

Abstract
Chronic exposure to industrial solvent and water pollutant trichloroethylene (TCE) in female MRL+/+ mice generates disease similar to human autoimmune hepatitis. The current study was initiated to investigate why TCE-induced autoimmunity targeted the liver. Compared to other tissues the liver has an unusually robust capacity for repair and regeneration. This investigation examined both time-dependent and dose-dependent effects of TCE on hepatoprotective and pro-inflammatory events in liver and macrophages from female MRL+/+ mice. After a 12-week exposure to TCE in drinking water a dose-dependent decrease in macrophage production of IL-6 at both the transcriptional and protein level was observed. A longitudinal study similarly showed that TCE inhibited macrophage IL-6 production. In terms of the liver, TCE had little effect on expression of pro-inflammatory genes (Tnfa, Saa2 or Cscl1) until the end of the 40-week exposure. Instead, TCE suppressed hepatic expression of genes involved in IL-6 signaling (Il6r, gp130, and Egr1). Linear regression analysis confirmed liver histopathology in the TCE-treated mice correlated with decreased expression of Il6r. A toxicodynamic model was developed to estimate the effects of TCE on IL-6 signaling and liver pathology under different levels of exposure and rates of repair. This study underlined the importance of longitudinal studies in mechanistic evaluations of immuntoxicants. It showed that later-occurring liver pathology caused by TCE was associated with early suppression of hepatoprotection rather than an increase in conventional pro-inflammatory events. This information was used to create a novel toxicodynamic model of IL-6-mediated TCE-induced liver inflammation.
Keywords: Trichloroethylene, liver, IL-6, immunotoxicity, modeling
Introduction
Trichloroethylene (TCE) is a chlorinated hydrocarbon that has been used as a degreasing agent since the 1920s. Because of inappropriate disposal over the years TCE is now a common pollutant at Superfund toxic waste sites and at many industry and government facilities. It is found in soil and surface water as a result of direct discharges and in groundwater due to leaching from disposal operations. As noted by a recent National Research Council report evidence on human health hazards from TCE exposure, either occupational or environmental, has strengthened in recent years (Committee on Human Health Risks of Trichloroethylene, 2006). One of the predominant non-cancer outcome associated with TCE exposure in humans is immunotoxicity, most notably the development of hypersensitivity responses including autoimmune scleroderma and autoimmune liver diseases (Byers et al., 1988; Yanez Diaz et al., 1992; Hansen et al., 1988; Saihan et al., 1978; Flindt-Hansen et al., 1987; Czirjak et al., 1994; Lockey et al., 1997; Dubrow et al., 1987; Gist et al., 1995). TCE is still widely used in Asia, where it has become a serious work-related health concern due to the induction of dermal and systemic hypersensitivity diseases often accompanied by non-viral, immune-mediated hepatitis (Kamijima et al., 2008).
Using a mouse model we and others have found that long-term exposure to TCE in drinking water at concentrations lower than sanctioned occupational exposure levels generated a T cell-mediated liver disease commensurate with human idiopathic autoimmune hepatitis (AIH)(Griffin et al., 2000; Gilbert et al., 2008; Cai et al., 2008). This TCE-induced liver inflammation was not usually accompanied by markers of acute liver injury such as increased blood levels of alanine transaminase or liver fibrosis, but was associated with the development of antibodies specific for liver microsomal proteins similar to those in patients with type 2 AIH.
The development of toxicant-induced immune pathology such as the autoimmune hepatitis caused by TCE exposure is almost certainly a complex multifactorial process. Developing conceptual models can be a way to delineate and quantify the contribution of different toxicant-induced alterations to the actual pathology. As a first step in this direction a model was developed here to describe a specific part of the process, namely IL-6-mediated liver events. IL-6 is one of the most important regulators of hepatic inflammation. The pathogenesis of AIH requires circumvention of the well-known propensity of the liver to induce T cell tolerance (Carambia et al., 2010). Pre-existing inflammation in the liver may subvert its tolerogenicity and help sustain an immune response by entering T cells (Crispe, 2009). The ability of toxicant exposure to generate such inflammation depends on opposing forces of tissue injury and tissue repair. Distress signals triggered during initiation of toxicant-induced liver injury (e.g. lipid peroxidation, reactive intermediate formation) can promote inflammation. However, they also stimulate protective (anti-apoptotic) and regenerative (cell division) mechanisms in the liver. One of the mechanisms that determine whether toxicant exposure ultimately leads to tissue repair or to injury-induced inflammation is regulated by IL-6.
Treatments to prevent or reverse immunological liver injury in mouse models have been associated with an increase in liver expression of Il6 (Liu et al., 2006). Disruption of IL-6, or its receptors IL-6R or Gp130, has been shown to promote liver inflammation and/or mortality following partial hepatectomy (Wuestefeld et al., 2003), ethanol-induced liver disease (Gao, 2012), carbon tetrachloride-induced liver necrosis (Bansal et al., 2005), obesity-associated insulin resistance (Wunderlich et al., 2010), autoimmune cholangitis (Zhang et al., 2010), and Con A-induced hepatitis (Lutz et al., 2012). Thus, IL-6 appears to prevent immunological liver injury. In addition to its documented ability to promote liver regeneration and/or protection in the face of damage or trauma IL-6 also appears to be required for normal liver maintenance. Liver weight and total DNA and protein contents were decreased 26–28% in older (5–10month-old) female IL-6-deficient mice as compared to age-matched wild-type controls (Wallenius et al., 2001). This suggests that IL-6 is needed for normal hepatocyte turnover, and that over time a loss of this cytokine is detrimental to liver function.
In an attempt to define why TCE-induced autoimmunity targets the liver, mice exposed to a single dose of TCE for 4, 10, 16, 22, 28, 34 or 40 weeks were evaluated in the current study for time-dependent alterations in IL-6 as well as other pro-inflammatory mediators. This was complemented by a second study that examined the dose-dependent effects of TCE on these mediators at a single time point.
The development of autoimmune hepatitis in our mouse model of TCE exposure involves alterations in both the liver and the immune system. This multi-factorial process mimics the complicated etiologies of human autoimmune diseases. Developing conceptual models can be a way to delineate and quantify the contribution of different disease-induced alterations to actual pathology. As a first step in this direction the results obtained here were used to model the part of the TCE-induced disease process revealed in the current study, namely the effect of TCE on IL-6-mediated liver events. Taken together, the results suggest that later-occurring TCE-induced liver damage was due to an early decrease in IL-6-mediated hepatoprotection rather than an increase in pro-inflammatory events.
