

Abstract
The alphaherpesvirus varicella-zoster virus (VZV) causes chickenpox and shingles. Current treatments are acyclovir (ACV) and its derivatives, foscarnet and brivudine (BVdU). Additional antiviral compounds with increased potency and specificity are needed to treat VZV, especially to treat post-herpetic neuralgia. We evaluated β-L-1-[5-(E-2-Bromovinyl)-2-(hydroxymethyl)-1,3-(dioxolan-4-yl)] uracil (L-BHDU, 1) and 5′-O-valyl-L-BHDU (2) in three models of VZV replication: primary human foreskin fibroblasts (HFFs), skin organ culture (SOC) and in SCID-Hu mice with skin xenografts. The efficacy of L-BHDU in vivo and its drug-drug interactions were previously not known. In HFFs, 200 μM L-BHDU was noncytotoxic over 3 days, and L-BHDU treatment reduced VZV genome copy number and cell to cell spread. The EC50 in HFFs for L-BHDU and valyl-L-BHDU were 0.22 and 0.03 μM, respectively. However, L-BHDU antagonized the activity of ACV, BVdU and foscarnet in cultured cells. Given its similar structure to BVdU, we asked if L-BHDU, like BVdU, inhibits 5-fluorouracil catabolism. BALB/c mice were treated with 5-FU alone or in combination with L-BHDU or BVdU. L-BHDU did not interfere with 5-FU catabolism. In SCID-Hu mice implanted with human skin xenografts, L-BHDU and valyl-L-BHDU were superior to ACV and valacyclovir. The maximum concentration (Cmax) levels of L-BHDU were determined in mouse and human tissues at 2 h after dosing, and comparison of concentration ratios of tissue to plasma indicated saturation of uptake at the highest dose. For the first time, an L-nucleoside analog, L-BHDU, was found to be effective and well tolerated in mice.
Keywords: Varicella-zoster virus, Antiviral, Nucleoside analogue, Skin organ culture, SCID-Hu mouse, 5-Fluorouracil
1. Introduction
Varicella-zoster virus (VZV) is a human-restricted alphaherpesvirus. It causes varicella (chicken pox) upon primary infection and zoster (shingles) upon reactivation from latency. VZV disease is partially preventable by inoculation with the live, attenuated vaccine strain Oka-Merck (Oxman et al., 2005; Vazquez et al., 2004). Pediatric vaccination has reduced varicella cases in the United States (Seward et al., 2008), although the incidence of zoster is not likely to decline in the near future because in older adults the vaccine efficacy is approximately 50% (Holcomb and Weinberg, 2006). There will continue to be a demand for antiviral drugs for VZV due to natural and breakthrough cases and in immunocompromised patients that cannot receive live virus vaccines. Current treatments are nucleoside and pyrophosphate analogues that target the viral DNA polymerase and may depend on viral thymidine kinase activity (De Clercq, 2004). Acyclovir (ACV) and its acyclic derivatives of guanosine, such as valacyclovir (VACV), penciclovir (PCV) and famciclovir (FCV), are the most frequently prescribed drugs to treat VZV. These drugs are widely approved for use in the United States, Europe, and Asia. They are moderately effective against VZV, but for best results treatment should begin within 72 h of rash onset and resistance may arise during long-term administration to immunocompromised patients (Sampathkumar et al., 2009). In these patients, Foscarnet (phosphonoformate) delivered intravenously may be necessary to treat resistant VZV (Ahmed et al., 2007). The cyclic derivatives of uridine are another class of drugs currently used to treat VZV. Infections in the eye (herpes zoster ophthalmicus) can be treated with topical idoxuridine and trifluridine. Brivudine [BVdU, (E)-5-(2-bromovinyl)-2′-deoxyuridine] is approved for use in Europe and was the first bromovinyl nucleoside analog to show anti-herpesvirus activity (De Clercq et al., 1979). BVdU is phosphorylated by VZV thymidine kinase (TK) to both the 5′-monophosphate and 5′-diphosphate forms. Cellular kinases produce the 5′-triphosphate form (BVdU-TP). BVdU-TP interacts with the viral DNA polymerase either as a competitive inhibitor or an alternative substrate whereby it can be incorporated into DNA (reviewed in De Clercq, 2005). BVdU is more potent against VZV than acyclovir and its derivatives (Andrei et al., 1995; Shigeta et al., 1983). Another benefit of BVdU over acyclovir is the ease of dosing, making it appealing to elderly patients (De Clercq, 2005). The main drawback of BVdU is that it is cleaved into a metabolite of bromovinyluracil (BVU). BVU in turn inhibits dihydropyrimidine dehydrogenase, which is involved in the degradation of thymidine, uracil, and the commonly used cancer drug 5-fluorouracil (5-FU). Patients receiving this chemotherapy regimen should not be given BVdU as it may cause toxic accumulation of 5-FU and result in death (De Clercq, 2004; De Clercq, 2005; Diasio, 1998; Keizer et al., 1994).
The serious possible adverse effects of BVdU are the main reason why alternative antiviral uridine compounds have been sought. One approach has been to screen nucleosides in the L-configuration, which can be just as effective as the D-nucleoside counterparts (Chu et al., 1995; Spadari et al., 1992). The uridine derivative, β-L-1-[5-(E-2-Bromovinyl)-2-(hydroxymethyl)-1,3-(dioxolan-4-yl)] uracil (L-BHDU), exhibited potent anti-VZV activity in cultured cells and was noncytotoxic in HEL 299 cells up to 200 μM (Choi et al., 2000; Li et al., 2000). Efforts to elucidate the mechanism of action found that L-BHDU was phosphorylated by VZV TK but not further converted to the di- and triphosphate forms, suggesting a different antiviral mechanism than BVdU (Li et al., 2000). Their evidence pointed to the monophosphate form as the active moiety.
5′-O-Amino acid esters of nucleosides have been extensively used as prodrugs due to their well-established chemistry, structural diversity, and lack of toxicity concerns (Ibrahim et al., 1996). Using amino acids for a prodrug strategy provides several advantages, including increasing cellular transport and bio-availability. These prodrugs are at least as polar as the parent molecules, due to the α-amino group at physiological pH and carrier mediated transport. Prodrugs can be rapidly hydrolyzed to the parent drugs by intracellular hydrolysis. Indeed, the parent drugs show greatly increased cellular uptake when the prodrugs have amino acid esters. Furthermore, prodrugs of 5′-O-L-valyl esters improve pharmacokinetic properties without cellular toxicity in comparison to the parent nucleoside [Han et al., 1998(a); Han et al., 1998(b)]. In view of this information, the 5′-O-L-valyl ester of L-BHDU (2) was synthesized and evaluated for its anti-VZV activity in vitro and in vivo. Additionally, phosphoramidate prodrugs are known to increase nucleoside potency, presumably by increasing the intracellular concentration of nucleotides by bypassing the mono-phosphorylation step (Rawal et al., 2013). Therefore, we also investigated the phosphoramidates of L-BHDU (3 & 4).
