is underlined). The resultant 1150-bp product was digested with AflIII and BclI, and inserted into the NcoI/BamHI site of the vaccinia virus expression vector pTF7-ECM1 (a kind gift from Dr. R. Fridman, Wayne State University). pTF7-ECM1 contains the T7 promoter upstream of the NcoI/BamHI cloning site; therefore, transcription of the inserted gene requires the presence of the T7 polymerase, supplied by infecting the cells with recombinant vaccinia virus. Fidelity of the in-frame sequence encoding the PDGF D:HIS fusion protein was confirmed by DNA sequencing (Elim Biopharmaceuticals, Inc., San Francisco, CA). This plasmid is referred to as pTF7-PDGF D:HIS and was used to produce recombinant full-length PDGF D following established vaccinia virus protocols (14). In short, BS-C-1 (Green monkey kidney) cells were first infected with the recombinant vaccinia virus vTF7-3, which expresses the T7 RNA polymerase. After 30 min of infection, the cells were washed with PBS and then transfected with the plasmid pTF7-PDGF D:HIS, using Effectene reagent (Qiagen). Expression of PDGF D inserted into the pTF7 plasmid is reliant on infection of the cells by vTF7-3, which expresses the T7 RNA polymerase gene. Cell-host machinery then transcribes the gene of interest. Forty-eight h after co-infection/transfection with vaccinia virus and PDGF D, the serum-free conditioned medium (CM) was collected and cleared of cellular debris by a 5-min centrifugation at 2000 × g. The resultant CM containing recombinant full-length PDGF D was used in subsequent PDGF D processing experiments.
Construction of a Mammalian PDGF D Expression Vector
To insert PDGF D into the mammalian expression vector pcDNA3, pTF7-PDGF D:HIS (described in the preceding paragraph) was used as the template in a PCR reaction with the primers (forward, 5′-ctggtgcttaagatgcaccggctc-3′; and reverse, 5′-catgcctgatcaaatatggtgatggtgatgatgtcgaggtggtcttga-3′) to add an AflII site at the 5′ end and to restore the BclI site at the 3′ end of the PDGF D:HIS open reading frame. The subsequent 1150-bp PCR product was then digested with AflII and BclI and inserted into the AflII, BamHI site of the mammalian expression vector pcDNA3.1(+) (Invitrogen). This plasmid is referred to as pcDNA3-PDGF D:HIS and was used to create the stable transfected cell line LNCaP-PDGF D.
Cell Culture
Human prostate carcinoma LNCaP cells and resultant transfectant lines were cultured in a humidified 5% CO2 incubator with RPMI 1640 supplemented with 5% fetal bovine serum, 2 mM glutamine, 100 units/ml penicillin, and 100 mg/ml streptomycin (Life Technologies, Inc., Carlsbad, CA). Medium and incubator conditions were the same for the human prostate fibroblast cell line 1532-FTX, except that medium was supplemented with 10% fetal bovine serum.
Establishment of PDGF D-Overexpressing Human Prostate Carcinoma LNCaP Cell Line
LNCaP cells transfected with pcDNA3-PDGF D:HIS and pcDNA3 empty vector were selected using 350 μg/ml Geneticin (G418) in RPMI 1640 with 10% fetal bovine serum, pooled together, and are referred to as LNCaP-PDGF D and LNCaP-neo, respectively. Overexpression of PDGF D was confirmed by reverse transcription-PCR as follows: total RNA was extracted from the cells using Trizol reagent (Invitrogen). One μg of total RNA from each cell line was used to synthesize cDNA using Superscript RTII (Invitrogen). Resultant cDNA was used as template in a PCR reaction, using Taq polymerase (Promega, Valencia, CA) and the following primers: forward, 5′-gaacagctaccccaggaacc-3′; and reverse, 5′-cttgtgtccacaccatcgtc-3′. These primers amplify a 192-bp product that represents part of the CUB coding region of the PDGF D protein. This primer pair does not allow for discrimination between endogenously and exogenously expressed PDGF D. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression was used as a positive control with the primer pair 5′-atcaccatcttccaggagcga-3′ (forward) and 5′-gccagtgagcttcccgttca-3′ (reverse).
Custom Antibody (Ab) Raised against the Growth Domain of PDGF D
An Ab was raised against PDGF D in rabbits using synthetic peptides (N′-RKSKVDLDRLNDDAKRYS-C′) representing a portion of the growth factor domain of PDGF D (amino acids 254–272). The resultant Abs were affinity purified (Zymed Biomedical, South San Francisco, CA) and are referred to as anti-PDGF D/GD Ab.
PDGF D-Mediated Paracrine Activation of β-PDGFR and Extra-cellular Signal-Regulated Protein Kinases (ERKs)
LNCaP-PDGF D and LNCaP-neo cells were cultured in serum-free medium for 48 h. CM was collected and centrifuged at 2000 rpm for 5 min to remove cell debris. Serum-starved NIH3T3 cells were treated with CM collected from LNCaP-PDGF D or LNCaP-neo cells for 10 or 60 min and lysed in immunoprecipitation assay lysis buffer [0.5% sodium deoxycholate, 1% NP40, 50 mM Tris (pH 7.6), 2 mM EGTA, 2 mM EDTA, 150 mM NaCl, 2 mM sodium vanadate, 1 mM phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, and 10 μg/ml aprotinin]. The lysates were centrifuged for 20 min at 12,000 × g to remove debris, and protein concentrations were determined using the BCA Protein Assay kit (Pierce Biotechnology, Rockford, IL). Lysate (250 μg) was used for immunoprecipitation with an anti-β-PDGFR Ab [Santa Cruz Biotechnology (Santa Cruz, CA) or Oncogene Research Products (San Diego, CA)] and protein G-agarose beads (Pierce Biotechnology). Immunoprecipitates were washed five times with immunoprecipitation assay buffer and resolved by reducing SDS-PAGE. Tyrosine-phosphorylated β-PDGFR was detected by immunoblot using an anti-phosphotyrosine Ab (Upstate Biotechnology, Lake Placid, NY). Total levels of immunoprecipitated β-PDGFR were detected using the same Ab used for immunoprecipitation. To determine the total and activated levels of ERK1/2, 50 μg of lysate from each sample were resolved by reducing SDS-PAGE, and immunoblots were performed using anti-pERK1/2 or anti-ERK1/2 Abs according to the manufacturer’s instructions (Cell Signaling Technologies, Beverly, MA).