Materials and Methods
Mouse treatment
Eight week-old female MRL+/+ mice (Jackson Laboratories; Bar Harbor, ME) were housed in polycarbonate ventilated cages and provided with drinking water (ultrapure from Milli-Q Integral Water Purification System, Millipore) ad libitum. TCE (purity 99± %; Aldrich Chemical Co. Inc.; Milwaukee, WI) was suspended in drinking water with 1% emulsifier Alkamuls EL-620 from Rhone-Poulenc (Cranbury, NJ). Freshly made TCE-containing drinking water was provided every 2–3 days. In one experiment the mice (12 mice/group) received either 0, 0.02, 0.1 mg/ml or 0.5 mg/ml TCE in their drinking water for 12 weeks. In a second experiment the mice (8–9 mice/group) received 0 or 0.5 mg/ml TCE in their drinking water for 4, 10, 16, 22, 28, 34 or 40 weeks. The mice were weighed weekly and water consumption was monitored. All studies were approved by the Animal Care and Use Committee at the University of Arkansas for Medical Sciences. When the mice were sacrificed at the different time points adherent macrophages isolated from pooled peritoneal exudates from 2–3 mice (n=3–4/treatment group) were incubated for 20 hours in the presence or absence of LPS (1 μg/ml). Approximately 80% of adherent peritoneal exudate cells (PEC), regardless of treatment group, expressed the transmembrane protein F4/80, a marker of mature macrophages (data not shown). Culture supernatants from the peritoneal macrophages were then collected for cytokine evaluation. RLT Lysis Buffer (Qiagen Sciences, Germantown, MD) was then added directly to the remaining adherent cells before freezing for subsequent qRT-PCR analysis. Liver tissue harvested at the time of sacrifice was stained with H&E and examined for liver pathology. Liver and sections were examined microscopically and scored in a blinded manner by a veterinary pathologist for the severity of inflammation and fibrosis based on a four point scale (0 – 3), ranging from no change to severe, respectively) as described (Gilbert et al., 2008). Some liver tissue was instead frozen for subsequent qRT-PCR analysis.
qRT-PCR
Fluorescence-based quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) was conducted using RNA isolated (using RNeasy, Qiagen, Germantown, MD) from peritoneal macrophages or liver. Details of the technique as well as quality controls and rationale for choice of reference gene Eef2 (eukaryotic translation elongation factor 2) has been described (Gilbert et al., 2008).
Cytokine Analysis
The macrophage culture supernatants were examined using the Mouse IL-1β, IL-6 and TNF alpha Ready-Set-Go! ELISA kits from eBioscience, Inc. (San Diego, CA). The amounts of IL-6 produced by macrophages in the 12-week study were generally lower than those generated in the 40-week study. This is likely due to difference in the number of macrophages/well. In the 12-week study pooled peritoneal cells from 3 mice were used to generate four 1ml wells (two unstimulated and two LPS-treated) for an n=4 per treatment group. With 8–9 mice/group in the 40-week study pooled peritoneal cells from 2–3 mice were used to generate two 1ml wells (one unstimulated and one LPS-treated) for an n=3 group.
Levels of IL-6R in the livers of individual mice were examined by Western blotting using goat IgG anti-mouse IL-6Rα (R&D Systems, Minneapolis, MN). The results were represented as mean ± standard deviation of the densitometric analysis of the IL-6R compared to reference protein GAPDH (glyceraldehyde 3-phosphate dehydrogenase) run in the same lane, and detected by rabbit IgG anti-GAPDH (Trevigen, Gaithersberg, MD).
Antibody Production
Using previously described methodology (Gilbert et al., 2008), microsomal liver protein (30 μg) obtained from an untreated MRL+/+ mouse was separated on 12% SDS-PAGE, electrotransferred onto nitrocellulose, and subsequently probed with pooled sera (1:500) obtained from control or TCE-treated MRL+/+ mice followed by HRP-conjugated polyclonal goat anti-mouse IgG (1:4,000). Densitometric analysis of mouse myeloma IgG run in adjoining lanes was used to normalize exposure times for the individual Western blots.
Statistics
The data are presented as means ± standard deviations. Assays were conducted using samples from 8–12 individual mice per treatment group or samples from equal numbers of pooled cells for n=3 or 4 per treatment group. The threshold for statistical significance was set at α = 0.05. Differences between experimental groups were tested first with analysis of variance (ANOVA), and where the F test was significant, subsequent pairwise contrasts were tested using a two-sample t-test. Homogeneity of variance between groups was tested using studentized Breusch-Pagan, and normality of residuals using Shapiro-Wilk. Where significant deviations from homoscedasticity or normality were observed, the non-parametric Kruskal-Wallis and Wilcoxon rank sum tests were applied instead of ANOVA/t-test. For experiments involving multiple timepoints, ordinal logistic regression models were fitted and the significance of TCE exposure was computed using a likelihood-ratio test; this is a two-factor generalization of Kruskal-Wallis. Linear regressions were fitted to evaluate the relationship between gene expression and histopathology, with statistical significance judged using an F test. Macrophage concentration and gene expression values were right-skewed, and therefore these data were log-transformed for statistical analyses.
Mechanistic Toxicodynamic Modeling
The effects of IL-6 signaling on liver events in TCE-treated mice were modeled to link changes in TCE mediated IL-6 signaling outcomes to the observed pathology following low-dose chronic exposure to TCE. The objective in developing a mathematical model for chronic, low dose exposure to TCE was to provide a means to quantitatively describe the role of IL-6 as a maintenance mechanism and predict downstream effects, such as changes in pathology, due to modifications of this IL-6 repair pathway. To this end, a time-dependent mathematical description of the health state of discrete volumes of liver (“liver units”) and the IL-6 and TCE-dependent transition between these health states was developed. Using in vivo results reported in this study, this toxicodynamic model will create a link between TCE exposure and the resulting histopathology. While not measurable in vivo, the state and number density of individual liver units serve as an intermediate measure to quantify the relationship between impaired cytokine signaling and the resulting autoimmune hepatitis.
Liver unit health state model
For the purposes of mathematical modeling, the characteristics of the liver units (LUs) were as follows: the entire liver comprises LUs, each of which is of equal volume; an LU is relatively small in volume compared to that of the entire liver, but consists of a large enough number of cells to be represented as a continuum; each LU exists in one of three health states: healthy (H), compromised (C), and inflamed (I), and is homogeneous with respect to its properties and health state; and the health state of the entire liver may be estimated through a number-weighted average of the health states of the constituent LUs. Table 1 lists the characteristics and assumptions for each mathematical state.
Table 1.
Description of LU states
| LU State | Distinguishing Characteristics | Relevant Pathology Score (PS) |
|---|---|---|
| Healthy (H) | Normal liver function, homeostatic levels of IL-6 and IL-6r | 1 |
| Compromised (C) | Intermediate state, events initiate inflammatory pathways which are normally countered by IL-6 signaling. | 2 |
| Inflamed (I) | Contains markers for the early stages of auto-immune hepatitis, including inflammation and lymphoplasmacytic portal infiltration | 4 |
The resulting transition between health states is described by
| (1) |
where the k’s represent transition rates, the subscripts DAM and REP refer to damage- and repair-associated phenomena, respectively, and the subscripts H-C and C-I refer to their respective transition pathways.
The corresponding system of differential equations governing the time-dependent fraction of LUs in each state may be written as
| (2) |
Here, t is time and [H], [C], and [I] are the fractions of LUs in the healthy, compromised, and inflamed states, respectively. It is assumed that initially (t=0), [H]=1 and [C]=[I]=0.
To complete the mathematical description of this system, four major assumptions were made:
LUs normally exist in a state of IL-6-mediated hepatocyte turnover and protection.
Events such as TCE exposure can initiate inflammatory processes and move the LUs into the “C” state. However, protective mechanisms mediated by IL-6 normally restore the LUs from the “C” to the “H” state. The rates of repair, kREP,H-C and kREP,C-I, are dependent on the fraction of IL-6 produced from the macrophage and the IL-6r expressed by the hepatocyte. If homeostasis levels of IL-6 and IL-6r are present, these pathways operate at the optimal repair rates.