It was not known whether L-BHDU and its prodrugs were effective against VZV in vivo or if it inhibited 5-FU metabolism like BVdU. To address these questions, we evaluated L-BHDU (1), its 5′-O-L- valyl ester prodrug (2), and two phosphoramidate derivatives (3 & 4) in a range of models that address cytotoxicity and efficacy in culture and in vivo. We have developed systems for screening potential antiviral compounds against VZV that employ fully differentiated, intact human tissues and live animals in an attempt to more closely mimic what occurs during a natural infection (Rowe et al., 2010). The cytotoxic and antiviral effects of L-BHDU were first examined in a primary cell line, human foreskin fibroblasts (HFFs), and then ex vivo in a skin organ culture (SOC) model (Taylor and Moffat, 2005). The effects of L-BHDU and valyl-L-BHDU were tested against VZV in SCID-Hu mice with human skin xenografts (Moffat and Arvin, 1999). This screening process employs the recombinant strain VZV-BAC-Luc, which was selected for its expression of firefly luciferase that can be quantitatively measured by bioluminescence, as well as for its wild type virulence and tissue tropism (Zhang et al., 2007). We report that L-BHDU prevented VZV replication in HFFs, in skin explants, and in SCID-Hu mice. Importantly, L-BHDU did not affect 5-FU metabolism in mice. These results demonstrate the potential of L-BHDU as a novel and safe anti-VZV agent.
2. Materials and Methods
2.1. Propagation of cells and virus
Human foreskin fibroblasts (HFFs) (CCD-1137Sk; American Type Culture Collection, Manassas, VA), used prior to passage 20, were grown in Eagle minimum essential medium with Earle’s salts and L-glutamine (HyClone Laboratories, Logan, UT), supplemented with 10% heat-inactivated fetal bovine serum (Benchmark FBS; Gemini Bio Products, West Sacramento, CA), penicillin-streptomycin (5,000 IU/ml), amphotericin B (250 μg/ml), and nonessential amino acids (all Mediatech, Herndon, VA). VZV-BAC-Luc (Zhang et al., 2007) was derived from the Parental Oka strain, a wild type clinical isolate from Japan (Accession number: AB097933). Dr. Hua Zhu (University of Medicine and Dentistry of New Jersey) kindly provided a master stock of VZV-BAC-Luc (passage 10). VZV-BAC-Luc was stored at −80°C and grown on HFFs for up to 10 passages.
2.2. Preparation of drugs
L-BHDU (1) was synthesized as previously reported (Choi et al., 2000). The structures of valyl ester and phosphoramidate analogues of L-BHDU used in this study are shown (Fig. 1). 5′-O-L-valyl ester of L-BHDU (2) was synthesized by coupling Boc-L-valine with L-BHDU (1) in the presence of 4-dimethylaminopyridine (DMAP) and 1,3-diisopropylcarbodiimide (DIC) to provide the Boc-protected valyl esters of L-BHDU. Deprotection of the Boc group was achieved by treating with 50% trifluoroacetic acid (TFA) in dichloromethane (DCM). The volatiles were removed under reduced pressure and the residue obtained was purified by column chromatography to give 5′-O-L-valyl ester of L-BHDU (2) in good yield.
Figure 1.
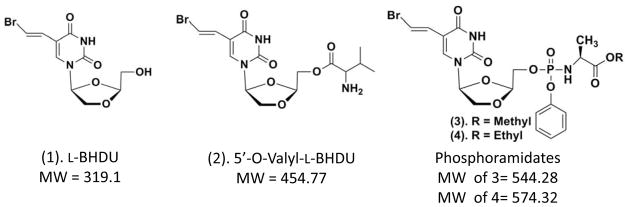
Chemical structures and molecular weights of L-BHDU and analogues
Phosphoramidates of L-BHDU (3 & 4) were synthesized according to the method described by McGuigan et al. (McGuigan, 1992). Phenol was first reacted with phosphoryl chloride in diethyl ether to give aryl dichlorophosphates. This product was then reacted with hydrochloride of amino acid alkyl ester in dichloromethane in the presence of triethylamine at −78°C to yield phosphorochloridate intermediates of the aryl alkoxy-amino acid. These phosphorochloridates, without further purification, were reacted with L-BHDU (1) in THF, in the presence of N-methylimidazole to provide the target compounds, aryl phosphoramidates of L-BHDU (3 & 4), in excellent yields.
Acyclovir (A669), (E)-5-(2-Bromovinyl)-2′-deoxyuridine (B9647), sodium phosphonoformate tribasic hexahydrate (PFA, P6801), and 5-fluorouracil (F6627) were purchased from Sigma Aldrich, St. Louis, MO. Stock solutions of all the compounds except 5-FU and PFA were prepared in dimethyl sulfoxide (DMSO, D2650; Sigma Aldrich), aliquoted and stored at −20°C. PFA and 5-FU stock solutions were made in water. Final drug dilutions used in all experiments were prepared fresh as indicated. Valacyclovir HCl (VACV, 500 mg tablet, GlaxoSmithKline) was crushed in a mortar and used in the in vivo studies. For in vivo studies, mice were administered drugs mixed in 0.4% sodium carboxymethylcellulose (CMC, C948, Sigma Aldrich).
2.3. Cytotoxicity and cell proliferation assay
The neutral red (NR) cytotoxicity assay was performed as described previously (Rowe et al., 2010). Cellular proliferation was evaluated by colorimetric MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) assay following the method developed by Mosmann (Mosmann, T.1983). Both these assays were used to determine the concentration causing a 50% reduction in the number of viable cells (CC50).
2.4. Dose-response studies
HFFs were seeded in clear bottom, black-sided, 6-well plates (W1150, Genetix, Molecular Devices) 24 h prior to infection. Subconfluent HFFs were infected with cell-associated VZV-BAC-Luc showing more than 80% cytopathic effect (CPE) at 1:100 ratio of infected to uninfected cells and adsorbed for 2 h at 37°C. Excess virus was removed and the cells were washed once with PBS. Medium containing either DMSO diluent or 2-fold dilutions of the test compounds at concentrations between 0.002 and 4.0 μM were added; this point was deemed time zero. Cells were treated for 48 hpi (hour post infection) and the medium containing the drug was changed after 24 h. VZV spread was measured by bioluminescence imaging of the same cultures at time zero and 48 hpi. VZV yield was determined daily by bioluminescence imaging (see below) using the IVIS® 50 instrument (Caliper Life Sciences/Xenogen, Hopkinton, MA) and expressed as Total Flux (photons/sec/cm2/steradian). The 50% effective concentration (EC50) values were calculated using two model systems, Yield-Density and Sigmoidal Models, by XLfit 5.3 software (ID Business Solution, www.idbs.com) and GraphPad Prism 5.02 for Windows (GraphPad Software, San Diego, CA, www.graphpad.com).
2.5. Quantitative Real time PCR
Cellular and viral DNA was isolated with DNeasy Blood & Tissue Kit (69506, Qiagen). Test samples and standards were analyzed in triplicate with iQ™ SYBR® Green Supermix (170–8880, Bio-Rad) and 2.5 μL template in a final volume of 25 μL. VZV genomes were amplified with this primer set: forward - AAGTTTCAGCCAACGTGCCAATAAA and reverse - AGACGCGCTTAACGGAAGTAACG (Hawrami and Breuer, 1999). The β-actin gene was amplified with this primer set: forward - TCACCCACACTGTGCCCATCTACGA and reverse - CAGCGGAACCGCTCATTGCCAATGG. Amplification conditions were: 1 cycle of 95°C (3 min) followed by 40 cycles of 95°C (10 s) and 55°C (30 s) using the Bio-Rad iCycler iQ. Data were analyzed with iQ5 optical software (Bio-Rad) and the absolute viral genome copy number was calculated based on a standard curve obtained from serial dilutions ranging from 0.04 pg/μL to 4 ng/μL of a plasmid containing a 647-bp fragment of VZV ORF38 (Taylor et al., 2004).