Tumor Growth in scid/scid Mice
LNCaP-neo and LNCaP-PDGF D cells were grown to 80–90% confluence, harvested by trypsin/EDTA digestion, and washed twice with PBS. Cells were resuspended in serum-free RPMI 1640, and viable cells were counted by trypan blue exclusion assay. One million cells were s.c. injected into 5-week-old male homozygous C.B.-17 scid/scid mice (Taconic Farms, Germantown, NY). For the 2-week experiment, each animal received one injection of 1 × 106 cells in the left flank. Tumor size was too small to measure reliably by caliper; therefore, animals were sacrificed at the end of 2 weeks to dissect out the developing tumors. For the 5-week experiment, six animals received two s.c. injections of 1 × 106 cells/injection, one injection in each flank, and tumor growth was assessed weekly by two-dimensional measurements (mm) using a caliper. Tumor volume was estimated by the formula a × b2/2, where a is the longest dimension, and b is the width. Statistical analysis of tumor volume (average and SE) was performed using the program GraphPad Instat Version 3 for Windows (GraphPad Software, San Diego, CA). Mice were maintained according to the guidelines for the care and use of experimental animals established by the NIH.
Wound Migration Assay
Cells were grown in complete medium in a 6-well plate to 90% confluence and pretreated with mitomycin C (25 μg/ml) for 30 min before an injury line was made using a 2-mm-wide plastic pipette tip. After rinsing with PBS, cells were allowed to migrate in appropriate medium, and photographs were taken (×40) at the indicated time points to assess cell motility.
Cell Migration Assay
Twenty-four-well Transwell units with polycarbonate filters (Corning Costar, Cambridge, MA) coated with type I collagen (Sigma, St. Louis, MO) were used for all cell migration assays. To determine 1532-FTX cell migration in the presence of CM, the lower compartments of the wells were filled with 600 μl of appropriate medium, and 5 × 104 cells were placed in the upper part of the Transwell and incubated for 12 h. To determine LNCaP-neo or LNCaP-PDGF D migration in the presence of 1532-FTX cells, 1532-FTX cells were grown to 90% confluence in a 24-well plate, and then medium was changed to serum-free medium. LNCaP-neo or LNCaP-PDGF D cells (5 × 104) were then placed in Transwell units and placed in the wells containing 1532-FTX cells in serum-free medium at the bottom of the well. Cells were co-cultured for 48 h. Filters from all assays performed were prepared in the following manner: cells were fixed with methanol and stained with hematoxylin for 10 min, followed briefly by eosin staining. Cells on the upper surface of the filter were removed mechanically by wiping with a cotton swab. The total number of cells migrating to the lower side of the filter was determined using microscopy at ×400.
Histology
Mice were sacrificed 5 weeks after injection of cells by cervical dislocation under anesthesia using isoflurane. The tumors and surrounding mouse tissues were dissected and fixed in 10% buffered formalin, embedded in paraffin, sectioned, and stained with H&E for histological analysis.
RESULTS
Human Prostate Carcinoma LNCaP Processes PDGF D into Active Growth Factor Domains
To examine the effects of PDGF D expression on human prostate carcinoma cell growth and tumorigenicity, LNCaP-PDGF D and LNCaP-neo cell lines were established as described in “Materials and Methods.” PDGF D overexpression in LNCaP-PDGF D was confirmed by reverse transcription-PCR analysis (Fig. 1A) as well as immunoblot analysis using anti-PDGF D/GD Ab (Fig. 1B). It was shown previously that when a PDGF D expression vector was transfected into human embryonic kidney (HEK 293) cells, only the full-length PDGF D protein was secreted from HEK 293 cells under serum-free conditions; HEK 293-secreted pro-PDGF D was found to be processed into an active growth factor dimer by unknown protease(s) in serum (10). Surprisingly, however, immunoblot analysis of secreted proteins collected from LNCaP-PDGF D cells in the absence of serum revealed that both the pro-form (Mr 50,000) and the processed forms (Mr 20,000 and 15,000), with sizes similar to what were reported to be growth factor monomers (9, 10), were detected under reducing condition using anti-PDGF D/GD Ab (Fig. 1B).
Fig. 1.

Overexpression and processing of platelet-derived growth factor (PDGF) D in LNCaP. A, RNA levels of PDGF D in parental LNCaP (Lane 1), LNCaP-neo (Lane 2), and LNCaP-PDGF D (Lane 3) cells were examined by reverse transcription-PCR using primers (see “Materials and Methods”) that amplify both endogenous and exogenous PDGF D. Negative control without template was included (Lane 4). B, LNCaP-neo and LNCaP-PDGF D cells were grown to 100% confluence in complete media, washed once with PBS, and then cultured in serum-free medium for 48 h. The resultant conditioned medium was collected, and proteins were resolved using SDS-PAGE under reducing conditions. PDGF D in the conditioned medium was detected by immunoblot with anti-PDGF D/GD antibody. FL-M, full-length PDGF D pro-protein monomer; GD-M, PDGF D growth factor domain monomer.
The above results suggest that the pro-PDGF D protein may be processed into the growth factor domain by protease(s) expressed in human prostate carcinoma LNCaP cells independent of serum. To examine PDGF D processing by proteinases produced by LNCaP cells, rPDGF D protein was produced using a vaccinia mammalian cell expression system as described in “Materials and Methods.” When BS-C-1 cells were cultured in serum-free medium after co-infection/transfection with vaccinia virus and pTF7-PDGF D:HIS (see “Materials and Methods”), only pro-PDGF D protein was detected, migrating as a dimer with a Mr of ~90,000 under nonreducing conditions and as a monomer with a Mr of ~50,000 under reducing conditions (Fig. 2A, left panel). In agreement with previous reports (9, 10), the pro-PDGF D protein was processed into the growth domain in the presence of serum, migrating as a dimer with Mr ~35,000 and as monomer species with Mr 20,000 and 15,000 (Fig. 2A, right panel). Using rPDGF D, we examined whether pro-PDGF D can be processed by secreted proteinase(s) or by cell surface/pericellular proteinase(s) produced from LNCaP cells. Whereas CM collected from LNCaP cells contained little proteolytic enzyme capable of processing rPDGF D (Fig. 2B, right panel), incubation of rPDGF D with LNCaP-neo cells resulted in cleavage of rPDGF D, suggesting that cell surface or pericellular proteinase(s) may be responsible for processing pro-PDGF D into the growth factor domain (Fig. 2B, left panel). It was noticed that rPDGF D was processed into several species with Mr ~25,000 by proteinase(s) produced by LNCaP-neo cells, whereas two major monomer species with Mr 20,000 and 15,000 are usually detected in CM collected from LNCaP-PDGF D cells (Fig. 1B). Next, we examined whether rPDGF D is more effectively processed into Mr 20,000 and 15,000 monomers in the presence of LNCaP-PDGF D cells compared with LNCaP neo cells. As shown in Fig. 2C, whereas rPDGF D was processed mostly to a Mr 25,000 fragment after 4 h of incubation with LNCaP-neo cells, it was more effectively processed into growth factor domains (Mr 25,000, 20,000, and 15,000) in the presence of LNCaP-PDGF D cells. This suggests that proteinase expression or its activation, responsible for PDGF D processing, was up-regulated by constitutive activation of PDGF D signaling in LNCaP-PDGF D cells.
