

Abstract
AIM: To detect germline mutations of MLH1, and investigate microsatellite instability and expression of MLH1 in tumor tissues of hereditary non-polyposis colorectal cancer (HNPCC) with two novel germline mutations, and further investigate the pathobiology of the two novel mutations of MLH1.
METHODS: RNA was extracted from the peripheral blood of 12 patients from 12 different families that fulfilled the Amsterdam II Criteria for HNPCC. Germline mutations of MLH1 were determined by RT-PCR, followed by cDNA sequencing analysis. PCR-GeneScan analysis was used to investigate microsatellite instability with a panel of five microsatellite markers (BAT26, BAT25, D5S346, D2S123 and mfd15), along with immunohistochemical staining to detect the expression of MLH1 protein in two patients' tumor tissues with novel mutations.
RESULTS: Three germline mutations were found in four patients, one of the mutations has previously been reported, but the other two, CGC→TGC at codon 217 of exon 8 and CCG→CTG at codon 581 of exon 16, have not been reported. The two patients' tumor tissues with novel mutations had high-frequency microsatellite instability that showed more than two unstable loci, and both tumors lost their MLH1 protein expression.
CONCLUSION: The two novel germline mutations of MLH1 in HNPCC families i.e. CGC→TGC at codon 217 of exon 8 and CCG→CTG at codon 581 of exon 16, are very likely to have pathological significance.
Keywords: Colorectal cancer, Hereditary non-polyposis colorectal cancer, MLH1 gene, Germline mutation, Microsatellite instability, Gene sequencing
INTRODUCTION
Colorectal cancer remains a serious public health challenge worldwide. According to the different molecular mechanisms, colorectal cancer is divided into two groups: sporadic and genetic. Hereditary non-polyposis colorectal cancer (HNPCC), the most common hereditary colon cancer syndrome[1], is a predominantly inherited disease associated with increased lifetime risk of a range of cancers, including colorectal and endometrial cancers, as well as extracolonic gastrointestinal, genitourinary, ovarian and brain cancers[2-9]. HNPCC development is associated with the functional deficiency of germline MMR genes. Up to now, seven MMR genes have been found, and among these, MLH1 and MSH2 are very closely associated with HNPCC[10-12]. Carriers of germline MMR mutations have a > 80% risk of cancer by the age of 75[13-15]. Twelve patients fulfilling Amsterdam Criteria II from China were explored in this study. Two novel MLH1 mutations were detected, and the pathobiology of the novel mutations was investigated.
MATERIALS AND METHODS
Subjects and samples
Twelve patients of Chinese descent from families fulfilling Amsterdam II Criteria were selected through the clinic at the Cancer Hospital of Fudan University, Shanghai, China. Personal and family cancer histories were obtained from the patients and participating relatives, and cancer diagnosis was confirmed by reviewing records and pathology reports. Informed consent was obtained from each participant. Three microliters of peripheral blood of each participant was taken. Total RNA of the peripheral blood was extracted using Trizol (Sigma), according to the manufacturer's instructions. Twenty unrelated volunteers from families without HNPCC were used as controls.
RT-PCR
cDNA was synthesized with RT (Roche Diagnostics), using 0.5 μg total RNA and specific primers complementary to the 3' end of the MLH1 (2484-TATGTTAAGACACATCTATTTATTTA-2459). cDNA of MLH1 was amplified in two overlapping fragments using primers (Table 1) that generated products of approximately 2000 bp. PCR was performed using Expand Long Template PCR (Roche Diagnostics): 94°C for 5 min; 10 cycles at 94°C for 30 s, 59°C for 30 s, and 68°C for 3 min; 32 cycles at 94°C for 30 s, 57°C for 30 s, 68°C for 3 min; and a final extension at 68°C for 7 min[16].
Table 1.
Sequences and localization of primers used for Amplification of cDNA of MLH1
| Sense | Antisense |
| MLH1-1F (1-18) | MLH1-5R (2198-2175) |
| CTTGGCTCTTCTGGCGCC | GAGCGCAAGGCTTTATAGACAATG |
| MLH1-4F (1333-1353) | MLH1-6R (2484-2459) |
| GCTGAAGTGGCTGCCAAAAAT | TATGTTAAGACACATCTATTTATTTA |
Sequencing
The purified PCR fragments were sequenced directly using a DNA sequencing kit with BigDye Terminators on an ABI3700 automated DNA sequencer (Applied Biosystems, Foster City, CA, USA), according to the manufacturer’s instructions. The cDNA of MLH1 (2484 bp) was sequenced in six overlapping fragments using primers described in Table 2.
