

Abstract
AIM: To evaluate the effect of inflammatory cytokines on arylamine N-acetyltransferase 1 (NAT1), which is a phase-II enzyme involved in the biotransformation of aromatic and heterocyclic amines found in food, drugs and the environment.
METHODS: Human cholangiocarcinoma KKU-100 cells were treated with a mixture of proinflammatory cytokines (interferon-γ, interleukin-1β, and tumor necrosis factor-α) for 48 h, and the effect on NAT1 activity was assessed by high performance liquid chromatography, while NAT1 expression was determined by reverse-transcription polymerase chain reaction. The oxidative stress on the cells was examined by the formation of nitric oxide, superoxide anion and glutathione (GSH) levels. The cells were also treated with S-nitroso-glutathione (GSNO), a nitric oxide donor, to see if the responses were similar to those obtained with the inflammatory cytokines.
RESULTS: Cytokines suppressed NAT1 activity, reducing the Vmax without affecting the Km. Cytokines also had a significant impact on the induction of nitric oxide production and in reducing the redox ratios of glutathione (GSH) and GSH disulfide. Treatment with GSNO for 2-48 h reduced NAT1 activity without affecting the GSH ratio. Moreover, inflammatory cytokines and GSNO suppressed NAT1 mRNA expression.
CONCLUSION: These findings indicate an association between inflammation and suppression of NAT1, which perhaps contributes to chemical-mediated toxicity and carcinogenesis.
Keywords: Arylamine N-acetyltransferase 1, Phase II drug-metabolizing enzyme, Inflammatory cytokine, Oxidative stress, Cholangiocarcinoma
INTRODUCTION
Arylamine N-acetyltransferases (NATs) are well known polymorphic phase-II drug-metabolizing enzymes. Human NAT1 and NAT2 are encoded by two closely related NAT1 and NAT2 genes[1,2]. NAT1 mRNA and protein are expressed in a wide range of tissues, whereas NAT2 mRNA and protein are present mainly in the liver and the gastrointestinal tract[3]. Biliary epithelial cells do not express NAT2 but retain NAT1 activity[4].
NATs are important enzymes capable of acetylation reactions, which are involved in the detoxification and metabolic activation of various chemicals in drugs, food and the environment[5]. N-acetylation is usually considered a detoxification process, because it renders the nitrogen atom less susceptible to oxidation, a process primarily mediated by cytochrome P450 1A2[6]. In contrast, O-acetylation is an activation step for heterocyclic and aromatic amines.
Genetic polymorphisms of NAT1 have been identified, but the relationship between genotypes and phenotypes is not clear[7]. Several studies have suggested that NAT1 and NAT2 acetylation polymorphism plays a role in carcinogenesis in humans exposed to certain carcinogenic chemicals[5]. Our previous studies suggested that NAT1 and NAT2 polymorphisms may be modifiers of individual risk for cholangiocarcinoma, a cancer of the biliary epithelium[8]. Overexpression of NAT1 in breast tumors is associated with growth properties, as well as chemotherapeutic drug resistance[9] and drug allergy, in particular, cutaneous drug reactions associated with sulfonamides, whereas inactivation of the enzyme may contribute to drug toxicity and cancer risk[10,11]. More recently, NAT1 activity was shown to be suppressed by oxidant species such as hydrogen peroxide (H2O2) and peroxynitrite[12,13]. Peroxynitrite and H2O2 irreversibly inactivate NAT1 by oxidation of its conserved catalytic cysteine residue to sulfinic or sulfonic forms[12,13].
Chronic infection and inflammation are important risks factors for several cancers, including cholangiocarcinoma, a highly malignant adenocarcinoma originating from the cholangiocytes. The highest incidence of cholangiocarcinoma worldwide is seen in Northeast Thailand[14]. Infection of the biliary system with liver fluke (Opisthorchis viverrini) and possibly exposure to carcinogenic chemicals are believed to be causally related to cholangiocarcinoma[15,16]. The association of cholangiocarcinoma and liver fluke was observed in hospital case series which showed an excessively increased risk in patients with liver fluke infestation. Inflammation of the biliary tract caused by mechanical injury and the release of metabolic products from the flukes, together with the damaging effects of reactive metabolites from endogenous and environmental chemicals have been proposed as the responsible factors[17], which induce alterations in gene expression resulting to cellular hyper-proliferation and development of neoplasia[15,16]. In an animal model of cholangiocarcinogenesis, hamster livers infected with liver flukes showed inflammation of the bile duct epithelium, and contained 8-oxo-deoxyguanosine and 8-nitroguanine adducts, which are biomarkers of DNA attack by reactive oxygen and nitrogen species[18]. Thus, inflammatory processes can cause oxidative stress and thereby affect NAT1 activity.
In the present study, we examined the effect of a combination of proinflammatory cytokines on KKU-100 cholangiocarcinoma cells. We also assessed the effect of cytokines on NAT1 activity and the development of oxidative stress; some of these effects were reproduced by a nitric oxide donor.