TCE initiates inflammatory processes (e.g. redox disequilibrium) that move the LUs from the “H” to “C” state. It also decreases the protective effects of IL-6-signaling that would normally restore the LUs to the “H” state. These dual effects of TCE allow the inflammatory processes to progress and to move the LUs from the “C” to the “I” state.
Autoimmune hepatitis pathology does not occur without TCE, even if the IL-6 pathway is impaired in some other way.
Consistent with these assumptions, the rate terms above can be further specified as follows:
| (3) |
where k1, k2, k3, and k4 are constants to be estimated using experimental data, fTCE represents the administered TCE dose normalized by the upper dose used in this study (0.5 mg/ml or a time-weighted average of approximately 54 mg/kg/day), and fIL6 is the fraction of IL-6 expressed by the macrophage compared to control levels. To express the dependence of fIL6 on TCE dose, a sub-model based on a saturation mechanism was used:
| (4) |
where α and β are constants to be derived from experimental data.
Predicting liver pathology scores
To compute overall liver pathology scores, the [H], [C], and [I] calculated from equations (2), (3), and (4) at the desired time point were used as weighting factors for the individual PS values corresponding to each of the model states. Mathematically, this can be expressed as
| (5) |
where PSs is the pathology score of a LU in state s (see Table 1).
Software and modeling tools
The system of differential equations were solved using a fourth-order Runge-Kutta method implemented in the Python programming language (v2.7.6) [https://www.python.org/]. Parameter estimation was conducted using lsqfit (v4.6.1) [https://github.com/gplepage/lsqfit], a software package for non-linear least-squares fitting of noisy data.
Results
Dose-dependent effects of TCE on peritoneal macrophage activity
Since autoimmune diseases and hypersensitivity disorders in humans involve an ill-defined genetic component, we use young “autoimmune-prone” female MRL+/+ mice to study the immunotoxicity of TCE. As observed previously, TCE exposure did not alter weight gain or water consumption (data not shown). Peritoneal macrophages from the mice exposed to different concentrations of TCE for 12 weeks were examined for the production of macrophage-derived cytokines IL-6 and IL-1β. Macrophage secretion of IL-1β was unchanged by exposure to TCE (Figure 1). The peritoneal macrophages collected from control mice secreted low but measurable levels of IL-6 even in the absence of LPS. Stimulation with LPS increased IL-6 production in all groups. However, both LPS-dependent and LPS-independent IL-6 production was suppressed in a dose-dependent manner in peritoneal macrophages from mice treated for 12 weeks with TCE. For example, LPS-induced IL-6 production in mice exposed to 0.5 mg/ml TCE was 70% lower than that of controls.
Figure 1. TCE inhibits macrophage IL-6 production in dose-dependent manner.
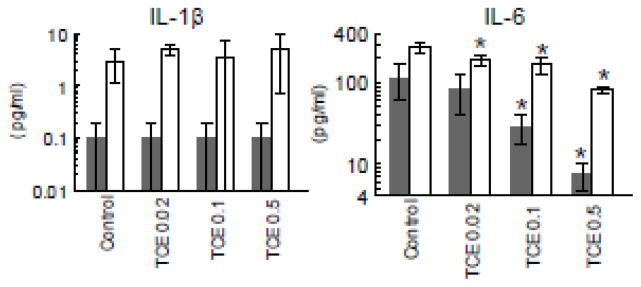
Peritoneal macrophages were incubated with (open bars) or without LPS (shaded bars) following isolation from untreated control mice or from mice exposed to TCE at different concentrations for 12 weeks. Culture supernatants were examined for cytokines (mean ± SD). *Significantly different (α<0.05) compared to control values.
IL-6 was also inhibited at the transcriptional level in macrophages from TCE-treated mice (Figure 2). Although LPS stimulation increased Il6 expression, this effect was significantly suppressed in macrophages from mice treated with 0.1 or 0.5 mg/ml TCE as compared to control mice. Once again the suppressive effects of TCE were confined to IL-6, and did not encompass expression of genes for other macrophage-derived cytokines, including Lt-α, IL-12, or IL-10. Taken together, a 12-week exposure to TCE selectively suppressed IL-6 gene expression and protein production by peritoneal macrophages in a dose-dependent manner. The ability of TCE to alter expression of genes for other macrophage-derived cytokines was intermittent and not dose-dependent.
Figure 2. TCE inhibits macrophage Il6 expression in dose-dependent manner.
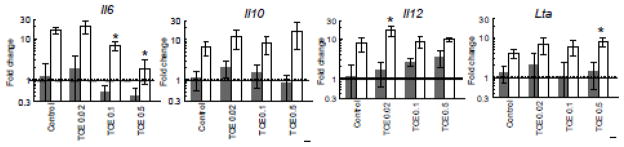
Cytokine gene expression was examined in peritoneal macrophages incubated with (open bars) or without LPS (shaded bars) after isolation from untreated control mice or from mice exposed to TCE for 12 weeks. The data represents the mean ± SD. *Significantly different (α<0.05) compared to control values.
Time-dependent effects of TCE on peritoneal macrophage gene expression
In a second study designed to examine time-dependency of TCE-induced effects mice were given drinking water alone or with 0.5 mg/ml TCE for 4, 10, 16, 22, 28, 34 or 40 weeks. TCE exposure did not alter the number of PEC recovered at any of the time points (data not shown). Once again TCE suppressed production of IL-6 (Figure 3). Also evident, but as yet unexplained, was the general time-dependent decrease in IL-6 production in both treatment and control groups. Production of TNF-α was not affected by TCE exposure. A longitudinal evaluation of cytokine gene expression showed that the TCE-induced decrease in Il6 expression by peritoneal macrophages was evident by 16 weeks of exposure (Figure 4). The time-dependent expression of several other genes for macrophage-derived cytokines, IL1b, Il12, and Mmp12 was for the most part unaltered by exposure to TCE (Figure 4 and data not shown). Thus, the primary effects of exposure to TCE on peritoneal macrophages was a decrease in Il6 that was maintained for the duration of the study.
Figure 3. TCE inhibition IL-6 production is maintained over time.

Peritoneal macrophages were incubated with LPS following isolation from untreated control mice or from mice exposed to TCE (0.5 mg/ml) for up to 40 weeks. Culture supernatants were examined for cytokines (mean ± SD). *Significantly different (α<0.05) compared to control values.
Figure 4. TCE inhibition of Il6 expression is maintained over time.
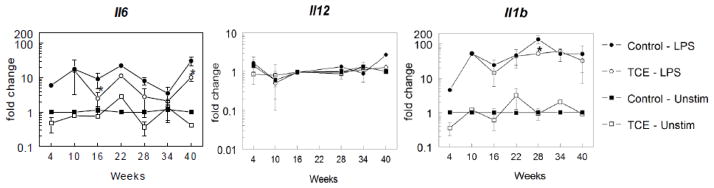
Cytokine gene expression was examined in peritoneal macrophages incubated with or without LPS after isolation from untreated control mice or from mice exposed to TCE (0.5 mg/ml) for up to 40 weeks. The data represents the mean ± SD. *Significantly different (α<0.05) compared to control values.