Table 2.
MLH1 primers used for sequencing of cDNA
| Sense | Antisense |
| MLH1-1F CCTGGCTCTTCTGGCGCC | MLH1-1R CTTTTCTCCTCGTGGCTATGTTGT |
| MLH1-2F ATGTGCTGGCAATCAAGGGA | MLH1-2R GGTGCACATTAACATCCACATTCT |
| MLH1-3F CCAAAAACACACACCCATTCCT | MLH1-3R CCTTTGTTGTATCCCCCTCCA |
| MLH1-4F GCTGAAGTGGCTGCCAAAAAT | MLH1-4R CATCTTCCTCTGTCCAGCCACTC |
| MLH1-5F TTGCCATGCTTGCCTTAGATAGTC | MLH5R GAGCGCAAGGCTTTATAGACAATG |
| MLH1-6F GCTCCATTCCAAACTCCT | MLH1-6R TATGTTAAGACACATCTATTTATTTA |
Microdissection and minimal amount of DNA extraction
One 5-μm and four 7-μm paraffin-embedded sections of tumor tissues were deparaffinized. The 5-μm sections were stained with hematoxylin and eosin and served as controls. The 7-μm sections were lightly stained with hematoxylin for microdissection. The microdissection was performed under a dissection microscope with a scalpel. Tumor cells should account for at least 80% of the total cells isolated. The microdissected tissues were transferred directly into an Eppendorf tube with 150 μL cell lysis buffer (0.5 mol/L Tris, 20 mmol/L EDTA, 10 mmol/L NaCl, 10 g/L SDS, 0.5 g/L Proteinase K). The subsequent DNA extraction was performed according to the protocol of the DNA extraction kit (Daxia Biotech, Shanghai, China). Genomic DNA was also extracted from peripheral white blood cells.
Microsatellite instability (MSI) analysis
Matched normal and tumor DNA was investigated with a panel of microsatellite markers (mononucleotide repeats BAT26 and BAT25, dinucleotide repeats D5S346, D2S123 and mfd15)[17]. The primer pairs were synthesized by Shenyou Biotech (Shanghai, China). Each forward primer was labeled with a fluorescent dye at the 5' end to enable the PCR products to be detected by an ABI 310 automated DNA sequencer. After successful amplification, the 2-μL PCR products were mixed with 12.5 μL deionized formamide and 2 μL 350 Rox Sizer. The mixture was denatured, snap-cooled and electrophoresed on an ABI 310 automated DNA sequencer according to the manufacturer's recommendation. The electrophoresis results were analyzed by GeneScan Software (Applied Biosystems). MSI was determined according to the method of Gebert et al[18]. Additional peaks (bands) at microsatellite loci in the tumor compared with normal tissue from the same patient were interpreted as MSIs. Cases with MSIs in more than two loci were interpreted as exhibiting high MSI.
Immunostaining for MLH1
A monoclonal antibody against MLH1 (Pharmingen, San Diego, CA, USA) was prepared at a 1:40 dilution. The antibody was detected by the EnVision method. Diminished expression of MLH1 in cancer tissues was demonstrated when there was complete absence of detectable nuclear staining of neoplastic cells. Infiltrating lymphocytes, as well as normal colonic crypt epithelium next to the tumor area, served as internal positive controls[19,20].
RESULTS
Germline mutations of MLH1
Four germline mutations were detected at three different sites of MLH1, involving four patients, which were at 649 codon 217 exon 8: CGC→TGC in family H2, at 1742 codon 581 exon 16: CCG→CTG in family H31, and at 1151 codon 384 exon 12: GTT→GAT in family H109 and H114. All three were missense mutations (Table 3, Figures 1 and 2). Their polymorphism possibilities were excluded by visiting the mutation database of MMR genes (www.INSIGHT-group.org). The mutation in families H109 and H114 has been reported to be pathogenic, while the mutations in H2 and H31 are not. The three abnormalities in MLH1 were not found in the control group.