MATERIALS AND METHODS
Human CCA cell line
The human biliary epithelial cell line KKU-100, derived from intrahepatic cholangiocarcinoma, was established in our institute[19]. The cells were cultured in Ham’s F12 containing 4 mmol/L L-glutamine, 1 mmol/L Na-pyruvate, 100 U/mL penicillin, and 100 μg/mL streptomycin and 10% fetal bovine serum and maintained under an atmosphere of 5% CO2 at 37°C. The media was renewed every 3 d. The cells were trypsinized with 0.25% trypsin-ethylenediamine tetraacetic acid (EDTA) and subcultured in the same media. Twenty four hours after subculture, cells at approximately 70% confluence were exposed to a combination of inflammatory cytokines consisting of human interleikin-1β (IL-1β) (1 ng/mL), interferon-γ (IFN-γ) (400 U/mL), and tumor necrosis factor-α (TNF-α) (500 U/mL) (Biosource International Camarillo, CA) for 48 h or S-nitroso glutathione (GSNO, 100 μmol/L) for 2 or 48 h. In experiments determining nitrite production, KKU-100 cells were cultured in non-phenol red medium.
Biochemical assays
Nitrite assay: After treatment of the cell cultures, the accumulation of nitrite in the culture medium was assessed by mixing an equal volume of the medium with Griess reagent (containing 0.1% N-1-napthylethylenediamine in water and 1% sulfanilamide in 5% H3PO4). Absorbance was read at 540 nm with an ELISA plate reader.
Superoxide production: KKU-100 cells were cultured in 35-mm dishes with cytokines for 48 h. Cell cultures were washed with Tris-buffered saline (TBS) (10 mmol/L Tris HCl and 150 mmol/L NaCl, pH 7.3) and incubated for 30 min with phorbol-12-myristate-13-acetate (PMA) (0.68 μg/mL) or with NG-nitro-L-arginine methylester (L-NAME) (100 μmol/L) or for 5 min with NADPH (200 μmol/L). Lucigenin (100 µmol/L) was added to the culture dishes, and chemiluminescence was recorded using a luminometer (Luminometer model 20/20n, Turner Biosystems, CA).
Assay of GSH and glutathione disulfide: After treatment with cytokines, the cells were trypsinized and washed with cold tris buffer saline (TBS) by centrifugation at 1500 × g at 4°C for 10 min and resuspended in TBS buffer. One hundred microliters of cell suspensions were reacted with 10 μL of 1-methyl-2 vinylpyridinium triflate (M2VP) (3.3 mmol/L) as a GSH scavenger for assay of GSH disulfide (GSSG)[20] or with distilled water for assay of total GSH, and cell suspensions were stored frozen at -20°C until analysis. Total GSH and GSSG were assayed according to the Tietze method[21]. The amount of reduced GSH was calculated from total GSH and GSSG. Another aliquot of cell suspensions was used to determine protein content in a Bradford dye binding assay with bovine serum albumin as the standard.
Assay of NAT1 activity: NAT1 enzyme activity was assayed using high performance liquid chromatography according to a previously described method[4], with some modifications. Briefly, the cell cultures were washed with TBS and scraped into a microcentrifuge tube with lysis buffer (1X cell lysis buffer containing 1 mmol/L dithiothreitol [DTT] and 0.1 mmol/L phenylmethylsulfonyl fluoride [PMSF]). The cells were vortexed and centrifuged at 12 000 × g at 4°C for 30 min. The supernatant (cytosol) was stored in 10% (v/v) glycerol. The cytosol protein was used for assays of protein concentration and NAT1 activity. The reaction mixture consisted of 40 μL cytosol (final concentration 50 μg/mL), 20 μL acetyl CoA-regenerating system (DL-acetylcarnitine, 5.4 mg/mL, carnitine acetyltransferase (1 U/mL) in NAT assay buffer (225 mmol/L triethanolamine HCl, 4.5 mmol/L EDTA, and 4.5 mmol/L DTT, pH 7.5), and 20 μL acetylCoA (final concentration 100 μmol/L). The reaction was initiated by addition of 10 μL p-aminobenzoic acid (PABA) in 2.5% dimethylsulfoxide (final concentration 1.25-100 μmol/L) and incubated for 30 min. The reaction was stopped by addition of 10 μL of 15% perchloric acid and centrifuged at 12 000 × g at 4°C for 10 min. The supernatant was injected directly onto a high performance liquid chromatography column (YMC-PACK Pro-C18, 5 μm, 150 mm × 4.6 mm; YMC Co., Japan) and eluted with a mobile phase consisting of water:acetonitrile:acetic acid:triethylamine:tetrahydrofuran (90.2:8.5:1:0.05:0.25 v/v) at a flow rate of 1.0 mL/min. N-acetyl-aminobenzoic acid (Ac-PABA) was detected using a fluorescence detector (Waters 740 scanning fluorescence detector; Waters Corp, Milford) set for excitation at 270 nm and emission at 340 nm. Analytical precision was evaluated by intra- and inter-day assay validation. The coefficients of variation were less than 5% and 10%, respectively. The detection limit of Ac-PABA was less than 1 pmol.