2.6. Skin organ culture
Human fetal skin tissue (approximately 18 weeks gestational age, Advanced Bioscience Resources, Alameda, CA) was obtained in accordance with all local, state, and federal guidelines. Skin was divided into approximately 1-cm2 pieces, cultured on NetWells (Corning, NY), and inoculated with VZV-BAC-Luc by scarification with a 27-gauge needle (Rowe et al., 2010; Taylor and Moffat, 2005). For bioluminescence imaging, skin explants were submerged in D-luciferin (300 μg/mL in PBS) for 1 hour before scanning in the IVIS® 50. Skin cytotoxicity was also determined after 6 days of treatment with the drugs by hematoxylin and eosin staining of the uninfected skin pieces.
2.7. Drug synergy assay
The activity of L-BHDU in combination with ACV, BVdU and PFA was evaluated in vitro using the method of Prichard and Shipman (Prichard and Shipman. Jr. 1990). VZV yield was measured by bioluminescence imaging after 48 h. Results were analyzed using a modified version of MacSynergy II (http://medicine.uab.edu/Peds/54568/infectious/79968/).
2.8. Mass spectrometry analysis of 5-fluorouracil (5-FU) metabolism in mice
5-fluorouracil was dissolved in water and a dose of 30 mg/kg was administered to 5-week-old, male BALB/c mice (Taconic, Hudson, NY) by tail vein. BVdU and L-BHDU (10 mg/kg dose) were mixed in 0.4% sodium carboxymethylcellulose (CMC, C948, Sigma Aldrich) in sterile water and administered by oral gavage. Exactly 30 or 60 min after treatment, blood was collected by cardiac puncture causing euthanasia by exsanguination. Plasma was separated from the blood and prepared for LC-MS/MS analysis by adding 10× volume of acetonitrile (271004, Sigma Aldrich). The mixture was vortexed for 2 min, and then centrifuged at 20,000 × g at 4°C for 5 min. The supernatant was collected, lyophilized and then stored at −80°C for later analysis. The lyophilized samples were resuspended in 100 μL distilled, deionized water, centrifuged at 20,000 × g in a microfuge to pellet particulates, and the supernatant was transferred to an autosampler vial. An aliquot of the sample (10%) was injected in an LC-MS run, trained for absolute quantification of 5-FU, using triple quadrupole mass spectrometry. This instrument configuration consisted of a Thermo Quantum Access Max triple quadrupole mass spectrometer, with a Dionex Ultimate 3000 UPLC, using porous carbon graphite chromatography, on a HyperCarb column (100 × 2.1 mm) with a 5-micron particle size, and with the column oven set at 40°C. The solvent system consisted of Solvent A: 0.1% formic acid and Solvent B: 100% methanol. The LC-MS/MS run began with the column equilibrated at 10% Solvent B, which was held for 1 min after injection, and then an organic gradient was ramped to 95% Solvent B over 1 min, held at 95% Solvent B for 1 min, then returned to 10% Solvent B in 0.25 min, which was held for equilibration for 2 min, prior to the next injection. Raw data files from the LC-MS/MS run were imported into LCQUAN software (Thermo Scientific), including a standard curve spanning concentrations of 0.1 – 10 μM in water. Area under the curve (AUC) analysis was used to quantify the absolute concentration of the compound in unknown samples. The selected reaction monitoring (SRM) parameters for 5-FU were performed by infusion in the negative mode using 50% methanol with 0.1% formic acid. The parent ion m/z, fragment ion m/z, collision energy, and tube lens voltage for the compound was: 129 m/z, 42.6 m/z, 20v, 70v, respectively.
2.9. Animal procedures
Human fetal skin xenografts were introduced subcutaneously into 5-week-old, male scid-beige mice (C.B-Igh-1b/GbmsTac-Prkdcscid-Lystbg N7, CBSCBG-M, Taconic, Hudson, NY) as full-thickness dermal grafts as described previously with the exception that single implants were used instead of bilateral implants (Moffat et al., 1995). Three weeks after implantation, xenografts were inoculated with cell-associated VZV-BAC-Luc as in (Rowe et al., 2010), and virus growth was monitored daily using the IVIS® 200 apparatus for bioluminescence imaging. Drugs were administered by oral gavage daily for 6–7 days in 0.4% CMC (vehicle) suspension. ACV (120 mg/kg/day) and valacyclovir (VACV, 120 and 200 mg/kg/day) were controls. L-BHDU (8, 15 or 150 mg/kg/day) and valyl-L-BHDU (10 mg/kg/day or 30 mg/kg/day) were the test compounds. Mice were weighed on the day of inoculation and again upon termination of the experiment. Mice were euthanized for immediate collection of blood and tissues 2 h after final treatment, which were stored in liquid nitrogen until the steady state tissue distribution of L-BHDU was determined by HPLC analysis. The protocol was reviewed and approved by the Committee for Humane Use of Animals at SUNY Upstate Medical University.
2.10. HPLC Analysis
L-BHDU concentrations were determined by an HPLC-UV method. Plasma (100 μL) or tissue homogenates (200 μL) spiked with 10 μL triamterene (100 mg/L) were mixed with 300 μL acetonitrile and 600 μL ethyl acetate. Samples were then centrifuged for 10 min at 2000 × g and the upper organic phase was evaporated to dryness. The HPLC measurements were carried out using an Agilent 1100 series HPLC coupled with a UV detector. The chromatographic separation of L-BHDU was achieved using a Phenomenex Gemini column (5μ, C18, 110Å, 250 × 4.6 mm) coupled with a Phenomenex Security Guard column (C18) (Torrance, CA). The mobile phase was isocratic: A: 20 mM KH2PO4 (pH= 9.0): acetonitrile (10:1); B: methanol; A:B = 63:37. The mobile phase flow rate was 1.0 mL/min and the UV detection wavelength was set at 254 nm. Under the chromatographic conditions described, L-BHDU and triamterene eluted at 8.7 min and 10.2 min, respectively. Calibration curves were generated by using samples from spiked blank matrix to yield final calibration points of 0.1 –150 μg/mL with an internal standard concentration of 10 μg/mL in each sample. This method has been previously validated to show acceptable precision and accuracy over the calibration range of 0.1 to 150 μg/mL. Absolute recoveries were greater than 90% for all matrices. Data acquisition was performed using ChemStation chromatography software package (Agilent).
2.11. Bioluminescence imaging
Imaging was performed exactly as described in (Rowe et al., 2010). Briefly, cell and skin cultures were scanned with the IVIS® 50 instrument; mice were scanned with the IVIS® 200 instrument (Caliper Life Sciences/Xenogen, Hopkinton, MA). Cell and skin images were acquired for 1 min; in vivo images were acquired for an initial exposure time of 5 minutes; if pixels were saturated, additional images with shorter exposure times were acquired. Background signals were determined from mock-treated, uninfected cultures or in vivo by placing a 1-cm2 region of interest (ROI) on the head between the ears of mice with VZV-infected skin implants. The rate of VZV spread in the skin implants was calculated as the slope of a line with y = log10 photons/s, and x = day of treatment, where slope = VZV growth rate (log10 photons/s/day). The time interval was set as the day after treatment was initiated to the day it ended.
2.12. Statistical analysis
Data from mouse experiments were analyzed using one-way ANOVA and post hoc Dunnett’s Multiple Comparison Test. Calculations were made using GraphPad Prism 5.02 for Windows (GraphPad Software, San Diego, CA, www.graphpad.com). A p ≤ 0.0001 was considered statistically significant.