Fig. 2.

Platelet-derived growth factor (PDGF) D is processed at/near the cell surface. A, BS-C-1 cells were co-infected/transfected with vaccinia virus vTF7-3 and the plasmid pTF7-PDGF D:HIS. After co-infection/transfection, cells were grown either in serum-free medium or in fetal bovine serum-containing medium (as indicated) for 48 h. Conditioned medium (CM) from the infected cells was collected and resolved using SDS-PAGE under either reducing or nonreducing conditions, as indicated. For optimum resolution of bands, serum-free CM was run on a 10% SDS-PAGE gel, and fetal bovine serum-containing CM was run on a 14% SDS-PAGE gel. B, recombinant PDGF D (rPDGF D) was incubated with serum-starved LNCaP-neo cells (left panel) or with conditioned medium collected from serum-starved LNCaP-neo cells for 48 h (right panel) for 4 or 24 h at 37°C. Control experiments were included in the absence of rPDGF D (Lane 1 in both panels). All samples were resolved on a SDS-PAGE gel under reducing conditions and analyzed through immunoblot using an anti-PDGF D/GD antibody. C, serum-starved LNCaP-neo and LNCaP-PDGF D cells were cultured in the absence or presence of rPDGF D for 4 h, after which the medium was collected from the cells and resolved on a SDS-PAGE gel under reducing conditions, and the immunoblot was analyzed using an anti-PDGF D/GD antibody. **, intermediate forms of processed rPDGF D. FL-D, full-length PDGF D pro-protein dimer; FL-M, full-length PDGF D pro-protein monomer; GD-D, PDGF D growth factor domain dimer; GD-M, PDGF D growth domain monomer.
To test whether the processed forms of PDGF D are biologically active, serum-starved NIH3T3 cells were treated with CM collected from LNCaP-neo and LNCaP-PDGF D. The β-PDGFR protein was immunoprecipitated with an anti β-PDGFR Ab, and the active form was detected by immunoblot analysis using an anti-phosphotyrosine Ab. As a positive control, serum-starved NIH3T3 cells were also treated with recombinant PDGF BB protein at a concentration of 50 ng/ml. As shown in Fig. 3A, β-PDGFR was readily activated by treatment for 10 or 60 min with LNCaP-PDGF D CM as well as by recombinant PDGF BB protein treatment for 10 min, whereas no significant activation was detected with LNCaP CM. To further confirm β-PDGFR activation, we also examined activation of its downstream signaling molecule, ERKs. As shown in Fig. 3B, ERKs were phosphorylated on LNCaP-PDGF D CM treatment as effectively as PDGF BB treatment, whereas LNCaP-neo CM had little effect on ERK phosphorylation. These results demonstrate that the growth factor domain of PDGF D produced by LNCaP-PDGF D cells can activate its cognate receptor, β-PDGFR, transducing its downstream signaling pathway.
Fig. 3.

LNCaP-processed platelet-derived growth factor (PDGF) D is biologically active. A, serum-starved NIH3T3 cells were stimulated with 5 ml of conditioned medium (CM) from either LNCaP-PDGF D (Lanes 3 and 6) or LNCaP-neo (Lanes 4 and 7) for 10 or 60 min. Serum-starved NIH3T3 cells stimulated with 5 ml of serum-free medium supplemented with 25 ng/ml PDGF BB served as a positive control (Lane 1), and cells stimulated with serum-free medium (SF) served as a negative control (Lanes 2 and 5). β-PDGF receptor was immunoprecipitated from 250 μg of lysate, as described in “Materials and Methods.” Resultant immunoprecipitates were resolved by reducing SDS-PAGE and analyzed by immunoblot with the indicated antibodies (Abs). The blot was then stripped and reprobed with an anti-β-PDGF receptor Ab to determine efficiency of immunoprecipitation. B, 50 μg of lysate from NIH3T3 cells treated with 25 ng/ml PDGF BB (Lane 1), serum-free medium (Lane 2), LNCaP-PDGF D CM (Lane 3), or LNCaP-neo CM (Lane 4) were resolved on reducing SDS-PAGE, and the immunoblot was analyzed using an Ab specific to active extracellular signal-regulated protein kinase (ERK) 1/2 (pERK1/2). To determine basal levels of ERK1/2, the same blot was stripped and immunoblotted with an Ab that recognizes both active and inactive forms of ERK1/2.
PDGF D Enhances LNCaP Cell Proliferation
To assess the in vitro mitogenic activity of PDGF D through an autocrine mechanism, we examined the growth rates of LNCaP-neo and LNCaP-PDGF D under normal growth conditions. During the first 24 h of cultures, neither LNCaP-neo nor LNCaP-PDGF D significantly increased their cell numbers. However, drastic differences in cell numbers between LNCaP-neo and LNCaP-PDGF D were observed after the medium was conditioned by these cells for 48 h. Whereas LNCaP-neo cells barely doubled the cell number, LNCaP-PDGF D exhibited a 3–4-fold increase in cell numbers after 48 h in normal culture medium (Fig. 4), showing that PDGF D expression in prostate carcinoma cells has growth-promoting activity through an autocrine mechanism.
Fig. 4.

Platelet-derived growth factor D accelerates cell growth in an autocrine manner. Cells (6000 cells/well) were plated in a 96-well plate, and the relative cell number of LNCaP-PDGF D (■) and LNCaP-neo (
 ) was determined using the {4-[3-(4-iodophe-nyl)-2-(4-nitrophenyl)-2H-5-tetrazolio]-1–3 benzene disulfonate} (WST) assay (Roche Diagnostics GmbH, Mannheim, Germany) at the indicated time points.