RNA isolation and reverse transcription-polymerase chain reaction
Total RNA was extracted from KKU-100 cells using Trizol®LS reagent according to the manufacturer’s instructions. Total RNA (3 μg) was reverse-transcribed in a 20 μL volume containing 0.5 μg oligo(dT)15 primer, 20 units RNasin® ribonuclease inhibitor and ImProm-II TM reverse trancriptase (Promega, Madison, WI) in 10x polymerase chain reaction (PCR) buffer, 3 mmol/L MgCl2, and 1 mmol/L dNTPs. The first-strand cDNA was synthesized at 42°C for 60 min. Reverse transcription products were used as a template for PCR. PCR amplification was performed using specific primers for NAT1 and farnesyl-diphosphate farnesyltransferase1 (FDFT1) as an internal control. The PCR primer sequences were: NAT1 forward primers, 5’-CCTAGAAGACAGCAAATACCG-3’; NAT1 reverse primers, 5’-AGCCCACCAAACAGTGA-3’ (PCR product: 170 bp); FDFT1 forward primers, 5’-TTTAACTTC TGTGCTATTCCAC-3’; FDFT1 reverse primers, 5’-TCTCCAGTCTGAACATAGTC-3’ (size of PCR product: 325 bp).
PCR was performed in a final volume of 25 μL containing cDNA template, 1.5 μmol/L of each NAT1 primer or 0.4 μmol/L of each FDFT1 primer, 1 U Platinum® Tag DNA polymerase (Invitrogen, Carlsbad, CA), 3 mmol/L MgCl2, and 0.8 mmol/L dNTPs using a Px2 Thermal Cycle (Thermo Electron, Milford, MA ). After an initial denaturating step at 94°C for 5 min, 32 PCR cycles were performed for NAT1 and 28 cycles for FDFT1, as follows: denaturating for 1 min at 94°C, annealing for 1 min at 55°C, and extension for 1 min at 72°C. The final extension was performed at 72°C for 10 min. The PCR products were separated by electrophoresis on a 3% agarose gel containing ethidium bromide. Gels were visualized and photographed. Band density was analyzed with Gel-Pro3 software. The relative amount of NAT1 mRNA was expressed as a ratio of FDFT1 mRNA.
Statistical analysis
Data are expressed as mean ± SE of duplicate assays from three independent experiments. Student’s t-test was used to determine significant differences between each experimental group. The level of significance was set at P < 0.05.
RESULTS
Effect of the cytokine mixture on the kinetics of NAT1 acetylation
NAT1 activity of KKU-100 cells and Michaelis-Menten constants for PABA N-acetylation are shown in Figure 1A. Treatment with the mixture of inflammatory cytokines for 48 h did not affect cell viability (data not shown) but resulted in a significant decrease in Vmax(apparent) without affecting Km(apparent) (Figure 1A). This implies a change in the amount of enzyme but not in its affinity. The initial velocities of PABA N-acetylation by the cytosolic enzyme from KKU-100 cells are shown in Figure 1B.
Figure 1.
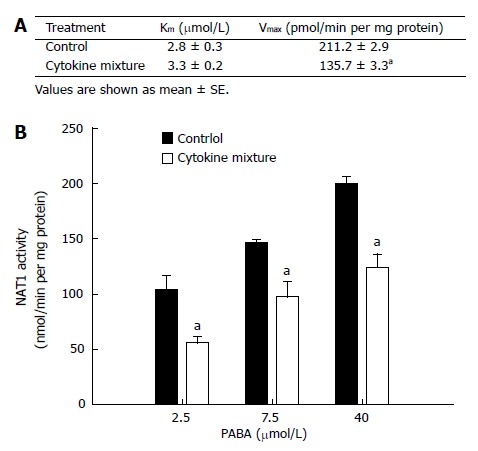
The effect of a mixture of inflammatory cytokines on NAT1 activity in KKU-100 cells. Assays were performed with the cytosol fraction extracted from KKU-100 cells and treated with a combination of cytokines for 48 h. A: Kinetic analysis of NAT1 activity; B: Activity of NAT1 at various substrate concentrations (PABA) of KKU-100 cells with or without treatment with cytokines. aP < 0.05 between cytokine treated and control groups.
Effect of the cytokine mixture on production of nitric oxide and superoxide
Since direct application of oxidant species has been reported recently to suppress NAT1 activity, our experiment showed that treatment with a mixture of cytokines could induce oxidative stress by overproduction of nitric oxide, detected by nitrite assay. Whereas basal production of nitric oxide by KKU-100 cells was nearly absent, production was greatly stimulated by inflammatory cytokines (Figure 2A).
Figure 2.