3. Results
3.1. Cytotoxicity and antiviral efficacy of L-BHDU (1) and its prodrug analogues (2, 3, 4)
To determine the cytotoxic effect of the L-dioxolane uracil analogues on low passage human foreskin fibroblast (HFFs) cells, neutral red dye uptake and MTT cell proliferation assays were performed after 72 h. All the test compounds were noncytotoxic (CC50 >200 μM) and did not affect cellular proliferation (Table 1). L-BHDU and its three prodrug analogues demonstrated good antiviral efficacy against VZV-BAC-Luc in two systems: HFFs and skin organ culture (SOC) (Table 1). Valyl-L-BHDU (2) was the most potent compound with an EC50 of 0.03 μM in cell culture (Table 1 and Fig 2B). Both L-BHDU (EC50 = 0.22 μM, Fig. 2A) and methyl-L-BHDU (EC50= 0.13 μM) were also potent against VZV-BAC-Luc in cell culture systems (Table 1). All the tested analogues of L-BHDU were superior to acyclovir (ACV) and phosphonoformate (PFA, Foscarnet), for which EC50 values of 5 μM and 40 μM, respectively, were obtained in the HFF culture system (data not shown).
Table 1.
Susceptibility of VZV-BAC-Luc to L-dioxolane uracil analogues in HFFs and skin organ culture
| Compounds | EC50 in HFFs (μM)a | CC50 (μM)b | SI (CC50/EC50)c | EC50 in SOC (μM)d |
|---|---|---|---|---|
| L-BHDU (1) | 0.22 ± 0.01 | >200 | >909 | 0.24 ± 0.02 |
| Valyl L-BHDU (2) | 0.03 ± 0.03 | >200 | >6667 | 0.04 ± 0.02 |
| Pro drug 3 | 0.13 ± 0.003 | >200 | >1538 | 0.19 ± 0.01 |
| Pro drug 4 | 0.35 ± 0.03 | >200 | >571 | Not Done |
50% inhibitory concentration 48 hpi determined by bioluminescence imaging, mean ± standard deviation from at least 3 experiments
50% cytotoxic concentration at 72 h determined by neutral red assay from 3 experiments
Selectivity index
50% inhibitory concentration 6 dpi determined by bioluminescence imaging, mean ± standard deviation from 6 replicates
Figure 2. Effects of L-BHDU (1) and valyl-L-BHDU (2) on VZV replication.

L-BHDU (A) and valyl-L-BHDU (B) were evaluated against VZV-BAC-Luc from 0.002 to 4 μM. VZV yield was determined by bioluminescence imaging. (C) VZV yield was determined by quantitative real-time PCR (genomes/ng β-actin). Each point represents the mean ± standard deviation of triplicate samples.
L-BHDU analogues (except the less potent ethyl-L-BHDU) were next evaluated in skin where VZV infects epidermal keratinocytes and dermal fibroblasts (Cohen et al., 2007; Sexton et al., 1992). We used a skin organ culture (SOC) model that provides the differentiated cell types and the tissue microenvironment that is highly suitable for VZV replication (Taylor and Moffat, 2005). SOC also provides the relevant conditions for evaluating potential antiviral compounds against VZV (Rowe et al., 2010). Previous studies showed that skin explants retained their integrity for at least 10 days and VZV replication was unrestricted (Taylor and Moffat, 2005). Triplicate skin samples were infected with VZV-BAC-Luc by scarification and treated for 6 days with 2-fold dilutions of the test compounds at concentrations between 0.008 and 4.0 μM (refreshed daily). The level of VZV infection was measured daily by bioluminescence imaging. Similar to the results in cultured HFFs, the antiviral effects of the analogues were dose-dependent in SOC (Table 1). Histopathological studies showed neither DMSO nor L-BHDU analogues treatment caused obvious detrimental effects to the tissue (data not shown).
3.2. L-BHDU antagonizes the antiviral activity of ACV, BVdU and PFA
A common approach to studying the mechanism of action of a compound is to evaluate its effects in combination with other drugs. If the drugs target different pathways, then synergy may occur. Conversely, if the drugs target the same pathway, then they may compete for the same substrates and antagonize. We used the MacSynergy platform to evaluate the interactions between L-BHDU and BVdU, ACV, and PFA, which are frequently prescribed for VZV infections (Prichard and Shipman, 1990). HFFs were inoculated with VZV-BAC-Luc, the drug combinations were added in the appropriate matrix, and the cultures were incubated for 48 h. Virus yield was measured by bioluminescence imaging. In all three cases, VZV yield was below the expected level of inhibition as compared to each drug alone, indicating that L-BHDU antagonized the activity of these drugs (Fig. 3). Interference between the drugs occurred at the highest concentration of L-BHDU (2 μM) and the approximate EC50 of the other drugs. It is possible that antagonism exhibited by the combination of L-BHDU with BVdU and ACV was due to competition for phosphorylation by VZV TK. Since PFA is not a substrate of TK, and its antiviral mechanism is inhibition of DNA polymerase, the antagonism with L-BHDU could have arisen from unknown effects of L-BHDU on VZV DNA Pol expression or activity.
Figure 3. Combined efficacy of L-BHDU with other common anti VZV drugs.

The plots generated by MacSynergy show the degree of antagonism or synergy (x-axis), and the concentrations of L-BHDU (y-axis) and the additional drug (z-axis). Inhibition values less than zero indicate antagonism between L-BHDU and BVdU (A), ACV (B) and PFA (C).
3.3. L-BHDU did not inhibit 5-fluorouracil metabolism in mice
L-BHDU has a similar structure to BVdU, which is highly active against VZV. BVdU is not approved for use in the U.S. because it may cause lethal drug interactions with the cancer drug 5-FU. The mechanism of this toxic interaction is the inhibition of dihydropyrimidine dehydrogenase by bromovinyluracil, a metabolite of BVdU, which results in toxic accumulation of 5-FU in a person treated with both drugs (De Clercq. 2004). The effects of L-BHDU on 5-FU levels in vivo were not known. Mice were administered 5-FU (30 mg/kg, i.v.). Two groups also received BVdU (10 mg/kg, p.o.) or L-BHDU (10 mg/kg, p.o.) immediately following. Blood was collected exactly 30 and 60 min later. Plasma was separated and 5-FU concentration was measured by LC-MS/MS. Mice treated with 5-FU alone eliminated the drug after 60 min (Fig 4A). The average AUC for the 5-FU group at 30 min was 1246 nM and at 60 min it was undetectable. As expected, 5-FU persisted in the mice that received BVdU, with the relative average concentration decreasing from 945 nM at 30 min to 685 nM at 60 min. In contrast, 5-FU concentration decreased 90% in mice treated with L-BHDU from an average concentration of 1720 nM to 180 nM. Thus L-BHDU did not affect 5-FU catabolism in vivo.
Figure 4. Effect of L-BHDU on 5-FU metabolism in vivo.

(A) The absolute concentration of 5-FU in plasma was measured by LC-MS/MS. Each point represents the average and standard deviation of 3 mice. (B) Representative MS chromatograms of individual mice show that BVdU interfered with 5-FU catabolism while L-BHDU did not. For each drug study, the intensity of each 5-FU assay run was normalized to the same signal intensity for the 30 and 60 min time points.