) was determined using the {4-[3-(4-iodophe-nyl)-2-(4-nitrophenyl)-2H-5-tetrazolio]-1–3 benzene disulfonate} (WST) assay (Roche Diagnostics GmbH, Mannheim, Germany) at the indicated time points.
PDGF D Accelerates Early Onset of Prostate Tumor Growth and Stimulates Prostate Carcinoma Cell Interactions with Stromal Cells in a Mouse Model
The above-mentioned studies showed that tumor cell-derived PDGF D enhances tumor cell growth in an autocrine manner. These effects may be transient in vitro or may result in profound differences in vivo for tumorigenicity and/or their interactions with stromal cells that express PDGFRs. To examine the in vivo effects of PDGF D expression in human prostate carcinoma, LNCaP-neo and LNCaP-PDGF D cells were injected s.c. in the flank of male SCID mice (1 injection/animal). When the formation of tumor nodule was examined in mice sacrificed 2 weeks after injection, only one of five injections developed from LNCaP-neo cells, whereas five of five animals developed lesions from LNCaP-PDGF D cells (20% versus 100%; Fig. 5A). To further examine the onset of tumor nodules and their growth, LNCaP-neo and LNCaP-PDGF D cells were s.c. injected into both the right and left flanks (2 injections/animal) of an additional six animals, and tumor incidence and size were monitored at weekly intervals. The differences in tumor incidence between LNCaP-neo and LNCaP-PDGF D became less significant after 5 weeks (80% versus 100%; Fig. 5A), and average tumor volume was similar between these two cell lines (Fig. 5B). These results indicate that whereas the onset of tumor nodules was shortened by 10–14 days in LNCaP-PDGF D tumors compared with LNCaP-neo tumors, overall tumor incidence and tumor volume were similar between these two cell lines.
Fig. 5.

Onset of LNCaP-PDGF D tumor formation is accelerated compared with LNCaP-neo tumors. A, 1 × 106 cells were injected s.c. in the flank of male severe combined immunodeficient mice, and tumor incidence was examined. To determine the 2 week incidence point, one injection was given per mouse, with a total of 5 mice/group (total, 5 injections/cell line). Because tumors were not palpable at 2 weeks, mice were sacrificed, and the skin was dissected away to note the appearance of small nodules. To determine tumor incidence over a 5-week period, a second set of mice were given 2 injections/mouse, 6 mice/group (total, 12 injections/cell line). Palpable tumors were examined at week 4 and 5. B, tumor growth is represented by average tumor volume. Error bars represent ±SE.
To examine the effects of tumor cell-derived PDGF D expression on host stromal cell responses in vivo, LNCaP-neo and LNCaP-PDGF D tumor tissues were harvested 5 weeks after injection, sectioned, and stained with H&E. Histological examination of LNCaP-neo tumors revealed a solid tumor mass that is well capsulated and separated from mouse stromal tissue (Fig. 6A). In contrast, LNCaP-PDGF D tumors showed a close interaction with the mouse stromal cells (Fig. 6B), suggesting a role for PDGF D in close reciprocal interplay between tumor cells and local mesenchymal cells. Among two different sections for each of eight individual LNCaP-neo and LNCaP-PDGF D xenografts examined, all of the LNCaP-PDGF D sections, but none of the LNCaP-neo sections, showed drastically increased carcinoma interactions with the stromal cells, an event known to be critical for cancer progression leading to metastasis. It should be mentioned that the average weight of xenografts formed by LNCaP-neo and LNCaP-PDGF D was comparable at the time of histological examination (1.5 versus 1.3 g, respectively), suggesting that PDGF D-mediated stromal-epithelial interactions are not directly associated with its growth-promoting activity.
Fig. 6.
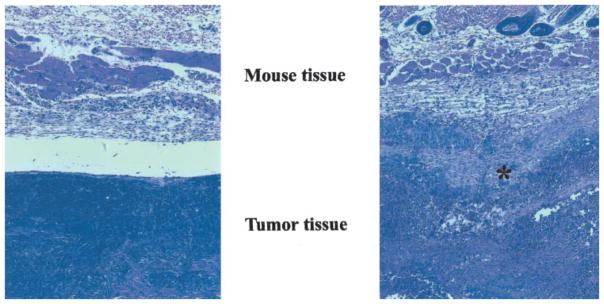
Increased tumor-stromal interactions observed in LNCap-PDGF D tumors compared with LNCaP-neo tumors. Tumors were harvested 5 weeks after injection, fixed, sectioned, and stained with H&E. A, LNCaP-neo tumor; B, LNCaP-PDGF D tumor. Asterisk indicates tumor-stromal interactions.
PDGF D Processed by LNCaP Enhances Cell Motility of Prostate Fibroblasts in a Paracrine Manner and Induces LNCaP Migration in a Secondary Paracrine Manner
We next investigated whether PDGF D stimulates prostate fibroblast cell migration in a paracrine manner and/or promotes migrative phenotype of human prostate cancer cells in an autocrine manner. As shown in Fig. 7A, cell motility of human prostate fibroblasts 1532-FTX was greatly induced in the presence of CM collected from LNCaP-PDGF D compared with treatment with CM collected from LNCaP-neo. To ensure that LNCaP-PDGF D CM-induced 1532-FTX migration does not result from increased cell proliferation rates, the wound migration assay was performed using cells pretreated with mitomycin C, a cell cycle blocker at the S phase (15). We then quantitated cell motility by a migration assay using a Transwell chamber. Whereas <2-fold induction of 1532-FTX cell migration was observed in the presence of LNCaP-neo CM compared with the serum-free medium condition, ~10-fold induction was detected in the presence of LNCaP-PDGF D CM (Fig. 7B). These results suggest that PDGF D-mediated stromal-prostate carcinoma interactions involve migration of surrounding mesenchymal cells in a paracrine manner.
Fig. 7.

Platelet-derived growth factor (PDGF) D induces cell motility of human prostate fibroblasts in a paracrine manner. A, wound healing assay. TFX-1532 cells were grown to 100% confluence and then pretreated with mitomycin C for 30 min to prevent cell proliferation. An injury line was made, and cells were cultured in the presence of either LNCaP-neo conditioned medium or LNCaP-PDGF D conditioned medium. Pictures were taken at indicated time points. B, 1532-FTX cell migration assay using a Transwell. Migrating cells were counted after 12 h of culture. Data points represent average total number of cells migrating to bottom of filter; experiments were done in triplicate. *, P < 0.05 as determined by Student’s t test.