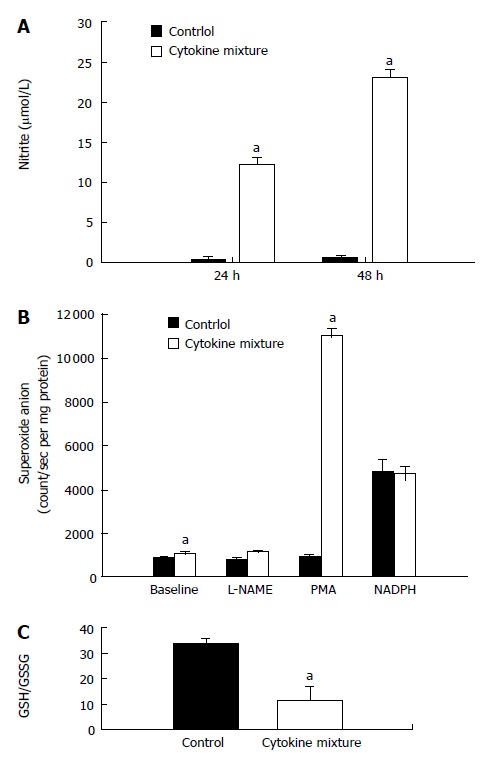
Oxidant status of KKU-100 cells after exposure to a mixture of inflammatory cytokines. A: Stimulation of nitric oxide production, assayed as nitrite levels. Cultured media was collected at 24 and 48 h after exposure; B: Superoxide formation. Cell cultures were washed and incubated with PMA (0.68 μg/mL), L-NAME (100 μmol/L), or NADPH (200 μmol/L), and superoxide production was measured using the chemiluminescence method; C: Redox status was assessed as the GSH and GSSG ratio. Results are presented as mean ± SE from 3 separate experiments. aP < 0.05 between cytokine treated and the respective control groups.
To determine if KKU-100 cells were capable of releasing superoxide anion, aggravating oxidative stress, the cells were cultured with a mixture of cytokines. The cells exhibited low basal release of superoxide; the levels increased slightly after cytokine treatment (Figure 2B). To determine whether the low levels of superoxide were due to scavenging subsequent to overproduction of nitric oxide, the cells were treated with a nitric oxide synthase inhibitor. Inhibition of nitric oxide synthases by L-NAME did not increase superoxide levels relative to the control or baseline values (Figure 2B). When the cells were incubated with NADPH, the substrate of NADPH oxidases, superoxide production in the control and cytokine-treated groups was greatly increased and reached similar levels. Furthermore, treatment with PMA, a protein kinase C activator, resulted in a marked increase in superoxide production in the cytokine-treated group but not in the controls (Figure 2B).
Effect of the cytokine mixture on GSH levels
Since treatment with inflammatory cytokines may result in the formation of free radicals, further experiments were performed to determine if treatment will alter GSH levels and the oxidative status. KKU-100 cells exposed to the cytokine combination for 48 h showed no significant change in the total GSH levels (control: 36.7 ± 2.8 nmol/mg protein, cytokine-treated: 31.9 ± 14.5 nmol/mg protein). However, there was a marked reduction in redox status (GSH/GSSG ratio; Figure 2C) of the treated cells.
Effect of GSNO on NAT1 activity and redox status
The above noted experiments demonstrated that the mixture of cytokines induced nitric oxide production and oxidative stress. The next experiment investigated whether a nitric oxide donor would produce similar results. Treatment with GSNO elicited a very large decrease in total GSH within 2 h; however, the GSH levels normalized to the control levels at 48 h (Figure 3A). There was no significant change in the GSH/GSSG ratio after GSNO treatment (Figure 3B). However, treatment with GSNO reduced NAT1 activity as early as 2 h, and the suppression persisted at 48 h (Figure 3C).
Figure 3.
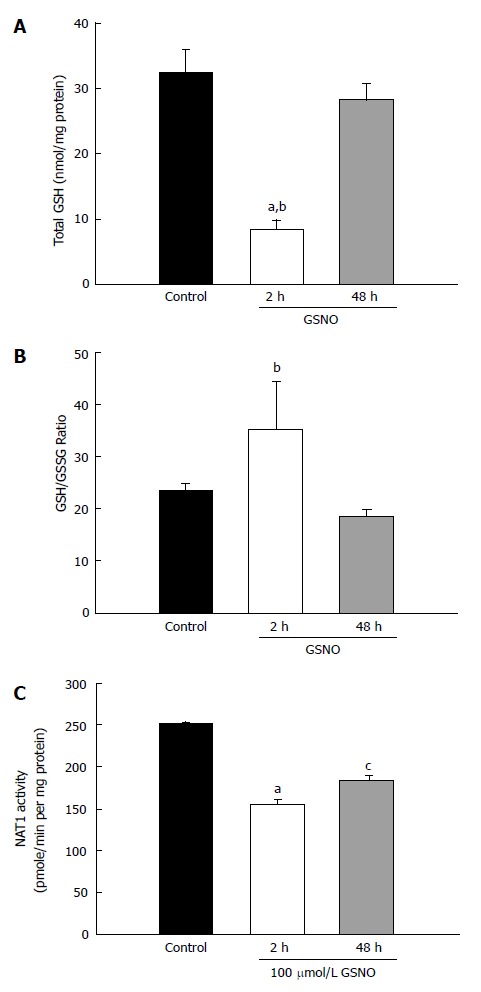
Effect of treatment with the nitric oxide donor GSNO on KKU-100 cells. Cell cultures were treated with 100 μmol/L GSNO for 2 and 48 h. A: Total GSH in the cells; B: GSH/GSSG ratio; C: Activity of NAT1 PABA-acetylation. Bars represent the mean ± SEM from 3 separate experiments. aP < 0.05 vs control groups, cP < 0.05 vs the 48-h treatment group.