Table 3.
Germline mutations of MLH1
| Families | Genes | Exon | Codons affected | DNA change | Amino acid change | Mutation types |
| H2 | MLH1 | 8 | 217 | C→T, at 649 | Arg→Cys | Missense |
| H31 | MLH1 | 16 | 581 | C→T, at 1742 | Pro→Leu | Missense |
| H109 | MLH1 | 12 | 384 | T→A, at 1151 | Val→Asp | Missense |
| H114 | MLH1 | 12 | 384 | T→A, at 1151 | Val→Asp | Missense |
Figure 1.
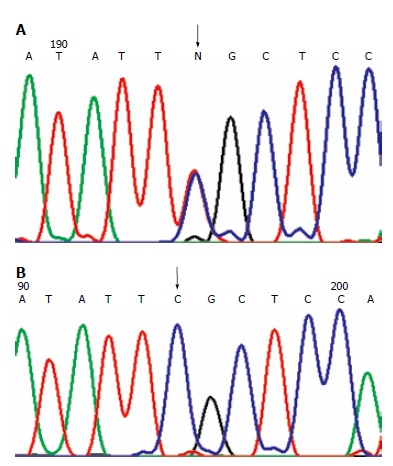
Missense mutations. (A): MLH1 mutation in H2 family at 649 codon 217 exon 8: CGC→TGC. The arrow shows the site of the mutation; (B): Wild-type sequence. The arrow shows the corresponding site.
Figure 2.
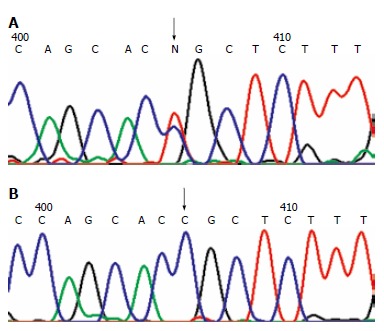
Missense mutation. A: MLH1 mutation in H31 family at 1742 odon 581 exon 16: CCG→CTG. The arrow shows the site of the mutation; B: Wild-type sequence. The arrow shows the corresponding site.
MSI analysis
Four loci in BAT25, BAT26, D2S123 and D5S346 showed MSI in the tumor tissue of the patient from the H2 family, and four loci in BAT25, BAT26, D2S123 and Mfd15 showed MSI in the tumor tissue of the patient from the H31 family. According to the criteria above for MSI, the tumor tissues of the two patients had high MSI (Figure 3).
Figure 3.
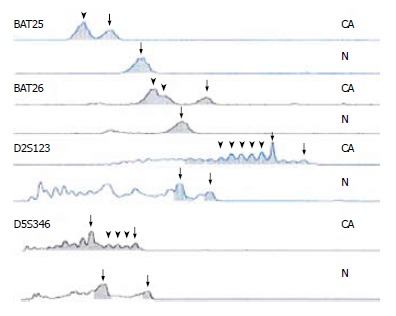
Four sites of MSI in H2 family. CA represents the tumor tissue, N represents the control tissue, the arrow-heads show new waves, and the arrows show wild ones.
Immunohistochemistry of MLH1
There was no expression of MLH1 protein in the tissues of the two patients. As a control, MLH1 protein was detected in infiltrating lymphocytes and the normal colonic crypt epithelium next to the tumor area in the patient from the H2 family, and in the stromal cells in the patient from the H31 family (Figures 4 and 5).
Figure 4.
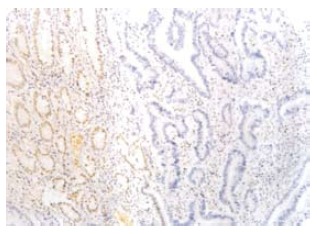
MLH1 protein in H2 was negative in the tumor glands (right), and positive in the mucous glands next to the tumor tissue (EnVison, × 200).
Figure 5.
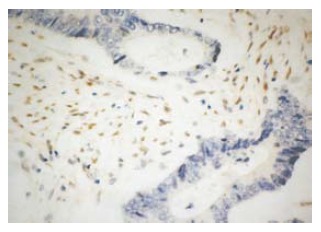
MLH1 protein in H31 was negative in the tumor glands, and positive in the stroma cells (EnVison, × 400).