When cell motility of LNCaP-neo and LNCaP-PDGF D was examined by a Transwell chamber assay (Fig. 8) as well as wound healing assay (data not shown), no significant difference was detected, suggesting that PDGF D expression has little effect on human prostate epithelial cell motility in an autocrine manner. Next, we asked whether prostate epithelial cell-produced PDGF D in the presence of prostate fibroblasts induces epithelial cell migration. To this end, cell migration of LNCaP-neo and LNCaP-PDGF D was examined by a Transwell chamber assay in the presence of prostate 1532-FTX fibroblasts on the bottom of the wells. Importantly, migration of LNCaP-PDGF D was induced significantly compared with migration of LNCaP-neo in the presence of 1532-FTX cells (Fig. 8). Taken together, these results suggest that prostate carcinoma-produced PDGF D induces PDGFR-mediated signaling transduction pathways in stromal cells, resulting in secretion of chemotactic molecule(s), which, in turn, induce prostate cell motility in a secondary paracrine manner.
Fig. 8.
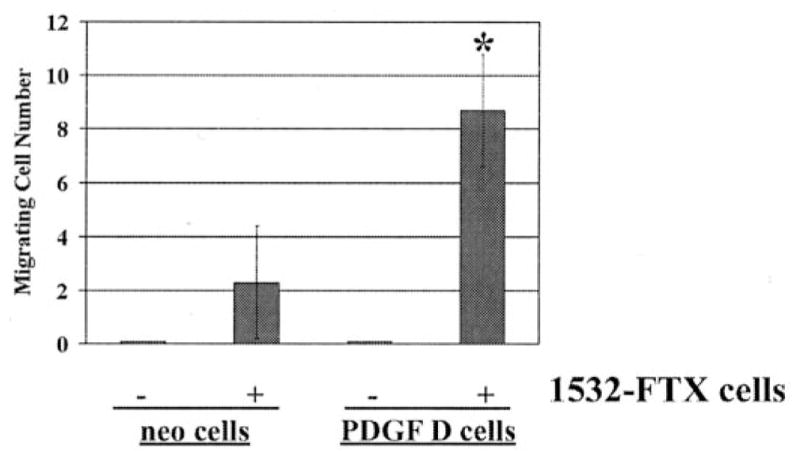
Platelet-derived growth factor D induces LNCaP cell motility in the presence of human prostate fibroblasts in a secondary paracrine manner. LNCaP-neo or LNCaP-PDGF D cells were placed on top of a Transwell chamber filter and cultured for 48 h in the absence or presence of 1532-FTX fibroblasts in serum-free medium. Data points represent average total number of cells migrating to bottom of filter; experiments were done in triplicate. *, P < 0.05 as determined by Student’s t test.
DISCUSSION
A causative role for PDGF in tumorigenesis was suggested when v-sis (the oncogene of simian sarcoma virus) was found to be 92% homologous to PDGF-B (6, 16, 17). Consistently, studies during the past two decades have clearly indicated the significance of PDGF in human tumors including glioma (18–20), dermatofibrosarcoma (21, 22), neurofibroma (23), myelomonocytic leukemia (24), and osteoblastoma and osteosarcoma (25). In vitro, overexpression of the v-sis oncogene product (p28v-sis) or PDGF B in cells that express their receptors enhances transformation, indicating an autocrine mechanism in tumorigenesis (26, 27). In addition to the autocrine mechanism, recent studies revealed a critical role for paracrine PDGF signaling in carcinogenesis involving epithelial-stromal interactions. Using nude mice, it was demonstrated that PDGFR activation of stromal cells results in tumorigenic conversion of immortal human keratinocytes (28). Mounting evidence suggests that aberrant interaction between stroma and epithelium is critical for the neoplastic progression of breast epithelium (29, 30), and PDGF was suggested to be among the critical mediators (31–34). Expression of PDGF B-protein and mRNA was restricted to the breast epithelium and tumor cells, whereas membranous PDGFR immunostaining was detected in stromal cell populations in all of the breast tissues examined (32). PDGFR staining was particularly localized in the periepithelial stroma of breast carcinoma, suggesting a paracrine stimulation of adjacent stromal tissue by breast tumor cells (32). Similar to breast cancer, paracrine mechanisms were also observed in colorectal cancer and small cell lung carcinoma (35, 36).
The present study reveals the potential oncogenic activity of PDGF D-mediated β-PDGFR in human prostate cancer in both autocrine and paracrine manners. In agreement with our hypothesis, previous studies showed that approximately 80% of prostate tumor tissues express PDGFRs at both primary and metastasized sites (7), and inhibition of PDGFR signaling using Gleevec significantly reduces prostate cancer bone metastasis in a mouse model (8). Interestingly, whereas increased expression levels of α-PDGFR and its cognate ligand, PDGF A, were detected in prostatic intraepithelial neoplasia and adenocarcinoma, only β-PDGFR (and not its ligand, PDGF B) was expressed in advancing stages of prostate cancer (37, 38). These prostate cancer cells were shown to exhibit phenotypes typical to β-PDGFR activation, suggesting that an alternative ligand, besides PDGF B, may play a critical role for β-PDGFR signaling during the progression of prostate cancer. Recent discovery of PDGF D as a specific activator of β-PDGFR and our present study suggest the involvement of PDGF D in human prostate cancer development and/or progression.