Effect of the cytokine mixture and GSNO on expression of NAT1
Reverse transcription PCR was used to assess the effects of proinflammatory cytokines and nitric oxide donors on the expression of NAT1 mRNA. The results are shown in Figure 4. Cytokine treatment elicited a decrease in NAT1 mRNA levels. Similarly, GSNO resulted in reduction of the NAT1 mRNA at 48 h, but not at 2 h.
Figure 4.
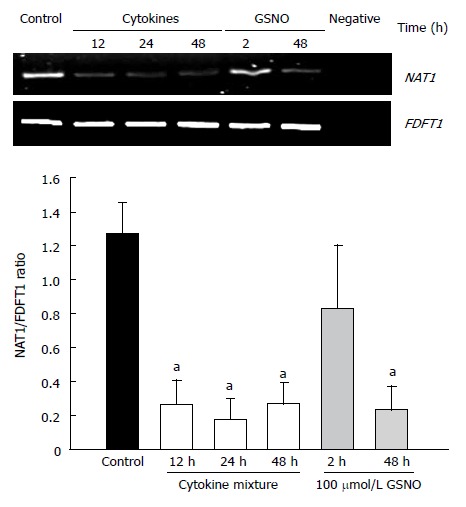
Effect of the cytokine mixture and a nitric oxide donor on the expression of NAT1 in KKU-100 cells. KKU100 cells were incubated with a mixture of cytokines for 48 h or with 100 μmol/L GSNO for 2-48 h. Cells were harvested, and RNA was extracted and analyzed by reverse transcription PCR using FDFT1 as an internal control. aP < 0.05 vs controls.
DISCUSSION
The catalytic activity of NAT enzymes is dependent on a reactive cysteine residue, since its activity is inhibited when this residue is modified by N-hydroxy-arylamine compounds, hydroxamic acids[22], enzyme substrates[10], as well as oxidant species[12,13,23]. Oxidative stress and chronic inflammation are inseparable such that inflammation inevitably produces oxidant species, resulting in tissue damage[24]. The present study shows that proinflammatory cytokines suppress NAT1 activity similar to treatment with oxidant species[12,13]. Kinetic analysis of NAT1 showed a significant decrease in Vmax, suggesting that the reduction of NAT1 activity was due to a decrease in the enzyme level. This alteration was probably due to enzyme inactivation or suppression of NAT1 expression.
Inflammatory cytokines induce the expression of inducible nitric oxide synthase (iNOS) and increase nitric oxide production in cholangiocytes[25]. Our results are consistent with this finding, in fact the basal nitric oxide production was very low but the production increased markedly after treatment with inflammatory cytokines. It is plausible that induction of nitric oxide formation is partly responsible for the suppression of NAT1 activity. Treatment with a nitric oxide donor inhibited NAT1 in a manner similar to cytokine treatment, supporting the possibility that nitric oxide and perhaps peroxynitrite play an important role in modulation of NAT1 activity.
A previous study showed that treatment of breast cancer cells with peroxynitrite irreversibly inactivated NAT1[12]. Peroxynitrite is formed by a reaction between nitric oxide and superoxide anion. In the present study, inflammatory cytokines did not induce a large increase in superoxide, even when nitric oxide formation was inhibited by L-NAME. However, NADPH oxidases, which are membrane bound enzymes that require assembly of subunits from cytosol to become fully functional, are responsible for the formation of superoxide in phagocytic and nonphagocytic cells[26]. They may be upregulated by inflammatory cytokines, as has been shown in smooth muscles and kidney cells[27]. The NADPH oxidases in KKU-100 cells may also be upregulated, however stimulation by PMA, a protein kinase C activator that mediates phosphorylation and recruitment of oxidase subunits[26] may be required to render the NAT1 enzyme fully functional. Together, these findings suggest that superoxide and perhaps peroxynitrite did not play a major role in the cytokine-induced suppression of NAT1 activity in KKU-100 cells. Nevertheless, the role of superoxide and peroxynitrite in vivo, a situation where infiltrating macrophages are present, has not been investigated.