3.4. L-BHDU (1) and valyl-L-BHDU (2) prevent VZV replication in SCID-Hu mice
The SCID-Hu mouse model is valuable for analyzing VZV pathogenesis and potential antiviral compounds, and so it was employed to evaluate the effectiveness of L-BHDU in vivo (Ku et al., 2004; Oliver et al., 2008; Rowe et al., 2010). SCID mice with xenografts of human fetal skin were inoculated by scarification with VZV-BAC-Luc. Virus spread in the skin implants was monitored by bioluminescence imaging starting at 2 days post infection (dpi). When VZV infection was established and the bioluminescence signal for an individual mouse crossed the background threshold of 2×104 Total Flux (usually at 2–3 dpi), the mouse was randomly assigned to either a treatment or control group. The control groups consisted of mice treated with 0.4% CMC (vehicle), acyclovir (120 mg/kg/day) or valacyclovir (120 or 200 mg/kg/day). The experimental groups were administered L-BHDU (8, 15, or 150 mg/kg/day) or valyl-L-BHDU (10 or 30 mg/kg/day) daily. All drugs were suspended in 0.4% CMC and administered by oral gavage. Representative images of mice from selected treatment groups at 5 dpi demonstrate the bioluminescence signal directly above the infected skin implant on the left flank (Fig. 5A). In mice treated with vehicle, virus growth was unrestricted and the bioluminescence signal increased more than 1000-fold over 7 days.
Figure 5. Evaluation of L-BHDU and valyl-L-BHDU in vivo.

Mice were treated with vehicle or drug and virus yield was measured by bioluminescence imaging. (A) Differences in VZV yield were visualized by overlaying an image of Total Flux over a photograph of the mouse (all images use the same scale, right). (B) The VZV growth rate for individual mice (symbols) and the average for the group (bars) are shown. Significant reductions in VZV growth rate compared to the vehicle group are indicated (*, p<0.0001, one-way ANOVA and post hoc Dunnett’s Multiple Comparison Test).
Treatment with L-BHDU (15 mg/kg/day) and valyl-L-BHDU (30 mg/kg/day) reduced virus spread after 4 days, resulting in significant reduction in bioluminescence signal (Fig. 5A) and VZV growth rate (Fig. 5B) by the end of the experiment. When the mice were treated with a higher dose of L-BHDU (150 mg/kg/day), virus growth was also severely limited. The rate of VZV spread in each skin xenograft was calculated from the day after treatment was initiated to the day it ended. The average VZV growth rate was then calculated for each group (Table 2). Combined data from two separate experiments demonstrate that the vehicle (n = 21) had no effect on the VZV growth rate, producing a rate of 0.43 ± 0.10 Log10 photons/s/day that corresponded to the typical results in cultured HFFs of 0.50 (½ Log10 per day). The average rate of VZV growth in the mice treated with 120 mg/kg/day acyclovir (n = 11) was 0.38 ± 0.07 log10 photons/s/day, which is nearly identical to that of the diluent treated group, thus there was no antiviral effect. Valacyclovir at 200 mg/kg/day (n=10) caused a significant reduction of the VZV growth rate (0.27 ± 0.07). Low doses of L-BHDU (8 mg/kg/day) and valyl-L-BHDU (10 mg/kg/day) caused an intermediate reduction in the VZV growth rate at 0.33 ± 0.03 and 0.34 ± 0.14 log10 photons/s/day. Higher doses of L-BHDU (15 and 150 mg/kg/day) prevented VZV replication and reduced the growth rate to 0.25 ± 0.9 and 0.22 ± 0.6 log10 photons/s/day respectively. Valyl-L-BHDU at 30 mg/kg/day (n=8) had a similar inhibitory effect as 200 mg/kg/day VACV in decreasing the VZV growth rate (0.26 ± 0.07). The reduction in VZV growth rates for the groups treated with 150 mg/kg/day L-BHDU and 30 mg/kg/day valyl-L-BHDU were significant compared to the vehicle group. No overt toxicity from L-BHDU was seen in vivo. L-BHDU and CMC both caused moderate weight loss, which did not correspond to dose, and mortality was 2/10 in 30 mg/kg/day valyl-L-BHDU group and 1/5 in the 8 mg/kg/day L-BHDU group. All groups lost weight during the studies due to the stress of surgery, oral gavage treatments, and handling. However, none of the treated groups lost >20% of body weight, suggesting that the mice tolerated these drugs.
Table 2.
Antiviral effects of L-BHDU and valyl-L-BHDU in SCID-Hu mice
| Treatment | Dose (mg/kg/day) | ABWDa,b (±SD) | Mortality | Ave. VZV Growth Rateb,c (± SD)c |
|---|---|---|---|---|
| Vehicle 0.4% CMC | 0 | −1.0 (1.3) | 2/21 | 0.43 (0.10) |
| ACV | 120 | −1.2 (0.9) | 0/11 | 0.38 (0.07) |
| VACV | 120 | −2.9 (1.2)* | 0/11 | 0.40 (0.09) |
| 200 | −3.6 (1.5)* | 0/10 | 0.27 (0.07) * | |
| L-BHDU (1) | 8 | −2.9 (3.0) | 1/5 | 0.33 (0.03) |
| 15 | −4.3 (1.9)* | 0/5 | 0.25 (0.09) * | |
| 150 | −1.5 (0.6) | 0/5 | 0.22 (0.06) * | |
| Valyl-L-BHDU (2) | 10 | −0.9 (0.8) | 0/10 | 0.34 (0.14) |
| 30 | −0.9 (2.1) | 2/10 | 0.26 (0.07) * |
Average Body Weight Difference from Days 0–7 in grams
SD, standard deviation. One-way ANOVA and post hoc Dunnett’s Multiple Comparison Test.
= p<0.0001 compared to vehicle group
VZV growth rate [Log10(photons/s/day)]
To determine the concentrations of L-BHDU in mouse organs and the human skin xenografts, preliminary pharmacokinetic analysis was performed. Tissue and plasma specimens were collected 2 h after the final treatment, and then drug concentrations were measured by HPLC. The maximum concentration (Cmax, μg/mL of plasma or μg/g of tissue) was determined and the ratio to plasma was calculated (Table 3). The ratio of L-BHDU to plasma was significantly higher at the lower dose for all tissues indicating dose dependent tissue distribution due, in part, to saturation of influx transporters. L-BHDU reached similar concentrations in the mouse skin and human skin xenografts. For the 150 mg/kg/day group, the amount of L-BHDU found in the skin implant was only slightly less than the concentration found in the plasma with a ratio to plasma of 0.88 ± 0.29. Drug levels in the heart were similar to plasma, while levels in the spleen and liver were slightly higher at 1.36 ± 0.23 and 1.47 ± 0.30, respectively. The kidney and lung had the highest ratio to plasma. Not unexpectedly, little drug was detected in the brain. The ratio to plasma of most tissues was greater than 1, indicating extensive tissue distribution of L-BDHU. In the mice given 8 mg/kg/day L-BHDU, the concentration in the human skin implants was 0.73 ± 0.08 μg/g, from which the approximate molar concentration of 2.3 μM can be calculated from the known molecular weight of 319.1 g/mol. This concentration is in the effective range determined in HFFs and SOC and exceeds the EC50 by 10-fold.
Table 3.