Many investigators have reported a more potent transforming signal by β-PDGFR than the signal transduced by α-PDGFR (26, 39, 40). Although both PDGF AA and BB are strong mitogens, only PDGF BB can induce phenotypic transformation in murine fibroblasts. A fundamental question in cell signaling is “how do two structurally related cell surface receptor tyrosine kinases (α- and β-PDGFRs) mediate differential cellular processes often using overlapping signaling pathways?” To address this issue, we previously established NIH3T3 clones in which α-PDGFR signaling is inhibited by a dominant-negative α-PDGFR or an antisense construct of α-PDGFR (41). We showed that inhibition of α-PDGFR signaling enhanced PDGF BB-mediated phenotypic transformation, suggesting that α-PDGFR antagonizes β-PDGFR-induced transformation. Whereas both α- and β-receptors effectively activate ERKs, α-PDGFR, but not β-PDGFR, activates stress-activated protein kinase-1/c-Jun NH2-terminal kinase-1 (JNK-1). Inhibition of JNK-1 activity using a dominant-negative JNK-1 mutant markedly enhanced PDGF BB-mediated anchorage-independent cell growth, demonstrating an antagonistic role for α-PDGFR-activated JNK-1 in PDGF-induced transformation. These results revealed a striking feature of PDGF signaling: the specificity and the strength of the PDGF growth signal is modulated by α-PDGFR-mediated simultaneous activation of growth-stimulatory and -inhibitory signals. With the discovery of new PDGF ligands, it has become clear that PDGFR signaling is finely modulated not only by PDGF A activation of α-PDGFR and PDGF B activation of both α- and β-PDGFR, but also by PDGF C activation of α-PDGF and PDGF D activation of β-PDGFR. Recently, it was shown that PDGF DD dimer activates ββ-PDGFR, not αα-PDGFR, but it can activate α-PDGFR through its dimerization with β-PDGFR [αβ-PDGFR (9, 10)]. Thus, for better understanding of roles of PDGF signaling during human cancer progression, it would be of particular importance to unveil signal transductions pathways and oncogenic activities induced by PDGF DD in comparison with better characterized pathways induced by PDGF BB, which activates all three forms of PDGFRs (αα, αβ, and ββ).
The unique structure of PDGF D, which requires proteolytic cleavage of the CUB domain to allow the growth factor domain to activate β-PDGFR, implements another level of regulation in PDGF signaling. The present study demonstrates that human prostate carcinoma LNCaP is capable of processing full-length PDGF D into a biologically active PDGF ligand for β-PDGFR activation and that this processing most likely occurs at or near the cell surface. This localized activation leads us to speculate a possible function of the CUB domain of the full-length PDGF DD. The CUB domain, composed of approximately 110 amino acids, shares sequence homology with the CUB domains of the complement subcomponents C1r/C1s, urchin epidermal growth factor-like protein (Uegf), and bone morphogenetic protein 1 (Bmp1; Ref. 42). The CUB domain is found in several extracellular proteins and is thought to mediate protein-protein or protein-carbohydrate interactions (43–45). Thus, the CUB domain may have unique regulatory functions toward PDGF D activity in addition to its activity in inhibiting PDGF D binding to the PDGFR. The CUB domain interaction with unidentified proteins may be critical for sequestration of the full-length PDGF D protein in extracellular and/or pericellular matrix, thereby regulating PDGF D protein localization. Conversely, the CUB domain could facilitate activation of PDGF DD by interacting with a specific protein(s) near the cell surface, localizing PDGF DD in appropriate association with the protease(s); only on this association would the protease be able to activate PDGF DD. It is also possible that the CUB domain may not serve as a mere “anchor” but that its interaction with co-receptor-like proteins may alter PDGFR signaling pathways. Considering its sequence similarities with epidermal growth factor-like protein and bone morphogenetic protein 1, it may be also worthy of investigation to determine whether the CUB domain alone after proteolytic cleavage from the full-length PDGF D has biological activity. In relation to prostate cancer, the CUB domain’s homology with bone morphogenetic protein (a protein known to play role in bone growth) may have implications in prostate cancer metastasis to the bone. This is worthy of further investigation using in vivo models.
Recent reports showed that PDGF D induces phenotypic transformation of NIH3T3 cells in vitro and also promotes tumor formation in mice (12, 46). In this study, we show that although PDGF D promotes human prostate carcinoma cell growth in vitro, it accelerates tumor formation only at the early stages, with little effect on the overall tumor incidence or the size of the tumors in vivo. However, PDGF D drastically increased the ability of human prostate carcinoma cells to interact with host stromal components in mice, an event known to be critical for prostate cancer progression. Our in vitro studies suggest that PDGF D is sufficient to induce migration of fibroblasts but that PDGF D alone is not sufficient to induce epithelial migration. Rather, PDGF D induces an as yet unidentified signal from the fibroblasts, which then induces the epithelial cell migration in a secondary paracrine manner. Taking our results together with those of recent studies reporting the significance of PDGF signaling in the progression of prostate cancer as well as β-PDGF signaling in metastasis, we propose that prostate carcinoma cells auto-activate PDGF D and use its signaling in both autocrine and paracrine manners, leading to a more malignant phenotype.
Acknowledgments
Grant support: NIH/National Cancer Institute Grant CA64139 (to H-R. C. Kim).
We thank Dr. R. Fridman and Pam Osenkowski for providing technical assistance with the vaccinia virus system.
References