Time-dependent effects of TCE on liver events
Most of the protective and/or regenerative events in T cell-mediated liver injury are triggered by IL-6 signaling that is initiated when IL-6 binds to a complex comprised of the transmembrane protein gp130 and the IL-6R on hepatocytes (Klein et al., 2005). As shown in Figure 5 hepatic expression of Il6r was suppressed by TCE at several time points, and only approached control values at the last time point. Protein levels of IL-6R were also lower in the livers of the TCE-treated mice. The gene that encoded for the other subunit in the IL-6R family, Gp130, was suppressed by TCE at early time points. Expression of IL-6 itself in the liver was undetectable (data not shown). Another molecule important in hepatoprotection is the transcription factor EGR-1. EGR-1 binds to the promoter region of Il6 (Hoffmann et al., 2008), and reciprocally, is important in mediating signaling from the IL-6R/STAT3 pathway (Pritchard et al., 2011). Expression of egr1 in the liver was suppressed midway through the TCE exposure, but then rebounded at the final 40-week time point. Increased levels of pro-inflammatory cytokines/chemokines such as TNF-α, osteopontin, serum amyloid A (SAA) and CXCL1 have been implicated in the induction or progression of chronic liver inflammation (Iwamoto et al., 2013; Nagoshi, 2014; Gollaher et al., 1990; Zhang et al., 2012). Hepatic expression of these Saa2, Cxcl1 and Spp1 (encodes for osteopontin) were for the most part unchanged or decreased during all but the last 40-week time point of TCE exposure. Thus, unlike IL-6R associated genes hepatic expression of several pro-inflammatory cytokines and chemokines was primarily unchanged or decreased by TCE exposure until the last time point when expression was dramatically reversed in select TCE-treated mice. These results showed that during most of the exposure TCE appeared to negatively impact liver repair rather than directly promote inflammation. Only at the last time point was this reversed; several pro-inflammatory cytokines/chemokines increased expression while the negative effect on hepatoprotective genes was overturned.
Figure 5. TCE alters expression of hepatic genes over time.

A. Gene expression in individual liver tissue isolated from untreated control mice or from mice exposed to TCE (0.5 mg/ml) for up to 40 weeks. The data represents the mean ± SD from 6–8 individual mice/treatment/time point. *Significantly different (α<0.05) compared to control values. B. Relative protein levels (percentage reference protein GAPDH) of IL-6R in individual livers from untreated control mice or mice exposed to TCE (0.5 mg/ml) for 16 weeks (mean ± SD).
Histopathology in the form of lymphoplasmacytic portal infiltrate and lobular inflammation in the liver was not noted until week 28 of TCE exposure, and became more robust during the course of the 40-week experiment (Figure 6A). This pathology was characteristic of the early stages of autoimmune hepatitis; hepatocellular necrosis was only noted in a couple of instances. The mice were also examined for the generation of anti-liver antibodies as another readout of immune-mediated liver disease (Figure 6B). MRL+/+ mice are noted for their age-dependent increase in the production of autoantibodies such as anti-nuclear antibodies, even in the absence of toxicant exposure (Yoshida et al., 1989). In accord with this predisposition the baseline production of anti-liver antibodies became more abundant in control mice at the 40 week time point. However, exposure to TCE further increased the levels and diversity of the anti-liver antibodies. Thus, the MRL+/+ mice treated with TCE for 40 weeks demonstrated liver inflammation and anti-liver autoantibody production consistent with AIH.
Figure 6. TCE liver pathology correlates with loss of hepatic Il-6r expression.
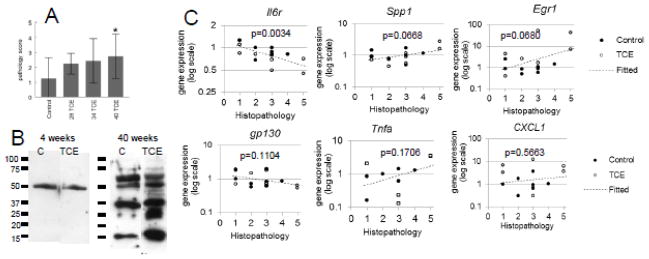
A. Liver pathology based on immune cell infiltration and inflammation was assessed in mice exposed to TCE (0.5mg/ml) for 28, 34 or40 weeks. B. Equal amounts of liver protein from an untreated mouse were separated in 4 lanes of SDS-PAGE, each of which were immunoblotted with pooled sera obtained from control MRL+/+ mice or mice treated with 0.5 mg/ml TCE for 4 or 40 weeks. C. Hepatic gene expression in from mice exposed to TCE (0.5 mg/ml) for 40 weeks was plotted against liver histopathology in the same mice. Gene expression values are shown in log scale because of right skewness. Regression p-values were computed using an F test of the null hypothesis of horizontal slope.
To help determine functional relevance hepatic gene expression in individual mice at the 40-week time point were plotted against liver pathology scores in the same mice. The linear regression showed that liver pathology was most closely correlated with a decrease in Il6r (p=0.003)(Figure 6C). Correlations between liver histopathology and expression of Egr1 and Spp1 were also moderately significant (p<0.07). There was no significant correlation between liver histopathology and expression of Tnfa or Cxcl1. This evaluation suggested that liver pathology was most closely correlated with a decrease in IL-6 signaling as reflected in a decrease in the expression of Il6r and Egr1 in the liver. There was also correlation with expression of Spp1, suggesting that the reversal of the TCE-induced decrease in OPN observed in the liver played a role in liver pathology.
Toxicodynamic model for liver response to TCE exposure
In order to develop a model to describe the effect of TCE on IL-6-mediated liver events certain required parameters were estimated based on the results described above.
Parameter estimation
In order to fit a curve that could be used to extrapolate IL-6 effects across a range of TCE doses values of α and β in the IL-6 submodel, Eq. (4), were estimated using a nonlinear least-squares approach with the non-LPS induced IL-6 results presented in Fig. 1. The resulting parameters values, mean (variance), were found to be α = 1.01 (0.01) and β = 0.071 (0.003). Figure 7A illustrates the resulting fit of the experimental data to the IL-6 submodel.
Figure 7. Submodel for parameter estimation.

A. An IL-6 submodel was developed for estimating dose-dependent reduction in the fraction of IL-6 expressed by the macrophage. Points and error bars represent data and uncertainty, while solid and dashed lines are the mean and 95% confidence intervals from model predictions. B. Time-course pathology scores were used to extrapolate liver pathology based on time of TCE exposure. Points and error bars represent data and uncertainty, while solid and dashed lines are the mean and 95% confidence intervals from model predictions.
It was similarly necessary to fit a curve to extrapolate liver pathology based on time of TCE exposure. The rate constants, ki, defined in Eq. (3), were estimated based on experimental time-course pathology scores (Figure 6A) to be k1 = 101.5 (98.0), k2 = 0.39 (0.18), k3 = 1.02 (0.08), and k4 = 0.21 (0.16). The resulting fit of the data to the mathematical model is depicted in Figure 7B. The uncertainty shown in model simulations results from both the uncertainty in the parameters associated with the IL-6 submodel and that from in vivo pathology scores.
Simulations of liver unit health states and the dose response
Following parameter estimation, simulations of time-course LU health were conducted. Figure 8 illustrates results from several such studies, where the fraction of LUs in a particular health state are shown as a function of time at the two highest doses used in the experimental study. For the 0.1 mg/ml dose (Figure 8A), almost all of the LUs are in a healthy state. However, as the external TCE dose is increased to 0.5 mg/ml (Figure 8B), the abundance of healthy LUs decreases while those in the compromised/inflamed state increase in a non-linear manner. At doses less than 0.1 mg/ml, simulations indicated that virtually all of the LUs were in the healthy (H) state over time.