DISCUSSION
Colorectal cancer is one of the most common malignant tumors; furthermore, its incidence is increasing conti-nuously. HNPCC, genetic colorectal cancer, accounts for about 10% of all colorectal cancer. Compared with sporadic colorectal cancer, HNPCC shows special characteristics associated with its molecular mechanisms and clinic features. HNPCC, as the most common hereditary colon cancer syndrome, is characterized by early onset of colorectal cancer, location of tumors in the proximal colon, and an increased risk of neoplasms of extracolonic organs, including endometrium, stomach, urothelium, small intestine, and ovary, multiple metachronous colorectal cancer[21-24], and better prognosis than that in sporadic cases[25-30]. Development of HNPCC is closely associated with deficiency or loss of MMR gene function. Identification of MMR gene germline mutations can have direct clinical implications in counseling and management of HNPCC families[31].
HNPCC has gained worldwide recognition, and several developed countries, such as the United States, Germany, Finland and the Netherlands, have established HNPCC genetic institutions, and several clinical criteria for the diagnosis of concerned families have been suggested. However, at present, there are only are a few institutions in China that are engaged in research on HNPCC. Our hospital collaborative group on HNPCC has been involved in the field for a few years, and has set up an HNPCC database. We detected the 12 random samples from our database that fulfilled Amsterdam Criteria II, using an mRNA-based sequencing technique, and three germline mutations of MLH1 were found. All three were missense mutations, two of which have not been reported previously. Diagnosis of HNPCC was based on finding the pathological germline mutation in MMR gene. In the present series, difficulties in the assessment of pathogenicity were mostly associated with missense mutations. Peltomaki et al thought the missense mutation meeting the following criteria was pathogenic (1) that it led to a nonconservative aminoacid change, (2) that the involved codon was evolutionarily conserved, (3) that the alteration did not occur in the normal population, and (4) that it cosegregated with the disease phenotype[11]. The patient in the H2 family was affected by colorectal cancer at 36 years of age, his mother was affected by endometrial cancer at 54 years of age, and one of his aunts was affected by colorectal cancer at 48 years of age. The patient in the H31 family was affected by colorectal cancer at 39 years of age, her father was affected by colorectal cancer at 50 years of age, and her uncle was affected by gastric carcinoma at 57 years of age. Their family histories suggested co-segregation of the mutations with the disease. Both of the novel mutations lead to amino acid changes, and the changed amino acids belong to the non-conserved ones. According to the criteria above, we estimated that the two novel mutations were pathogenic. In order to further evaluate the mutations, DNA were isolated from the two patients' tumor tissues, and GeneScan was employed for MSI analysis, and immunohistochemistry was used to detect the expression of MLH1 protein in tumor tissues[32,33]. The tumor tissues of the two patients showed high MSI and lack of MLH1 protein expression. Based on the above results, we concluded that the two novel mutations were very likely pathogenic.
Differentiating HNPCC from sporadic colorectal cancer has practical clinical value, and the identification of HNPCC depends on the detection of germline mutations of the MMR gene. More MMR genes should be investigated besides MLH1, and when a novel mutation is found, its pathogenic evaluation should be carried out so that more HNPCC can be identified.
COMMENTS
Background
Hereditary non-polyposis colorectal cancer (HNPCC) is one of the most common autosomal dominantly inherited cancer syndromes and accounts for 10% of all colorectal cancer. HNPCC shows its own characteristics associated with its molecular mechanisms, clinical features, method of treatment, and management of HNPCC families. It has gained worldwide recognition, and the International Collaborative Group on Hereditary Non-polyposis Colorectal Cancer (ICG-HNPCC) was founded in 1990. At present, there are a few ways to screen HNPCC families; however, only the pathological germline mutation in the mismatch repair (MMR) gene can be used for identifying HNPCC families.
Research frontiers
Development of HNPCC is closely associated with deficiency or loss of function of MMR genes. At least 5 MMR genes, MLH1, MSH2, MSH6, PMS1, and PMS2, have been implicated in HNPCC. Genetic linkage analysis showed that germline mutations of MLH1 and MSH2 account for nearly 90% of all the germline mutations found in HNPCC. Germline mutations of MLH1 were detected in this study.