The ability of inflammatory cytokines to induce oxidative stress was clearly evident in the present study, as there was a marked increase in pro-oxidant status, evidenced by a decrease in the redox ratio of GSH/GSSG in cultured cells exposed to the mixture of cytokines. In contrast, treatment with GSNO did not affect the redox ratio at any time point. However, total GSH, representing the major intracellular antioxidant pool, decreased substantially after GSNO treatment, especially at the 2-h time point. Altogether, the differences in the effects of cytokines and GSNO suggest that the effects of the cytokine mixture were mediated by nitric oxide and redox-sensitive pathways.
A recent report has shown that intrahepatic cholangiocarcinoma seen in liver fluke endemic areas is characterized by altered expression of drug metabolizing genes, whereas that from non-endemic areas such as Japan shows alteration in the growth factor signaling genes[17]. This may indicate that drug metabolizing genes are involved in the metabolism of potential carcinogenic chemicals. However, information regarding NAT1 expression is currently not available. Regulation of NAT1 expression has been under study for a long time[23,28]. Previous studies have shown that suppression of NAT1 activity by substrates or oxidant species is due to direct inhibition of the enzyme molecules[13,29], since the expression of NAT1 mRNA does not show any alteration[29]. Our study is the first to demonstrate that treatment with a mixture of cytokines suppresses NAT1 mRNA expression. Using deletion mutant constructs, a promoter site for basal NAT1 expression was identified[30] comprising of an activator protein 1 (AP-1) binding site. Transcription factor AP-1 is upregulated or downregulated by oxidative stress and inflammatory cytokines, depending upon the oxidant levels and the type of inflammatory cytokines[31,32]. Suppression of NAT1 expression in KKU-100 cells may involve down regulation or inactivation of AP-1, although specific evidence in this regard remains to be established. Direct inhibition of NAT1 by oxidant species (nitric oxide) cannot be ruled out, particularly in in vivo conditions; this may occur concurrently with suppression of expression.
In summary, treatment with inflammatory cytokines suppresses NAT1 activity and mRNA expression in cholangiocarcinoma KKU-100 cells. This suppression was associated with oxidative stress and nitric oxide production. These findings show that inflammation can suppress NAT1, a key cellular defense enzyme. Moreover, such a suppression may be implicated in drug toxicity and cancer risk.
ACKNOWLEDGMENTS
Auemduan Prawan was a research fellow of Thailand Research Fund through the Basic Research Grant scheme.
COMMENTS
Background
Arylamine N-acetyltransferases-1 (NAT1) is an important phase II drug metabolizing enzyme that is constitutively expressed in most tissues. Its activity is inactivated by oxidant species.
Research frontiers
NAT1 expression has been demonstrated in cholangiocarcinoma cells (CCA), and polymorphism of NAT genes has been implicated as a risk factor for cancer. Inflammation of the bile duct resulting from opisthorchiasis may alter the activity of the drug metabolizing enzymes including cytochrome P450 and NAT1. Modulation of NAT1 activity may be implicated in drug induced-toxicity and carcinogenesis.
Innovations and breakthroughs
Suppression of NAT1 activity and gene expression is associated with cytokine-induced oxidative stress. A combination of proinflammatory cytokines induces changes in cellular redox and nitric oxide production in CCA and these may be involved in down-regulation of NAT1.
Applications
NAT1 activity could be modulated by inflammation and this may be associated with drug induced toxicity and carcinogenesis.
Terminology
Opischorchiasis: liver fluke (Opisthorchis viverrini) infection of the bile duct.
Peer review
This manuscript describes the intriguing idea that inflammatory cytokines, which are known to be upregulated in cholangiocarcinoma, may function in part by suppressing the activity and/or expression of NAT1, an enzyme thought to be involved in the detoxification of xenobiotics. The authors also show a parallel increase in oxidative stress and nitric oxide after treatment with inflammatory cytokines. Treatment of the cell line with nitric oxide donors also shows a similar suppression of NAT1 activity and expression.