Maximum L-BHDU concentration two hours post final treatment
| Tissue | 150 mg/kg/day (n = 5) | 8 mg/kg/day (n = 4) | ||
|---|---|---|---|---|
| Max. Conc. | Ratio to Plasma | Max. Conc. | Ratio to Plasma | |
| Plasma | 13.9 (3.9)a | 1 | 0.3 (0.2) | 1 |
| Brain | 1.1 (0.3)b | 0.1 (0.03) | 0.1 (0.02) | 0.4 (0.3) |
| Skin (Human) | 11.3 (1.1)b | 0.9 (0.3) | 0.7 (0.1) | 3.1 (2.6) |
| Skin (Mouse) | 11.8 (1.0)b | 0.9 (0.2) | 0.7 (0.1) | 3.0 (2.5) |
| Heart | 13.7 (2.8)b | 1.0 (0.2) | 0.7 (0.1) | 2.9 (2.5) |
| Spleen | 18.8 (6.2)b | 1.4 (0.2) | 0.6 (0.2) | 2.1 (1.3) |
| Liver | 19.8 (4.1)b | 1.5 (0.3) | 1.4 (0.5) | 4.5 (1.1) |
| Kidney | 27.4 (3.5)b | 2.1 (0.4) | 1.3 (0.2) | 5.1 (3.1) |
| Lung | 30.4 (2.8)b | 2.3 (0.5) | 3.0 (0.8) | 11.2 (5.7) |
Mean (± Standard Deviation), μg/mL
Mean (± Standard Deviation), μg/g
4. Discussion
The results of this study confirmed the activity of L-BHDU against VZV in cultured cells and extended the evaluation in human skin ex vivo and engrafted in mice. L-BHDU was effective in all three systems at low micromolar concentrations, and the treatment was not toxic to cells, skin, or mice. We found that L-BHDU did not cause cell death or inhibit cell proliferation at 200 μM. Using 200 μM as an approximation of the CC50, the selective index (SI) in HFFs was estimated to be >909 for L-BHDU and >6667 for the valyl analogue. These values greatly exceed the SIs for acyclovir and foscarnet that we determined using the same culture system as 250 and 60, respectively (Rowe et al., 2010). Of all the compounds tested in HFFs, valyl-L-BHDU was the most potent with an EC50 of 0.03 μM. The EC50 of L-BHDU was 0.22 μM, which was higher than previously reported values of 0.055 and 0.07 μM in HEL cells using the VZV Ellen strain and measured by quantification of DNA hybridization (Choi et al., 2000; Li et al., 2000). This difference could be due to the cell type, the VZV strain, or measuring virus yield by bioluminescence. Interestingly we found that the EC50 of L-BHDU was approximately 0.07 μM when determined by qPCR, which was similar to the previously reported EC50 (Li et al., 2000). The difference between the bioluminescence and qPCR assay could be due to the fact that inhibition of DNA replication by L-BHDU would not immediately block transcription and translation of luciferase that is expressed from the SV40 immediate early promoter in VZV-BAC-Luc (Zhang et al., 2007). This continued protein synthesis prolongs bioluminescence even when L-BHDU concentrations are inhibitory to viral DNA synthesis. The long duration of bioluminescence from the VZV-BAC-Luc strain could also explain why the apparent virus growth rates in the mouse model were only reduced by half despite high concentrations of L-BHDU and valyl-L-BHDU in vivo.
L-BHDU proved to be equally effective in skin organ culture and the estimated EC50 was 0.24 μM. The concordance of EC50 in SOC and cell culture is another difference between L-BHDU and ACV. We found that 400 μM ACV was necessary to prevent VZV spread in SOC whereas 100 μM ACV was required in HFFs (Rowe et al., 2010). It is not known why ACV is less potent in full-thickness skin than in skin fibroblasts, but it could be due to the high levels of ATP in skin cells. It is well known that absorption of ACV is poor in the gastrointestinal tract, which limits its antiviral potency against VZV and HSV. However addition of the valine moiety to ACV increases its bioavailability by carrier-mediated transporters (MacDougall and Guglielmo, 2004). Similarly, valyl-L-BHDU showed a much lower EC50 of 0.04 μM in SOC. This bodes well for future studies with L-BHDU and valyl-L-BHDU, as their potency is not hampered by the increased complexity of the skin tissue.
VZV infection has been treated mainly with ACV, which has low oral bioavailability (Snoeck et al., 1994) as well as low specific activity against clinical isolates of VZV (Arvin AM. 1996). Though highly potent, BVdU is not approved by the FDA for use in the United States (De Clercq, 2005). Recently, interest in combination drug therapy has increased because the side effects and cytotoxicity may be reduced if lower doses are effective, and because this could also lower the probability that drug-resistant mutants will arise (Prichard and Shipman, 1990). Drug-drug interactions have been also used to study the mechanism of action (Tallarida, 2001). Since not all drug combinations result in synergism, it is important to have a clear understanding of drug combination pharmacology. For example, we found that L-BHDU combined with ACV or BVdU showed statistically significant antagonism, which may be due to competition for phosphorylation by VZV TK. Since BVdU is a better substrate for VZV TK than ACV (Karlström et al., 1986), we observed a robust antagonism with BVdU and L-BHDU. In support of this contention, James et al found that competition between cyclopropavir and ganciclovir for phosphorylation by the UL97 kinase of human cytomegalovirus was the basis of antagonism of these drugs (James et al., 2011).
In some European and South American countries, BVdU is used to treat HSV and VZV infections, and it is more potent in culture than L-BHDU (De Clercq, 2005). However, the potential lethal interactions between BVdU and 5-FU deserve caution. BVdU is efficiently catabolized by thymidine phosphorylases (TPases) to (E)-5-(2-bromovinyl)uracil (BVU) and 2-deoxyribose-1-phosphate. BVU is a potent inhibitor of dihydropyrimidine dehydrogenase (DPD), the enzyme which is responsible for the first step in pyrimidine catabolism. Inhibition of DPD increases the half life of 5-FU leading to elevated plasma concentration of 5-FU and increased toxicity leading to patient death (Reviewed by De Clercq, 2004). In contrast, we found that L-BHDU had no affect on 5-FU metabolism. A possible explanation is that L-nucleosides show different substrate specificities toward metabolic enzymes, resulting in reduced toxicity, increased potency, and metabolic stability (Choi et al., 2000). Due to the L-configuration, it could be similarly predicted that L-BHDU is not a substrate of TPases and the metabolized products of L-BHDU do not interact with DPD or interfere with 5-FU metabolism. Thus, L-BHDU may have safety advantages compared to BVdU.
The effects of L-BHDU in vivo were not known prior to this study. The rationale was to mimic natural infection, thus VZV infection was established in skin xenografts before treatment commenced. We have found that treating earlier than 2 dpi confounds the data analysis, as one cannot distinguish between failure of the virus inoculum to infect the implant or an effective antiviral response. Starting L-BHDU treatment before VZV infection, or simultaneously, would likely improve the drug efficacy, but this approach is less likely to provide data with clinical relevance. We found that ACV or VACV at a dose of 120 mg/kg/day had no effect on VZV replication. Others have also found that VACV has little to no effect on VZV replication in SCID-Hu mice (Oliver et al., 2008). In contrast, L-BHDU at 15 and 150 mg/kg/day and valyl-L-BHDU at 30 mg/kg/day significantly reduced VZV replication in vivo.