- 1.Sakr WA, Grignon DJ, Crissman JD, Heilbrun LK, Cassin BJ, Pontes JJ, Haas GP. High grade prostatic intraepithelial neoplasia (HGPIN) and prostatic adenocarcinoma between the ages of 20–69: an autopsy study of 249 cases. In Vivo. 1994;8:439–443. [PubMed] [Google Scholar]
- 2.Small EJ, Reese DM, Vogelzang NJ. Hormone-refractory prostate cancer: an evolving standard of care. Semin Oncol. 1999;26(Suppl 17):61–67. [PubMed] [Google Scholar]
- 3.Hugosson J, Aus G. Natural course of localized prostate cancer: a personal view with a review of published papers. Anticancer Res. 1997;17:1441–1448. [PubMed] [Google Scholar]
- 4.Rosenkranz S, Kazlauskas A. Evidence for distinct signaling properties and biological responses induced by the PDGF receptor α and β subtypes. Growth Factors. 1999;16:201–216. doi: 10.3109/08977199909002130. [DOI] [PubMed] [Google Scholar]
- 5.Yu J, Ustach C, Kim HR. Platelet-derived growth factor signaling and human cancer. J Biochem Mol Biol. 2003;36:49–59. doi: 10.5483/bmbrep.2003.36.1.049. [DOI] [PubMed] [Google Scholar]
- 6.Deuel TF. Polypeptide growth factors: roles in normal and abnormal cell growth. Annu Rev Cell Biol. 1987;3:443–492. doi: 10.1146/annurev.cb.03.110187.002303. [DOI] [PubMed] [Google Scholar]
- 7.Ko YJ, Small EJ, Kabbinavar F, Chachoua A, Taneja S, Reese D, DePaoli A, Hannah A, Balk SP, Bubley GJ. A multi-institutional Phase II study of SU101, a platelet-derived growth factor receptor inhibitor, for patients with hormone-refractory prostate cancer. Clin Cancer Res. 2001;7:800–805. [PubMed] [Google Scholar]
- 8.Uehara H, Kim SJ, Karashima T, Shepherd DL, Fan D, Tsan R, Killion JJ, Logothetis C, Mathew P, Fidler IJ. Effects of blocking platelet-derived growth factor-receptor signaling in a mouse model of experimental prostate cancer bone metastases. J Natl Cancer Inst (Bethesda) 2003;95:458–470. doi: 10.1093/jnci/95.6.458. [DOI] [PubMed] [Google Scholar]
- 9.Bergsten E, Uutela M, Li X, Pietras K, Ostman A, Heldin CH, Alitalo K, Eriksson U. PDGF-D is a specific, protease-activated ligand for the PDGF β-receptor. Nat Cell Biol. 2001;3:512–516. doi: 10.1038/35074588. [DOI] [PubMed] [Google Scholar]
- 10.LaRochelle WJ, Jeffers M, McDonald WF, Chillakuru RA, Giese NA, Lokker NA, Sullivan C, Boldog FL, Yang M, Vernet C, Burgess CE, Fernandes E, Deegler LL, Rittman B, Shimkets J, Shimkets RA, Rothberg JM, Lichenstein HS. PDGF-D, a new protease-activated growth factor. Nat Cell Biol. 2001;3:517–521. doi: 10.1038/35074593. [DOI] [PubMed] [Google Scholar]
- 11.Li X, Ponten A, Aase K, Karlsson L, Abramsson A, Uutela M, Backstrom G, Hellstrom M, Bostrom H, Li H, Soriano P, Betsholtz C, Heldin CH, Alitalo K, Ostman A, Eriksson U. PDGF-C is a new protease-activated ligand for the PDGF α-receptor. Nat Cell Biol. 2000;2:302–309. doi: 10.1038/35010579. [DOI] [PubMed] [Google Scholar]
- 12.LaRochelle WJ, Jeffers M, Corvalan JR, Jia XC, Feng X, Vanegas S, Vickroy JD, Yang XD, Chen F, Gazit G, Mayotte J, Macaluso J, Rittman B, Wu F, Dhanabal M, Herrmann J, Lichenstein HS. Platelet-derived growth factor D: tumorigenicity in mice and dysregulated expression in human cancer. Cancer Res. 2002;62:2468–2473. [PubMed] [Google Scholar]
- 13.Uutela M, Lauren J, Bergsten E, Li X, Horelli-Kuitunen N, Eriksson U, Alitalo K. Chromosomal location, exon structure, and vascular expression patterns of the human PDGFC and PDGFC genes. Circulation. 2001;103:2242–2247. doi: 10.1161/01.cir.103.18.2242. [DOI] [PubMed] [Google Scholar]
- 14.Fuerst T, Earl P, Moss B. Use of a hybrid vaccinia virus-T7 RNA polymerase system for expression of target genes. Mol Cell Biol. 1987;7:2538–2544. doi: 10.1128/mcb.7.7.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jozaki K, Marucha PT, Despins AW, Kreutzer DL. An in vitro model of cell migration: evaluation of vascular endothelial cell migration. Anal Biochem. 1990;190:39–47. doi: 10.1016/0003-2697(90)90130-2. [DOI] [PubMed] [Google Scholar]
- 16.Doolittle RF, Hunkapiller MW, Hood LE, Devare SG, Robbins KC, Aaronson SA, Antoniades HN. Simian sarcoma virus onc gene, v-sis, is derived from the gene (or genes) encoding a platelet-derived growth factor. Science (Wash DC) 1983;221:275–277. doi: 10.1126/science.6304883. [DOI] [PubMed] [Google Scholar]
- 17.Waterfield MD, Scrace GT, Whittle N, Stroobant P, Johnsson A, Wasteson A, Westermark B, Heldin CH, Huang JS, Deuel TF. Platelet-derived growth factor is structurally related to the putative transforming protein p28sis of simian sarcoma virus. Nature (Lond ) 1983;304:35–39. doi: 10.1038/304035a0. [DOI] [PubMed] [Google Scholar]
- 18.Hermanson M, Funa K, Koopmann J, Maintz D, Waha A, Westermark B, Heldin CH, Wiestler OD, Louis DN, von Deimling A, Nister M. Association of loss of heterozygosity on chromosome 17p with high platelet-derived growth factor α receptor expression in human malignant gliomas. Cancer Res. 1996;56:164–171. [PubMed] [Google Scholar]
- 19.Plate KH, Breier G, Farrell CL, Risau W. Platelet-derived growth factor receptor-β is induced during tumor development and upregulated during tumor progression in endothelial cells in human gliomas. Lab Investig. 1992;67:529–534. [PubMed] [Google Scholar]
- 20.Westermark B, Heldin CH, Nister M. Platelet-derived growth factor in human glioma. Glia. 1995;15:257–263. doi: 10.1002/glia.440150307. [DOI] [PubMed] [Google Scholar]
- 21.Greco A, Roccato E, Miranda C, Cleris L, Formelli F, Pierotti MA. Growth-inhibitory effect of STI571 on cells transformed by the COL1A1/PDGFB rearrangement. Int J Cancer. 2001;92:354–360. doi: 10.1002/ijc.1190. [DOI] [PubMed] [Google Scholar]
- 22.Shimizu A, O’Brien KP, Sjoblom T, Pietras K, Buchdunger E, Collins VP, Heldin CH, Dumanski JP, Ostman A. The dermatofibrosarcoma protuber-ans-associated collagen type Iα1/platelet-derived growth factor (PDGF) B-chain fusion gene generates a transforming protein that is processed to functional PDGF-BB. Cancer Res. 1999;59:3719–3723. [PubMed] [Google Scholar]
- 23.Kadono T, Kikuchi K, Nakagawa H, Tamaki K. Expressions of various growth factors and their receptors in tissues from neurofibroma. Dermatology. 2000;201:10–14. doi: 10.1159/000018421. [DOI] [PubMed] [Google Scholar]