Figure 8. Liver unit state predictions based on the model.
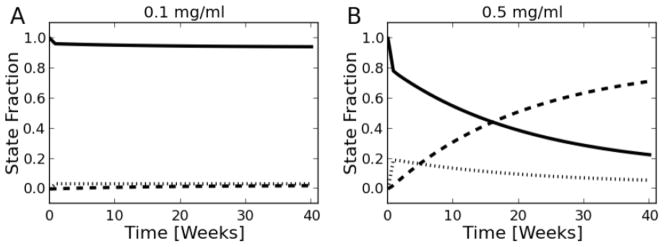
Fraction of liver units in each state for the 0.1 (A) and 0.5 (B) mg/ml experimental doses. This reflects the overall health of the liver rather than specific changes in IL-6 production. Solid lines represent the H state, while vertical and dashed lines correspond to the C and I states, respectively.
One of the benefits of the mathematical model is the prediction of system variables and endpoints not directly measured during the course of the studies. For instance, using 40-week pathology scores as an endpoint, the model was used to generate a dose-response curve (Figure 9). This curve can be used to relate this endpoint to any dose within the predicted range. As an example, for a benchmark response level (BML) corresponding to mild inflammation of 25% or less of the portal regions of the liver (PS=2), the benchmark dose (BMD) was estimated to be fTCE = 0.55, corresponding to a 0.27 mg/ml dose, or approximately 37 mg/kg/day, of continuous TCE exposure.
Figure 9. Dose response curve for current study.

Predicted dose response curves for pathology scores (PS) 40 weeks post TCE exposure. The mean values and 95% confidence intervals are shown as solid and dashed lines, respectively. The point represents the value of the benchmark dose (BMD) corresponding to the benchmark response level (BMR) described in the text.
Examining the impact of varying relative rates of damage and repair
Another important benefit to the mathematical modeling is the ability to vary system parameters and observe the effects on system states of interest. Here, the effects of varying the relative rates of damage and repair in the H-C and C-I state transitions were investigated through a parametric study. Since each damage and repair pathway is first order with respect to the abundance of LUs, ratios of rate constants were defined:
| (1) |
By conducting simulations with varying values for κ (Figure 10), the effects of relative rates of repair and damage in the system could be examined. If κ ≫ 1, the repair mechanism dominates and the LUs tend toward a relatively low value of PS, even at higher TCE dose; conversely, if κ ≪ 1, the damage mechanism dominates for the given pathway and LUs may acquire high values of PS values, even at relatively low doses. As can be seen, the shape and nonlinearity of the dose-response curve is highly dependent on the relative rates of repair and damage in both of the health state transitions. Overall, such predictions can help in the understanding of the interactions in this system and lend insight into the effects of non-TCE mediated events, such as additional stress from other hepatotoxicants or further impaired IL-6 repair mechanisms.
Figure 10. Simulations illustrate the effects of relative rates of repair and damage on liver damage.
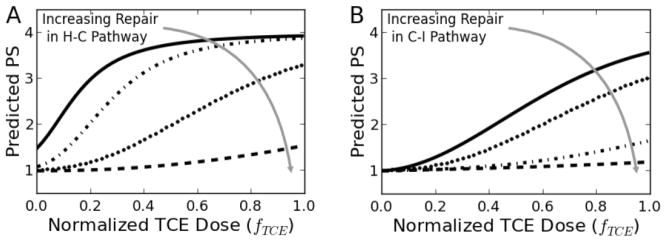
Model predictions for varying relative levels of repair in (A) the H-C pathway (κH-C=[1, 10, 100, 1000], κC-I=1) and (B) the C-I pathway (κH-C=100, κC-I=[0.1, 1, 10, 100]). κH-C=100 and κC-I=1 are the values that that relate to the pathology scores from the current study. The two dotted lines represent the kappas (k) used in this study (k1=100 and k2=1). Dashed lines are representative of hypothetical pathways with an order of magnitude increase in repair mechanisms. Conversely, solid lines represent scenarios where external factors result in an order of magnitude decrease in repair.
DISCUSSION
MRL+/+ mice can spontaneously develop autoimmune diseases such as lupus nephritis, pancreatitis, and Sjogren’s syndrome late in life (after 1-year of age)(Kanno et al., 1992; Toda et al., 1999). However, before they reach one year of age most female MRL+/+ mice do not exhibit autoimmune tissue pathology, and are often used to examine the autoimmune-promoting capacity of a toxicant such as TCE. Based on water consumption and TCE degradation in the water bottles, the mice given water containing TCE at 0.02, 0.1 or 0.5 mg/ml for 12 weeks were exposed to TCE at time-weighted levels of approximately 3, 14 or 64 mg/kg/day, respectively. Even the highest exposure is lower than the current 8-hour Permissible Exposure Limit [established by the Occupational Safety and Health Administration (OSHA)] for TCE of 100 ppm or approximately 76 mg/kg/day.
The effects of TCE on macrophage activity have been primarily studied in inhalation models. Inhalation exposure to TCE was shown to increase susceptibility to respiratory bacterial infection in mice, and to suppress phagocytosis in lung macrophages (Selgrade et al., 2010). Similarly, multiple inhalation exposures to TCE lowered resistance to respiratory streptococcus infection (Aranyi et al., 1986). Although the mechanism for this suppressive effect of inhaled TCE on macrophages was not defined, others have shown that an IL-6 deficiency increases susceptibility to viral and bacterial respiratory infections (Murphy et al., 2008; Jones et al., 2006). The results of the current study showed that oral exposure to TCE suppressed IL-6 at the level of protein production and gene expression in macrophages.
IL-6 is a pleiotropic cytokine, which can make it difficult to predict the cumulative impact of its altered production. Elevated levels of IL-6 in the blood have been observed in a number of pathological conditions associated with chronic inflammation including rheumatoid arthritis (Gottenberg et al., 2012), systemic lupus erythematosus (Chun et al., 2007), and active disease in Guillain-Barre syndrome (Weller et al., 1991). IL-6 did not reach detectable levels in the blood of control or TCE-treated mice in the current study. Circulating levels of IL-6 are increased in children with AIH type 1, but not with AIH type 2 (Maggiore et al., 1995), the type of AIH that most closely resembles TCE-induced disease in MRL+/+ mice. Some studies of idiopathic autoimmune liver disease in humans have found increased levels of IL-6 in liver biopsies (Zhao et al., 2011), while other studies of autoimmune hepatitis have demonstrated decreased expression of hepatic Il6 in the liver (Tovey et al., 1991). On the other hand, treatments to prevent or reverse immunological liver injury in mouse models have been associated with an increase in liver expression of Il6 (Liu et al., 2006). Thus, the majority of studies suggest that in the liver IL-6 is primarily protective. Increases in hepatic levels of IL-6 in some humans with AIH may represent a compensatory rather than pathological mechanism. Alternatively, changes in IL-6 may be specific for a certain stage of disease development, type of autoimmune hepatitis (e.g. type 1 vs type 2) (Maggiore et al., 1995), or cell type (e.g. peritoneal exudate macrophages vs Kupffer cells). Deletion of IL-6 in mice deficient in TGF-β receptor II improved colitis but exacerbated autoimmune cholangitis in association with increased numbers of activated T cells (Zhang et al., 2010).