Determining the minimum effective dose in the SCID-Hu mouse model may show that these compounds are more potent than we report here. Studies in animals and humans showed that bioavailability of ACV was reduced as doses increased, suggesting a saturable absorptive process (MacDougall and Guglielmo, 2004). This could be the case in the treatment group of 150 mg/kg/day L-BHDU. Due to higher bioavailability of valyl-L-BHDU we see a comparable antiviral effect at 30 mg/kg/day. Drug absorption can be measured by sampling mouse skin, since we found that the maximum concentration of L-BHDU in mouse skin was comparable to the levels in human skin xenografts. Interestingly, the 8 mg/kg/day dose of L-BHDU in this study yielded approximately 0.7 μg/g (human skin), which is similar to the brivudine Cmax of 1.7 μg/mL (plasma) in herpes zoster patients given a dose of 125 mg/day (Keam et al., 2004). Thus these related uridine analogues might have similar bioavailability in vivo.
There are several questions that deserve further investigation. Further studies would be necessary to understand the molecular mechanism of action of L-BHDU. Overall, L-bromovinyl uracil nucleoside analogues were potent, safe and well tolerated making them a good choice for drug therapy.
Acknowledgments
We thank Wanda Coombs for excellent technical assistance and Jenny Rowe for her help with the in vivo evaluation. LC-MS/MS assays to measure 5-FU in plasma were performed at the University of Rochester Proteomics Center. J.F.M. is supported in part by the contract HHSN272201000023I from the Division of Microbiology and Infectious Diseases, NIAID.
Contributor Information
Chandrav De, Email: dec@upstate.edu.
Dongmei Liu, Email: liud@upstate.edu.
Bo Zheng, Email: bozheng@buffalo.edu.
Uma S. Singh, Email: ussingh@rx.uga.edu.
Satish Chavre, Email: schavre@rx.uga.edu.
Catherine White, Email: cwhite@rx.uga.edu.
Robert D. Arnold, Email: rda0007@auburn.edu.
Fred K. Hagen, Email: fred_hagen@urmc.rochester.edu.
Chung K. Chu, Email: dchu@mail.rx.uga.edu.
Jennifer F. Moffat, Email: moffatj@upstate.edu.
References
- Ahmed AM, Brantley JS, Madkan V, Mendoza N, Tyring SK. Managing herpes zoster in immunocompromised patients. Herpes. 2007;14(2):32–6. [PubMed] [Google Scholar]
- Andrei G, Snoeck R, Reymen D, Liesnard C, Goubau P, Desmyter J, De Clercq E. Comparative activity of selected antiviral compounds against clinical isolates of varicella-zoster virus. Eur J Clin Microbiol Infect Dis. 1995;14(4):318–29. doi: 10.1007/BF02116525. [DOI] [PubMed] [Google Scholar]
- Arvin AM. Varicella-zoster virus. Clin Microbiol Rev. 1996;9(3):361–81. doi: 10.1128/cmr.9.3.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y, Li L, Grill S, Gullen E, Lee CS, Gumina G, Tsujii E, Cheng YC, Chu CK. Structure-activity relationships of (E)-5-(2-bromovinyl)uracil and related pyrimidine nucleosides as antiviral agents for herpes viruses. J Med Chem. 2000;43(13):2538–46. doi: 10.1021/jm990543n. [DOI] [PubMed] [Google Scholar]
- Chu CK, Ma T, Shanmuganathan K, Wang C, Xiang Y, Pai SB, Yao GQ, Sommadossi JP, Cheng YC. Use of 2′-fluoro-5-methyl-beta-L-arabinofuranosyluracil as a novel antiviral agent for hepatitis B virus and Epstein-Barr virus. Antimicrob Agents Chemother. 1995;39(4):979–81. doi: 10.1128/aac.39.4.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JI, Straus SE, Arvin AM. Varicella-zoster virus replication, pathogenesis, and management. In: Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE, editors. Fields Virology. 5. Vol. 2. Lippincott-Williams and Wilkens; Philadelphia: 2007. pp. 2773–2818. [Google Scholar]
- De Clercq E. Antiviral drugs in current clinical use. J Clin Virol. 2004;30(2):115–33. doi: 10.1016/j.jcv.2004.02.009. [DOI] [PubMed] [Google Scholar]
- De Clercq E, Descamps J, De Somer P, Barr PJ, Jones AS, Walker RT. (E)-5-(2-Bromovinyl)-2′-deoxyuridine: a potent and selective anti-herpes agent. Proc Natl Acad Sci U S A. 1979;76(6):2947–51. doi: 10.1073/pnas.76.6.2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Clercq E. Recent highlights in the development of new antiviral drugs. Current Opinion in Microbiology. 2005;8(5):552–60. doi: 10.1016/j.mib.2005.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diasio RB. Sorivudine and 5-fluorouracil; a clinically significant drug-drug interaction due to inhibition of dihydropyrimidine dehydrogenase. Br J Clin Pharmacol. 1998;46(1):1–4. doi: 10.1046/j.1365-2125.1998.00050.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han HK, de Vrueh RLA, Rhie JK, Covitz KMY, Smith PL, Lee CP, Oh DM, Sadee W, Amidon GL. 5′-Amino acid esters of antiviral nucleosides, acyclovir, and AZT are absorbed by the intestinal PEPT1 peptide transporter. Pharmaceut Res. 1998;15(8):1154–9. doi: 10.1023/a:1011919319810. (a) [DOI] [PubMed] [Google Scholar]
- Han HK, Oh DM, Amidon GL. Cellular uptake mechanism of amino acid ester prodrugs in Caco-2/hPEPT1 cells overexpressing a human peptide transporter. Pharmaceut Res. 1998;15(9):1382–6. doi: 10.1023/a:1011945420235. (b) [DOI] [PubMed] [Google Scholar]
- Hawrami K, Breuer J. Development of a fluorogenic polymerase chain reaction assay (TaqMan) for the detection and quantitation of varicella zoster virus. J Virol Methods. 1999;79(1):33–40. doi: 10.1016/s0166-0934(98)00176-1. [DOI] [PubMed] [Google Scholar]
- Holcomb K, Weinberg JM. A novel vaccine (Zostavax) to prevent herpes zoster and postherpetic neuralgia. J Drugs Dermatol. 2006;5(9):863–6. [PubMed] [Google Scholar]
- Ibrahim SS, Boudinot FD, Schinazi RF, Chu CK. Physicochemical properties, bioconversion and disposition of lipophilic prodrugs of 2′,3′-dideoxycytidine. Antiviral Chem & Chemother. 1996;7:167–172. [Google Scholar]
- James SH, Hartline CB, Harden EA, Driebe EM, Schupp JM, Engelthaler DM, Keim PS, Bowlin TL, Kern ER, Prichard MN. Cyclopropavir inhibits the normal function of the human cytomegalovirus UL97 kinase. Antimicrob Agents Chemother. 2011;55(10):4682–91. doi: 10.1128/AAC.00571-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlström AR, Källander CF, Abele G, Larsson A. Acyclic guanosine analogs as substrates for varicella-zoster virus thymidine kinase. Antimicrob Agents Chemother. 1986;29(1):171–4. doi: 10.1128/aac.29.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keam SJ, Chapman TM, Figgitt DP. Brivudin (bromovinyl deoxyuridine) Drugs. 2004;64(18):2091–7. doi: 10.2165/00003495-200464180-00011. discussion 2098–9. [DOI] [PubMed] [Google Scholar]