- 24.Golub TR, Barker GF, Lovett M, Gilliland DG. Fusion of PDGF receptor β to a novel ets-like gene, tel, in chronic myelomonocytic leukemia with t(5;12) chromosomal translocation. Cell. 1994;77:307–316. doi: 10.1016/0092-8674(94)90322-0. [DOI] [PubMed] [Google Scholar]
- 25.Sulzbacher I, Traxler M, Mosberger I, Lang S, Chott A. Platelet-derived growth factor-AA and -α receptor expression suggests an autocrine and/or paracrine loop in osteosarcoma. Mod Pathol. 2000;13:632–637. doi: 10.1038/modpathol.3880109. [DOI] [PubMed] [Google Scholar]
- 26.Beckmann MP, Betsholtz C, Heldin CH, Westermark B, Di Marco E, Di Fiore PP, Robbins KC, Aaronson SA. Comparison of biological properties and transforming potential of human PDGF-A and PDGF-B chains. Science (Wash DC) 1988;241:1346–1349. doi: 10.1126/science.2842868. [DOI] [PubMed] [Google Scholar]
- 27.Uhrbom L, Hesselager G, Ostman A, Nister M, Westermark B. Dependence of autocrine growth factor stimulation in platelet-derived growth factor-B-induced mouse brain tumor cells. Int J Cancer. 2000;85:398–406. doi: 10.1002/(sici)1097-0215(20000201)85:3<398::aid-ijc17>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 28.Skobe M, Fusenig NE. Tumorigenic conversion of immortal human keratinocytes through stromal cell activation. Proc Natl Acad Sci USA. 1998;95:1050–1055. doi: 10.1073/pnas.95.3.1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moinfar F, Man YG, Arnould L, Bratthauer GL, Ratschek M, Tavassoli FA. Concurrent and independent genetic alterations in the stromal and epithelial cells of mammary carcinoma: implications for tumorigenesis. Cancer Res. 2000;60:2562–2566. [PubMed] [Google Scholar]
- 30.Zoltowska A. Pathogenesis of breast carcinoma. Immunohistochemical study. Arch Immunol Ther Exp (Warsz ) 1997;45:101–108. [PubMed] [Google Scholar]
- 31.Bronzert DA, Pantazis P, Antoniades HN, Kasid A, Davidson N, Dickson RB, Lippman ME. Synthesis and secretion of platelet-derived growth factor by human breast cancer cell lines. Proc Natl Acad Sci USA. 1987;84:5763–5767. doi: 10.1073/pnas.84.16.5763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Coltrera MD, Wang J, Porter PL, Gown AM. Expression of platelet-derived growth factor B-chain and the platelet-derived growth factor receptor β subunit in human breast tissue and breast carcinoma. Cancer Res. 1995;55:2703–2708. [PubMed] [Google Scholar]
- 33.Palman C, Bowen-Pope DF, Brooks JJ. Platelet-derived growth factor receptor (β-subunit) immunoreactivity in soft tissue tumors. Lab Investig. 1992;66:108–115. [PubMed] [Google Scholar]
- 34.Seymour L, Bezwoda WR. Positive immunostaining for platelet derived growth factor (PDGF) is an adverse prognostic factor in patients with advanced breast cancer. Breast Cancer Res Treat. 1994;32:229–233. doi: 10.1007/BF00665774. [DOI] [PubMed] [Google Scholar]
- 35.Kawai T, Hiroi S, Torikata C. Expression in lung carcinomas of platelet-derived growth factor and its receptors. Lab Investig. 1997;77:431–436. [PubMed] [Google Scholar]
- 36.Sundberg C, Branting M, Gerdin B, Rubin K. Tumor cell and connective tissue cell interactions in human colorectal adenocarcinoma. Transfer of platelet-derived growth factor-AB/BB to stromal cells. Am J Pathol. 1997;151:479–492. [PMC free article] [PubMed] [Google Scholar]
- 37.Fudge K, Bostwick DG, Stearns ME. Platelet-derived growth factor A and B chains and the α and β receptors in prostatic intraepithelial neoplasia. Prostate. 1996;29:282–286. doi: 10.1002/(SICI)1097-0045(199611)29:5<282::AID-PROS2>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 38.Fudge K, Wang CY, Stearns ME. Immunohistochemistry analysis of platelet-derived growth factor A and B chains and platelet-derived growth factor α and β receptor expression in benign prostatic hyperplasias and Gleason-graded human prostate adenocarcinomas. Mod Pathol. 1994;7:549–554. [PubMed] [Google Scholar]
- 39.Heldin CH, Westermark B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev. 1999;79:1283–1316. doi: 10.1152/physrev.1999.79.4.1283. [DOI] [PubMed] [Google Scholar]
- 40.Bejcek BE, Hoffman RM, Lipps D, Li DY, Mitchell CA, Majerus PW, Deuel TF. The v-sis oncogene product but not platelet-derived growth factor (PDGF) A homodimers activate PDGF α and β receptors intracellularly and initiate cellular transformation. J Biol Chem. 1992;267:3289–3293. [PubMed] [Google Scholar]
- 41.Yu J, Deuel TF, Kim HR. Platelet-derived growth factor (PDGF) receptor-α activates c-Jun NH2-terminal kinase-1 and antagonizes PDGF receptor-β-induced phenotypic transformation. J Biol Chem. 2000;275:19076–19082. doi: 10.1074/jbc.M910329199. [DOI] [PubMed] [Google Scholar]
- 42.Bork P. Complement components C1r/C1s bone morphogenic protein 1Xenopus laevis developmentally regulated protein UVS.2 share common repeats. FEBS Lett. 1991;282:9–12. doi: 10.1016/0014-5793(91)80433-4. [DOI] [PubMed] [Google Scholar]
- 43.Nakamura F, Tanaka M, Takahashi T, Kalb RG, Strittmatter SM. Neuropilin-1 extracellular domains mediate semaphorin D/III-induced growth cone collapse. Neuron. 1998;21:1093–1100. doi: 10.1016/s0896-6273(00)80626-1. [DOI] [PubMed] [Google Scholar]
- 44.Kristiansen M, Kozyraki R, Jacobsen C, Nexo E, Verroust PJ, Moestrup SK. Molecular dissection of the intrinsic factor-vitamin B12 receptor, cubilin, discloses regions important for membrane association and ligand binding. J Biol Chem. 1999;274:20540–20544. doi: 10.1074/jbc.274.29.20540. [DOI] [PubMed] [Google Scholar]
- 45.Thielens NM, Bersch B, Hernandez JF, Arlaud GJ. Structure and functions of the interaction domains of C1r and C1s: keystones of the architecture of the C1 complex. Immunopharmacology. 1999;42:3–13. doi: 10.1016/s0162-3109(99)00019-3. [DOI] [PubMed] [Google Scholar]
- 46.Li H, Fredriksson L, Li X, Eriksson U. PDGF-D is a potent transforming and angiogenic growth factor. Oncogene. 2003;22:1501–1510. doi: 10.1038/sj.onc.1206223. [DOI] [PubMed] [Google Scholar]