Cytokine production by macrophages from MRL+/+ mice is reportedly aberrant even in the absence of TCE exposure. LPS-induced production of IL-6, IL-1β, TNF-α, and IL-12 by macrophages from untreated MRL+/+ mice were all significantly decreased in comparison to macrophages from C57BL/6, BALB/c or A/J mice(Hartwell et al., 1995; Alleva et al., 2000). Of these macrophage-derived cytokines only IL-6 was found in the present study to be further decreased by TCE exposure.
In addition to a decrease in macrophage-derived IL-6, TCE suppressed liver expression of Il6r and gp130, the dual components of the IL-6R. This TCE-induced decrease would seem to further ensure the lack of IL-6 signaling in the liver. The IL-6-induced liver protection to T cell-mediated liver injury has been attributed to a downstream increase in acute phase protein serum amyloid A2, (SAA2)(Klein et al., 2005). TCE suppressed hepatic expression of Saa2 at two time points late in the exposure period, thus seeming to prevent the upregulation of this molecules needed for liver regeneration. Egr1 is a transcription factor required for wound healing, and which has been identified as a negative regulator of carbon tetrachloride-induced hepatotoxicity (Pritchard et al., 2010). Egr1 has been described as both a trigger and a target for IL-6 (Zhang et al., 2013; Maekawa et al., 2010). Only at the final time point did TCE increase expression of Egr1 and Saa2. It is not known why the earlier TCE-induced suppression was reversed, but presumably the late recovery of these genes was not sufficient to protect against liver damage.
The contribution of TCE to AIH in the present model is multidimensional; the healthy-to-inflamed state model described here can be amended to include more immune parameters such as the contribution of CD4+ T cells as they are characterized. However, even in its present state, the model facilitated point-of-departure predictions based on dose-dependent changes in liver pathology. The model stemmed from the linear regression analyses showing that liver pathology in TCE-treated mice was best correlated with the decreased liver expression of macrophage Il-6r. We now have the tools to predict liver pathology based on relative rates of liver repair and damage. In addition to its predicted effect on IL-6 signaling the model also infers that TCE initiates inflammatory processes that transition LUs from “H” to “C”. These processes were not investigated in this study, but probably include, but are not restricted to, alterations in redox equilibrium. In a previous study, a metabolomics analysis following chronic 32 week exposure to 0.5 mg/ml in MRL+/+ mice revealed significant alterations in several metabolites (e.g., cystathionine) involved in the generation of glutathione, which functions as the major intracellular antioxidant against oxidative stress and plays an important role in the detoxification of reactive oxygen species and subsequent oxidative damage from pro-oxidant environmental exposures. Others have shown the functional significance of oxidative stress in TCE-induced liver pathology (Wang et al., 2007; Wang et al., 2013). IL-6 has been shown to inhibit oxidative stress and steatosis in the liver (El-Assal et al., 2004). Consequently, a TCE-induced loss of IL-6 signaling in the liver would be expected to exacerbate associated oxidative-stress and resulting inflammation. The first stage model development described here (i.e. generation of equations and description of parameters) was based on data from two different experiments, albeit with some differences in experimental design. Obtaining new data to validate and extend this model will be included in the design of future chronic TCE exposure studies.
Highlights.
We developed a toxicodynamic model to study effects of trichloroethylene on liver.
We examined protective as well as pro-inflammatory events in the liver.
Trichloroethylene inhibits IL-6 production by macrophages.
Trichloroethylene inhibits components of the IL-6R in the liver.
Trichloroethylene inhibits events associated with IL-6-mediated hepatoprotection.
Acknowledgments
Funding
This work was supported by grants to Dr. K. Gilbert from the Arkansas Biosciences Institute, the National Institutes of Health (R01ES017286, R01ES021484-02), and the Organic Compounds Property Contamination class action settlement (CV 1992-002603).
We would like to gratefully acknowledge the excellent technical assistance of Brannon Broadfoot, Kirk West, Rachel Lee and the UAMS Translational Research Institute (National Institutes of Health UL1RR029884).
Abbreviations
- TCE
trichloroethylene
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Brad Reisfeld, Email: brad.reisfeld@colostate.edu.
Todd Zurlinden, Email: tjzurlin@rams.colostate.edu.
Meagan N. Kreps, Email: MNKreps@uams.edu.
Stephen W. Erickson, Email: serickson@uams.edu.
Sarah J. Blossom, Email: blossomsarah@uams.edu.
Reference List
- 1.Alleva DG, Pavlovich RP, Grant C, Kaser SB, Beller DI. Aberrant macrophage cytokine production is a conserved feature among autoimmune-prone mouse strains: elevated interleukin (IL)-12 and an imbalance in tumor necrosis factor-alpha and IL-10 define a unique cytokine profile in macrophages from young nonobese diabetic mice. Diabetes. 2000;49:1106–1115. doi: 10.2337/diabetes.49.7.1106. [DOI] [PubMed] [Google Scholar]
- 2.Aranyi C, O’Shea WJ, Graham JA, Miller FJ. The effects of inhalation of organic chemical air contaminants on murine lung host defenses. Fundam Appl Toxicol. 1986;6:713–720. doi: 10.1016/0272-0590(86)90184-3. [DOI] [PubMed] [Google Scholar]
- 3.Bansal MB, Kovalovich K, Gupta R, Li W, Agarwal A, Radbill B, Alvarez CE, Safadi R, Fiel MI, Friedman SL, Taub RA. Interleukin-6 protects hepatocytes from CCl4-mediated necrosis and apoptosis in mice by reducing MMP-2 expression. J Hepatol. 2005;42:548–556. doi: 10.1016/j.jhep.2004.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Byers VS, Levin AS, Ozonoff DM, Baldwin RW. Association between clinical symptoms and lymphocyte abnormalities in a population with chronic domestic exposure to industrial solvent-contaminated domestic water supply and a high incidence of leukemia. Cancer Immunol Immunother. 1988;27:77–82. doi: 10.1007/BF00205762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cai P, Konig R, Boor PJ, Kondraganti S, Kaphalia BS, Khan MF, Ansari GA. Chronic exposure to trichloroethene causes early onset of SLE-like disease in female MRL +/+ mice. Toxicol Appl Pharmacol. 2008;228:68–75. doi: 10.1016/j.taap.2007.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carambia A, Herkel J. CD4 T cells in hepatic immune tolerance. J Autoimmun. 2010;34:23–28. doi: 10.1016/j.jaut.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 7.Chun HY, Chung JW, Kim HA, Yun JM, Jeon JY, Ye YM, Kim SH, Park HS, Suh CH. Cytokine IL-6 and IL-10 as biomarkers in systemic lupus erythematosus. J Clin Immunol. 2007;27:461–466. doi: 10.1007/s10875-007-9104-0. [DOI] [PubMed] [Google Scholar]