- Keizer HJ, De Bruijn EA, Tjaden UR, De Clercq E. Inhibition of fluorouracil catabolism in cancer patients by the antiviral agent (E)-5-(2-bromovinyl)-2′-deoxyuridine. J Cancer Res Clin Oncol. 1994;120(9):545–9. doi: 10.1007/BF01221032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku CC, Zerboni L, Ito H, Graham BS, Wallace M, Arvin AM. Varicella-zoster virus transfer to skin by T Cells and modulation of viral replication by epidermal cell interferon-alpha. J Exp Med. 2004;200(7):917–25. doi: 10.1084/jem.20040634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Dutschman GE, Gullen EA, Tsujii E, Grill SP, Choi Y, Chu CK, Cheng YC. Metabolism and mode of inhibition of varicella-zoster virus by L-beta-5-bromovinyl-(2-hydroxymethyl)-(1,3-dioxolanyl)uracil is dependent on viral thymidine kinase. Mol Pharmacol. 2000;58(5):1109–14. doi: 10.1124/mol.58.5.1109. [DOI] [PubMed] [Google Scholar]
- MacDougall C, Guglielmo BJ. Pharmacokinetics of valacyclovir. J Antimicrob Chemother. 2004;53(6):899–901. doi: 10.1093/jac/dkh244. [DOI] [PubMed] [Google Scholar]
- McGuigan C, Pathirana RN, Mahmood N, Devine KG, Hay A. Aryl phosphate derivatives of AZT retain activity against HIV1 in cell lines which are resistant to the action of AZT. J Antivir Res. 1992;17(4):311–21. doi: 10.1016/0166-3542(92)90026-2. [DOI] [PubMed] [Google Scholar]
- Moffat JF, Stein MD, Kaneshima H, Arvin AM. Tropism of varicella-zoster virus for human CD4+ and CD8+ T lymphocytes and epidermal cells in SCID-hu mice. Journal of Virology. 1995;69(9):5236–42. doi: 10.1128/jvi.69.9.5236-5242.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffat JF, Arvin AM. Varicella-zoster virus infection of T cells and skin in the SCID-hu mouse model. In: Zak O, Sande MA, editors. Handbook of Animal Models of Infection. Academic Press; San Diego: 1999. pp. 973–980. [Google Scholar]
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Oliver SL, Zerboni L, Sommer M, Rajamani J, Arvin AM. Development of recombinant varicella-zoster viruses expressing luciferase fusion proteins for live in vivo imaging in human skin and dorsal root ganglia xenografts. J Virol Methods. 2008;154(1–2):182–93. doi: 10.1016/j.jviromet.2008.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxman MN, Levin MJ, Johnson GR, Schmader KE, Straus SE, Gelb LD, Arbeit RD, Simberkoff MS, Gershon AA, Davis LE, Weinberg A, Boardman KD, Williams HM, Zhang JH, Peduzzi PN, Beisel CE, Morrison VA, Guatelli JC, Brooks PA, Kauffman CA, Pachucki CT, Neuzil KM, Betts RF, Wright PF, Griffin MR, Brunell P, Soto NE, Marques AR, Keay SK, Goodman RP, Cotton DJ, Gnann JW, Jr, Loutit J, Holodniy M, Keitel WA, Crawford GE, Yeh SS, Lobo Z, Toney JF, Greenberg RN, Keller PM, Harbecke R, Hayward AR, Irwin MR, Kyriakides TC, Chan CY, Chan IS, Wang WW, Annunziato PW, Silber JL. A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N Engl J Med. 2005;352(22):2271–84. doi: 10.1056/NEJMoa051016. [DOI] [PubMed] [Google Scholar]
- Prichard MN, Shipman C., Jr A three-dimensional model to analyze drug-drug interactions. Antiviral Res. 1990;14:181–205. doi: 10.1016/0166-3542(90)90001-n. [DOI] [PubMed] [Google Scholar]
- Rawal RK, Singh US, Chavre SN, Wang JN, Sugiyama M, Hung W, Govindarajan R, Korba B, Tanaka Y, Chu CK. 2′-Fluoro-6′-methylene-carbocyclic adenosine phosphoramidate (FMCAP) prodrug: in vitro anti-HBV activity against the lamivudine-entecavir resistant triple mutant and its mechanism of action. Bioorg Med Chem Lett. 2013;15; 23(2):503–6. doi: 10.1016/j.bmcl.2012.11.027. [DOI] [PubMed] [Google Scholar]
- Rowe J, Greenblatt RJ, Liu D, Moffat JF. Compounds that target host cell proteins prevent varicella-zoster virus replication in culture, ex vivo, and in SCID-Hu mice. Antiviral Res. 2010 doi: 10.1016/j.antiviral.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampathkumar P, Drage LA, Martin DP. Herpes zoster (shingles) and postherpetic neuralgia. Mayo Clin Proc. 2009;84(3):274–80. doi: 10.4065/84.3.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seward JF, Marin M, Vazquez M. Varicella vaccine effectiveness in the US vaccination program: a review. J Infect Dis. 2008;197(Suppl 2):S82–9. doi: 10.1086/522145. [DOI] [PubMed] [Google Scholar]
- Sexton CJ, Navsaria HA, Leigh IM, Powell K. Replication of varicella zoster virus in primary human keratinocytes. J Med Virol. 1992;38(4):260–4. doi: 10.1002/jmv.1890380406. [DOI] [PubMed] [Google Scholar]
- Shigeta S, Yokota T, Iwabuchi T, Baba M, Konno K, Ogata M, De Clercq E. Comparative efficacy of antiherpes drugs against various strains of varicella-zoster virus. J Infect Dis. 1983;147(3):576–84. doi: 10.1093/infdis/147.3.576. [DOI] [PubMed] [Google Scholar]
- Snoeck R, Andrei G, De Clercq E. Chemotherapy of varicella zoster virus infections. International Journal of Antimicrobial Agents. 1994;4(3):211–226. doi: 10.1016/0924-8579(94)90011-6. [DOI] [PubMed] [Google Scholar]
- Spadari S, Maga G, Focher F, Ciarrocchi G, Manservigi R, Arcamone F, Capobianco M, Carcuro A, Colonna F, Iotti S, et al. L-thymidine is phosphorylated by herpes simplex virus type 1 thymidine kinase and inhibits viral growth. J Med Chem. 1992;35(22):4214–20. doi: 10.1021/jm00100a029. [DOI] [PubMed] [Google Scholar]
- Tallarida RJ. Drug synergism: its detection and applications. J Pharmacol Exp Ther. 2001;298(3):865–72. [PubMed] [Google Scholar]
- Taylor SL, Moffat JF. Replication of varicella-zoster virus in human skin organ culture. J Virol. 2005;79(17):11501–6. doi: 10.1128/JVI.79.17.11501-11506.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SL, Kinchington PR, Brooks A, Moffat JF. Roscovitine, a cyclin dependent kinase inhibitor, prevents replication of varicella-zoster virus. Journal of Virology. 2004;78(6):2853–2862. doi: 10.1128/JVI.78.6.2853-2862.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez M, LaRussa PS, Gershon AA, Niccolai LM, Muehlenbein CE, Steinberg SP, Shapiro ED. Effectiveness over time of varicella vaccine. Jama. 2004;291(7):851–5. doi: 10.1001/jama.291.7.851. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Rowe J, Wang W, Sommer M, Arvin A, Moffat J, Zhu H. Genetic analysis of varicella-zoster virus ORF0 to ORF4 by use of a novel luciferase bacterial artificial chromosome system. J Virol. 2007;81(17):9024–33. doi: 10.1128/JVI.02666-06. [DOI] [PMC free article] [PubMed] [Google Scholar]