Footnotes
Supported by Khon Kaen University Research Fund, Grant from National Science and Technology Development Agency through the Research-Team-Strenghtening Grant Scheme 2006
S- Editor Zhu LH L- Editor Anand BS E- Editor Wang HF
References
- 1.Sim E, Payton M, Noble M, Minchin R. An update on genetic, structural and functional studies of arylamine N-acetyltransferases in eucaryotes and procaryotes. Hum Mol Genet. 2000;9:2435–2441. doi: 10.1093/hmg/9.16.2435. [DOI] [PubMed] [Google Scholar]
- 2.Grant DM, Hughes NC, Janezic SA, Goodfellow GH, Chen HJ, Gaedigk A, Yu VL, Grewal R. Human acetyltransferase polymorphisms. Mutat Res. 1997;376:61–70. doi: 10.1016/s0027-5107(97)00026-2. [DOI] [PubMed] [Google Scholar]
- 3.Windmill KF, Gaedigk A, Hall PM, Samaratunga H, Grant DM, McManus ME. Localization of N-acetyltransferases NAT1 and NAT2 in human tissues. Toxicol Sci. 2000;54:19–29. doi: 10.1093/toxsci/54.1.19. [DOI] [PubMed] [Google Scholar]
- 4.Kukongviriyapan V, Phromsopha N, Tassaneeyakul W, Kukongviriyapan U, Sripa B, Hahnvajanawong V, Bhudhisawasdi V. Inhibitory effects of polyphenolic compounds on human arylamine N-acetyltransferase 1 and 2. Xenobiotica. 2006;36:15–28. doi: 10.1080/00498250500489901. [DOI] [PubMed] [Google Scholar]
- 5.Hein DW. N-Acetyltransferase genetics and their role in predisposition to aromatic and heterocyclic amine-induced carcinogenesis. Toxicol Lett. 2000;112-113:349–356. doi: 10.1016/s0378-4274(99)00226-x. [DOI] [PubMed] [Google Scholar]
- 6.Hein DW. Molecular genetics and function of NAT1 and NAT2: role in aromatic amine metabolism and carcinogenesis. Mutat Res. 2002;506-507:65–77. doi: 10.1016/s0027-5107(02)00153-7. [DOI] [PubMed] [Google Scholar]
- 7.Kukongviriyapan V, Prawan A, Warasiha B, Tassaneyakul W, Aiemsa-ard J. Polymorphism of N-acetyltransferase 1 and correlation between genotype and phenotype in a Thai population. Eur J Clin Pharmacol. 2003;59:277–281. doi: 10.1007/s00228-003-0630-2. [DOI] [PubMed] [Google Scholar]
- 8.Prawan A, Kukongviriyapan V, Tassaneeyakul W, Pairojkul C, Bhudhisawasdi V. Association between genetic polymorphisms of CYP1A2, arylamine N-acetyltransferase 1 and 2 and susceptibility to cholangiocarcinoma. Eur J Cancer Prev. 2005;14:245–250. doi: 10.1097/00008469-200506000-00008. [DOI] [PubMed] [Google Scholar]
- 9.Adam PJ, Berry J, Loader JA, Tyson KL, Craggs G, Smith P, De Belin J, Steers G, Pezzella F, Sachsenmeir KF, et al. Arylamine N-acetyltransferase-1 is highly expressed in breast cancers and conveys enhanced growth and resistance to etoposide in vitro. Mol Cancer Res. 2003;1:826–835. [PubMed] [Google Scholar]
- 10.Butcher NJ, Ilett KF, Minchin RF. Inactivation of human arylamine N-acetyltransferase 1 by the hydroxylamine of p-aminobenzoic acid. Biochem Pharmacol. 2000;60:1829–1836. doi: 10.1016/s0006-2952(00)00501-3. [DOI] [PubMed] [Google Scholar]
- 11.Bhaiya P, Roychowdhury S, Vyas PM, Doll MA, Hein DW, Svensson CK. Bioactivation, protein haptenation, and toxicity of sulfamethoxazole and dapsone in normal human dermal fibroblasts. Toxicol Appl Pharmacol. 2006;215:158–167. doi: 10.1016/j.taap.2006.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dairou J, Atmane N, Rodrigues-Lima F, Dupret JM. Peroxynitrite irreversibly inactivates the human xenobiotic-metabolizing enzyme arylamine N-acetyltransferase 1 (NAT1) in human breast cancer cells: a cellular and mechanistic study. J Biol Chem. 2004;279:7708–7714. doi: 10.1074/jbc.M311469200. [DOI] [PubMed] [Google Scholar]
- 13.Atmane N, Dairou J, Paul A, Dupret JM, Rodrigues-Lima F. Redox regulation of the human xenobiotic metabolizing enzyme arylamine N-acetyltransferase 1 (NAT1). Reversible inactivation by hydrogen peroxide. J Biol Chem. 2003;278:35086–35092. doi: 10.1074/jbc.M303813200. [DOI] [PubMed] [Google Scholar]
- 14.Sriplung H, Wiangnon S, Sontipong S, Sumitsawan Y, Martin N. Cancer incidence trends in Thailand, 1989-2000. Asian Pac J Cancer Prev. 2006;7:239–244. [PubMed] [Google Scholar]
- 15.Thamavit W, Bhamarapravati N, Sahaphong S, Vajrasthira S, Angsubhakorn S. Effects of dimethylnitrosamine on induction of cholangiocarcinoma in Opisthorchis viverrini-infected Syrian golden hamsters. Cancer Res. 1978;38:4634–4639. [PubMed] [Google Scholar]