- 8.Committee on Human Health Risks of Trichloroethylene, N. R. C. Assessing the Human Health Risks of Trichloroethylene: Key Scientific Issues. The National Academies Press; 2006. [Google Scholar]
- 9.Crispe IN. The liver as a lymphoid organ. Annu Rev Immunol. 2009;27:147–63. doi: 10.1146/annurev.immunol.021908.132629. [DOI] [PubMed] [Google Scholar]
- 10.Czirjak L, Pocs E, Szegedi G. Localized scleroderma after exposure to organic solvents. Dermatology. 1994;189:399–401. doi: 10.1159/000246888. [DOI] [PubMed] [Google Scholar]
- 11.Dubrow R, Gute DM. Cause-specific mortality among Rhode Island jewelry workers. Am J Ind Med. 1987;12:579–593. doi: 10.1002/ajim.4700120511. [DOI] [PubMed] [Google Scholar]
- 12.El-Assal O, Hong F, Kim WH, Radaeva S, Gao B. IL-6-deficient mice are susceptible to ethanol-induced hepatic steatosis: IL-6 protects against ethanol-induced oxidative stress and mitochondrial permeability transition in the liver. Cell Mol Immunol. 2004;1:205–211. [PubMed] [Google Scholar]
- 13.Flindt-Hansen H, Isager H. Scleroderma after occupational exposure to trichloroethylene and trichlorethane. Toxicol Lett. 1987;95:173–181. [PubMed] [Google Scholar]
- 14.Gao B. Hepatoprotective and anti-inflammatory cytokines in alcoholic liver disease. J Gastroenterol Hepatol. 2012;27(Suppl 2):89–93. doi: 10.1111/j.1440-1746.2011.07003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gilbert KM, Przybyla B, Pumford NR, Han T, Fuscoe J, Schnackenberg LK, Holland RD, Doss JC, MacMillan-Crow LA, Blossom SJ. Use of transcriptomics and metabolomics to delineate liver events associated with trichloroethylene-induced autoimmune hepatitis. Chem Res Tox. 2008;22:626–632. doi: 10.1021/tx800409r. [DOI] [PubMed] [Google Scholar]
- 16.Gist GL, Burg JR. Trichloroethylene--a review of the literature from a health effects perspective. Toxicol Ind Health. 1995;11:253–307. doi: 10.1177/074823379501100301. [DOI] [PubMed] [Google Scholar]
- 17.Gollaher CJ, Bausserman LL. Hepatic catabolism of serum amyloid A during an acute phase response and chronic inflammation. Proc Soc Exp Biol Med. 1990;194:245–250. doi: 10.3181/00379727-194-43086. [DOI] [PubMed] [Google Scholar]
- 18.Gottenberg JE, Dayer JM, Lukas C, Ducot B, Chiocchia G, Cantagrel A, Saraux A, Roux-Lombard P, Mariette X. Serum IL-6 and IL-21 are associated with markers of B cell activation and structural progression in early rheumatoid arthritis: results from the ESPOIR cohort. Ann Rheum Dis. 2012;71:1243–1248. doi: 10.1136/annrheumdis-2011-200975. [DOI] [PubMed] [Google Scholar]
- 19.Griffin JM, Gilbert KM, Lamps LW, Pumford NR. CD4+ T cell activation and induction of autoimmune hepatitis following trichloroethylene treatment in MRL+/+ mice. Toxicol Sci. 2000;57:345–352. doi: 10.1093/toxsci/57.2.345. [DOI] [PubMed] [Google Scholar]
- 20.Hansen BL, Isager H. A scleroderma-resembling disease-exposure to trichloroethylene and trichloroethane, is there a causal connection? Ugeskr Laeger. 1988;150:805–808. [PubMed] [Google Scholar]
- 21.Hartwell DW, Fenton MJ, Levine JS, Beller DI. Aberrant cytokine regulation in macrophages from young autoimmune-prone mice: evidence that the intrinsic defect in MRL macrophage IL-1 expression is transcriptionally controlled. Mol Immunol. 1995;32:743–751. doi: 10.1016/0161-5890(95)00036-e. [DOI] [PubMed] [Google Scholar]
- 22.Hoffmann E, Ashouri J, Wolter S, Doerrie A, Dittrich-Breiholz O, Schneider H, Wagner EF, Troppmair J, Mackman N, Kracht M. Transcriptional regulation of EGR-1 by the interleukin-1-JNK-MKK7-c-Jun pathway. J Biol Chem. 2008;283:12120–12128. doi: 10.1074/jbc.M800583200. [DOI] [PubMed] [Google Scholar]
- 23.Iwamoto S, Kido M, Aoki N, Nishiura H, Maruoka R, Ikeda A, Okazaki T, Chiba T, Watanabe N. TNF-alpha is essential in the induction of fatal autoimmune hepatitis in mice through upregulation of hepatic CCL20 expression. Clin Immunol. 2013;146:15–25. doi: 10.1016/j.clim.2012.10.008. [DOI] [PubMed] [Google Scholar]
- 24.Jones MR, Quinton LJ, Simms BT, Lupa MM, Kogan MS, Mizgerd JP. Roles of interleukin-6 in activation of STAT proteins and recruitment of neutrophils during Escherichia coli pneumonia. J Infect Dis. 2006;193:360–369. doi: 10.1086/499312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kamijima M, Wang H, Huang H, Li L, Shibata E, Lin B, Sakai K, Liu H, Tsuchiyama F, Chen J, Okamura A, Huang X, Hisanaga N, Huang Z, Ito Y, Takeuchi Y, Nakajima T. Trichloroethylene causes generalized hypersensitivity skin disorders complicated by hepatitis. J Occup Health. 2008;50(4):328–338. doi: 10.1539/joh.l8013. [DOI] [PubMed] [Google Scholar]
- 26.Kanno H, Nose M, Itoh J, Taniguchi Y, Kyogoku M. Spontaneous development of pancreatitis in the MRL/Mp strain of mice in autoimmune mechanism. Clin Ex Immunol. 1992;89:68–73. doi: 10.1111/j.1365-2249.1992.tb06879.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klein C, Wustefeld T, Assmus U, Roskams T, Rose-John S, Muller M, Manns MP, Ernst M, Trautwein C. The IL-6-gp130-STAT3 pathway in hepatocytes triggers liver protection in T cell-mediated liver injury. J Clin Invest. 2005;115:860–869. doi: 10.1172/JCI200523640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu DF, Wei W, Song LH. Protective effect of paeoniflorin on immunological liver injury induced by bacillus Calmette-Guerin plus lipopolysaccharide: modulation of tumour necrosis factor-alpha and interleukin-6 MRNA. Clin Exp Pharmacol Physiol. 2006;33:332–339. doi: 10.1111/j.1440-1681.2006.04371.x. [DOI] [PubMed] [Google Scholar]
- 29.Lockey JE, Kelly CR, Cannon GW, Colby TV, Aldrich V, Livingston GK. Progressive systemic sclerosis associated with exposure to trichloroethylene. J Occup Med. 1997;29:493–496. [PubMed] [Google Scholar]
- 30.Lutz HH, Sackett SD, Kroy DC, Gassler N, Trautwein C. Deletion of gp130 in myeloid cells modulates IL-6-release and is associated with more severe liver injury of Con A hepatitis. Eur J Cell Biol. 2012;91:576–581. doi: 10.1016/j.ejcb.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 31.Maekawa T, Takahashi N, Honda T, Yonezawa D, Miyashita H, Okui T, Tabeta K, Yamazaki K. Porphyromonas gingivalis antigens and interleukin-6 stimulate the production of monocyte chemoattractant protein-1 via the upregulation of early growth response-1 transcription in human coronary artery endothelial cells. J Vasc Res. 2010;47:346–354. doi: 10.1159/000265568. [DOI] [PubMed] [Google Scholar]
- 32.Maggiore G, De BF, Massa M, Pignatti P, Martini A. Circulating levels of interleukin-6, interleukin-8, and tumor necrosis factor-alpha in children with autoimmune hepatitis. J Pediatr Gastroenterol Nutr. 1995;20:23–27. doi: 10.1097/00005176-199501000-00005. [DOI] [PubMed] [Google Scholar]
- 33.Murphy EA, Davis JM, Brown AS, Carmichael MD, Ghaffar A, Mayer EP. Effect of IL-6 deficiency on susceptibility to HSV-1 respiratory infection and intrinsic macrophage antiviral resistance. J Interferon Cytokine Res. 2008;28:589–595. doi: 10.1089/jir.2007.0103. [DOI] [PubMed] [Google Scholar]
- 34.Nagoshi S. Osteopontin: Versatile modulator of liver diseases. Hepatol Res. 2014;44:22–30. doi: 10.1111/hepr.12166. [DOI] [PubMed] [Google Scholar]
- 35.Pritchard MT, Malinak RN, Nagy LE. Early growth response (EGR)-1 is required for timely cell-cycle entry and progression in hepatocytes after acute carbon tetrachloride exposure in mice. Am J Physiol Gastrointest Liver Physiol. 2011;300:G1124–G1131. doi: 10.1152/ajpgi.00544.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pritchard MT, Nagy LE. Hepatic fibrosis is enhanced and accompanied by robust oval cell activation after chronic carbon tetrachloride administration to Egr-1-deficient mice. Am J Pathol. 2010;176:2743–2752. doi: 10.2353/ajpath.2010.091186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saihan EM, Burton JL, Heaton KW. A new syndrome with pigmentation, scleroderma, gynaecomastia, Raynaud’s phenomenon and peripheral neuropathy. Br J Dermatol. 1978;99:437–440. doi: 10.1111/j.1365-2133.1978.tb06184.x. [DOI] [PubMed] [Google Scholar]
- 38.Selgrade MK, Gilmour MI. Suppression of pulmonary host defenses and enhanced susceptibility to respiratory bacterial infection in mice following inhalation exposure to trichloroethylene and chloroform. J Immunotoxicol. 2010;7:350–356. doi: 10.3109/1547691X.2010.520139. [DOI] [PubMed] [Google Scholar]
- 39.Toda I, Sullivan BD, Rocha EM, Da Silveira LA, Wickham LA, Sullivan DA. Impact of gender on exocrine gland inflammation in mouse models of Sjogren’s syndrome. Exp Eye Res. 1999;69:355–366. doi: 10.1006/exer.1999.0715. [DOI] [PubMed] [Google Scholar]
- 40.Tovey MG, Gugenheim J, Guymarho J, Blanchard B, Vanden Broecke C, Gresser I, Bismuth H, Reynes M. Genes for interleukin-1, interleukin-6, and tumor necrosis factor are expressed at markedly reduced levels in the livers of patients with severe liver disease. Autoimmunity. 1991;10:297–310. doi: 10.3109/08916939109001904. [DOI] [PubMed] [Google Scholar]
- 41.Wallenius V, Wallenius K, Hisaoka M, Sandstedt J, Ohlsson C, Kopf M, Jansson JO. Retarded liver growth in interleukin-6-deficient and tumor necrosis factor receptor-1-deficient mice. Endocrinology. 2001;142:2953–2960. doi: 10.1210/endo.142.7.8270. [DOI] [PubMed] [Google Scholar]
- 42.Wang G, Cai P, Ansari GA, Khan MF. Oxidative and nitrosative stress in trichloroethene-mediated autoimmune response. Toxicology. 2007;229:186–193. doi: 10.1016/j.tox.2006.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang G, Wang J, Ma H, Ansari GA, Khan MF. N-Acetylcysteine protects against trichloroethene-mediated autoimmunity by attenuating oxidative stress. Toxicol Appl Pharmacol. 2013;273:189–195. doi: 10.1016/j.taap.2013.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weller M, Stevens A, Sommer N, Melms A, Dichgans J, Wietholter H. Comparative analysis of cytokine patterns in immunological, infectious, and oncological neurological disorders. J Neurol Sci. 1991;104:215–221. doi: 10.1016/0022-510x(91)90313-v. [DOI] [PubMed] [Google Scholar]
- 45.Wuestefeld T, Klein C, Streetz KL, Betz U, Lauber J, Buer J, Manns MP, Muller W, Trautwein C. Interleukin-6/glycoprotein 130-dependent pathways are protective during liver regeneration. J Biol Chem. 2003;278:11281–11288. doi: 10.1074/jbc.M208470200. [DOI] [PubMed] [Google Scholar]
- 46.Wunderlich FT, Strohle P, Konner AC, Gruber S, Tovar S, Bronneke HS, Juntti-Berggren L, Li LS, van RN, Libert C, Berggren PO, Bruning JC. Interleukin-6 signaling in liver-parenchymal cells suppresses hepatic inflammation and improves systemic insulin action. Cell Metab. 2010;12:237–249. doi: 10.1016/j.cmet.2010.06.011. [DOI] [PubMed] [Google Scholar]
- 47.Yanez Diaz S, Moran M, Unamuno P, Armijo M. Silica and trichloroethylene-induced progressive systemic sclerosis. Dermatol. 1992;184:98–102. doi: 10.1159/000247513. [DOI] [PubMed] [Google Scholar]
- 48.Yoshida A, Hosokawa T, Nishi Y, Koyama K, Nakamura K, Marui N, Rokutan K, Aoike A, Kawai K. Studies on age-related functional changes in regulatory T cells and B cells involved in the autoantibody production of MRL/MpJ- +/+ mice. Mech Ageing Dev. 1989;50:179–192. doi: 10.1016/0047-6374(89)90013-4. [DOI] [PubMed] [Google Scholar]
- 49.Zhang J, Xie S, Ma W, Teng Y, Tian Y, Huang X, Zhang Y. A newly identified microRNA, mmu-miR-7578, functions as a negative regulator on inflammatory cytokines tumor necrosis factor-alpha and interleukin-6 via targeting Egr1 in vivo. J Biol Chem. 2013;288:4310–4320. doi: 10.1074/jbc.M112.351197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang W, Tsuda M, Yang GX, Tsuneyama K, Rong G, Ridgway WM, Ansari AA, Flavell RA, Coppel RL, Lian ZX, Gershwin ME. Deletion of interleukin-6 in mice with the dominant negative form of transforming growth factor beta receptor II improves colitis but exacerbates autoimmune cholangitis. Hepatology. 2010;52:215–222. doi: 10.1002/hep.23664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang Y, Chen L, Gao W, Hou X, Gu Y, Gui L, Huang D, Liu M, Ren C, Wang S, Shen J. IL-17 neutralization significantly ameliorates hepatic granulomatous inflammation and liver damage in Schistosoma japonicum infected mice. Eur J Immunol. 2012;42:1523–1535. doi: 10.1002/eji.201141933. [DOI] [PubMed] [Google Scholar]
- 52.Zhao L, Tang Y, You Z, Wang Q, Liang S, Han X, Qiu D, Wei J, Liu Y, Shen L, Chen X, Peng Y, Li Z, Ma X. Interleukin-17 contributes to the pathogenesis of autoimmune hepatitis through inducing hepatic interleukin-6 expression. PLoS One. 2011;6:e18909. doi: 10.1371/journal.pone.0018909. [DOI] [PMC free article] [PubMed] [Google Scholar]