- 16.Sripa B, Kaewkes S, Sithithaworn P, Mairiang E, Laha T, Smout M, Pairojkul C, Bhudhisawasdi V, Tesana S, Thinkamrop B, et al. Liver fluke induces cholangiocarcinoma. PLoS Med. 2007;4:e201. doi: 10.1371/journal.pmed.0040201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jinawath N, Chamgramol Y, Furukawa Y, Obama K, Tsunoda T, Sripa B, Pairojkul C, Nakamura Y. Comparison of gene expression profiles between Opisthorchis viverrini and non-Opisthorchis viverrini associated human intrahepatic cholangiocarcinoma. Hepatology. 2006;44:1025–1038. doi: 10.1002/hep.21330. [DOI] [PubMed] [Google Scholar]
- 18.Pinlaor S, Hiraku Y, Ma N, Yongvanit P, Semba R, Oikawa S, Murata M, Sripa B, Sithithaworn P, Kawanishi S. Mechanism of NO-mediated oxidative and nitrative DNA damage in hamsters infected with Opisthorchis viverrini: a model of inflammation-mediated carcinogenesis. Nitric Oxide. 2004;11:175–183. doi: 10.1016/j.niox.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 19.Sripa B, Leungwattanawanit S, Nitta T, Wongkham C, Bhudhisawasdi V, Puapairoj A, Sripa C, Miwa M. Establishment and characterization of an opisthorchiasis-associated cholangiocarcinoma cell line (KKU-100) World J Gastroenterol. 2005;11:3392–3397. doi: 10.3748/wjg.v11.i22.3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Somparn N, Kukongviriyapan U, Tassaneeyakul W, Jetsrisuparb A, Kukongviriyapan V. Modification of CYP2E1 and CYP3A4 activities in haemoglobin E-beta thalassemia patients. Eur J Clin Pharmacol. 2007;63:43–50. doi: 10.1007/s00228-006-0224-x. [DOI] [PubMed] [Google Scholar]
- 21.Tietze F. Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Anal Biochem. 1969;27:502–522. doi: 10.1016/0003-2697(69)90064-5. [DOI] [PubMed] [Google Scholar]
- 22.Wang H, Wagner CR, Hanna PE. Irreversible inactivation of arylamine N-acetyltransferases in the presence of N-hydroxy-4-acetylaminobiphenyl: a comparison of human and hamster enzymes. Chem Res Toxicol. 2005;18:183–197. doi: 10.1021/tx049801w. [DOI] [PubMed] [Google Scholar]
- 23.Rodrigues-Lima F, Dupret JM. Regulation of the activity of the human drug metabolizing enzyme arylamine N-acetyltransferase 1: role of genetic and non genetic factors. Curr Pharm Des. 2004;10:2519–2524. doi: 10.2174/1381612043383845. [DOI] [PubMed] [Google Scholar]
- 24.Hensley K, Robinson KA, Gabbita SP, Salsman S, Floyd RA. Reactive oxygen species, cell signaling, and cell injury. Free Radic Biol Med. 2000;28:1456–1462. doi: 10.1016/s0891-5849(00)00252-5. [DOI] [PubMed] [Google Scholar]
- 25.Jaiswal M, LaRusso NF, Burgart LJ, Gores GJ. Inflammatory cytokines induce DNA damage and inhibit DNA repair in cholangiocarcinoma cells by a nitric oxide-dependent mechanism. Cancer Res. 2000;60:184–190. [PubMed] [Google Scholar]
- 26.Hancock JT, Desikan R, Neill SJ. Role of reactive oxygen species in cell signalling pathways. Biochem Soc Trans. 2001;29:345–350. doi: 10.1042/0300-5127:0290345. [DOI] [PubMed] [Google Scholar]
- 27.Moe KT, Aulia S, Jiang F, Chua YL, Koh TH, Wong MC, Dusting GJ. Differential upregulation of Nox homologues of NADPH oxidase by tumor necrosis factor-alpha in human aortic smooth muscle and embryonic kidney cells. J Cell Mol Med. 2006;10:231–239. doi: 10.1111/j.1582-4934.2006.tb00304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barker DF, Husain A, Neale JR, Martini BD, Zhang X, Doll MA, States JC, Hein DW. Functional properties of an alternative, tissue-specific promoter for human arylamine N-acetyltransferase 1. Pharmacogenet Genomics. 2006;16:515–525. doi: 10.1097/01.fpc.0000215066.29342.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Butcher NJ, Ilett KF, Minchin RF. Substrate-dependent regulation of human arylamine N-acetyltransferase-1 in cultured cells. Mol Pharmacol. 2000;57:468–473. doi: 10.1124/mol.57.3.468. [DOI] [PubMed] [Google Scholar]
- 30.Butcher NJ, Arulpragasam A, Pope C, Minchin RF. Identification of a minimal promoter sequence for the human N-acetyltransferase Type I gene that binds AP-1 (activator protein 1) and YY-1 (Yin and Yang 1) Biochem J. 2003;376:441–448. doi: 10.1042/BJ20030650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kamata H, Hirata H. Redox regulation of cellular signalling. Cell Signal. 1999;11:1–14. doi: 10.1016/s0898-6568(98)00037-0. [DOI] [PubMed] [Google Scholar]
- 32.Jana M, Anderson JA, Saha RN, Liu X, Pahan K. Regulation of inducible nitric oxide synthase in proinflammatory cytokine-stimulated human primary astrocytes. Free Radic Biol Med. 2005;38:655–664. doi: 10.1016/j.freeradbiomed.2004.11.021. [DOI] [PubMed] [Google Scholar]