Innovations and breakthroughs
There are a few ways to screen HNPCC families, however, the most specific method is to detect germline mutations of MMR genes. In present study, a new method, RNA-based sequencing analysis, was used. Two novel germline mutations in MLH1 were found, and their possible pathobiology was investigated by PCR-GeneScan analysis and immunohistochemical staining.
Applications
Identification of pathological germline mutations in MMR genes is the gold standard for HNPCC. Differentiating HNPCC from sporadic colorectal cancer has direct clinical implications for counseling and management of HNPCC family members.
Peer review
HNPCC is different from sporadic colorectal cancer(SCRC), and to differentiate HNPCC from SCRC owns practical clinical implications. The study detected germline mutations of MLH1 with a new method, and investigated the pathobiology of the detected novel mutations in MLH1. The study is a valuable contribution with potential clinical importance.
Footnotes
Supported by the Key Project of Shanghai Medical Subjects, No.05 III 004 and Shanghai Pujiang Program, No. 06PJ14019
S- Editor Liu Y L- Editor Kerr C E- Editor Li HY
References
- 1.Kadiyska TK, Kaneva RP, Nedin DG, Alexandrova AB, Gegova AT, Lalchev SG, Christova T, Mitev VI, Horst J, Bogdanova N, et al. Novel MLH1 frameshift mutation in an extended hereditary nonpolyposis colorectal cancer family. World J Gastroenterol. 2006;12:7848–7851. doi: 10.3748/wjg.v12.i48.7848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Plaschke J, Linnebacher M, Kloor M, Gebert J, Cremer FW, Tinschert S, Aust DE, von Knebel Doeberitz M, Schackert HK. Compound heterozygosity for two MSH6 mutations in a patient with early onset of HNPCC-associated cancers, but without hematological malignancy and brain tumor. Eur J Hum Genet. 2006;14:561–566. doi: 10.1038/sj.ejhg.5201568. [DOI] [PubMed] [Google Scholar]
- 3.Huang D, Chen C, Sun W, Strom CM, Bender RA. High-throughput gene sequencing assay development for hereditary nonpolyposis colon cancer. Clin Colorectal Cancer. 2004;4:275–279. doi: 10.3816/ccc.2004.n.027. [DOI] [PubMed] [Google Scholar]
- 4.Kámory E, Tanyi M, Kolacsek O, Olasz L, Tóth L, Damjanovich L, Csuka O. Two germline alterations in mismatch repair genes found in a HNPCC patient with poor family history. Pathol Oncol Res. 2006;12:228–233. doi: 10.1007/BF02893417. [DOI] [PubMed] [Google Scholar]
- 5.Chen LM, Yang KY, Little SE, Cheung MK, Caughey AB. Gynecologic cancer prevention in Lynch syndrome/hereditary nonpolyposis colorectal cancer families. Obstet Gynecol. 2007;110:18–25. doi: 10.1097/01.AOG.0000267500.27329.85. [DOI] [PubMed] [Google Scholar]
- 6.Renkonen-Sinisalo L, Bützow R, Leminen A, Lehtovirta P, Mecklin JP, Järvinen HJ. Surveillance for endometrial cancer in hereditary nonpolyposis colorectal cancer syndrome. Int J Cancer. 2007;120:821–824. doi: 10.1002/ijc.22446. [DOI] [PubMed] [Google Scholar]
- 7.Okamoto H, Mineta T, Nakahara Y, Ichinose M, Shiraishi T, Tabuchi K. Molecular analysis of astrocytoma associated with Turcot syndrome type 1--case report. Neurol Med Chir (Tokyo) 2004;44:124–128. doi: 10.2176/nmc.44.124. [DOI] [PubMed] [Google Scholar]
- 8.Tamiya T, Hamazaki S, Ono Y, Tokunaga K, Matsumoto K, Furuta T, Ohmoto T. Ganglioglioma in a patient with Turcot syndrome. Case report. J Neurosurg. 2000;92:170–175. doi: 10.3171/jns.2000.92.1.0170. [DOI] [PubMed] [Google Scholar]
- 9.Hartmann A, Cheville JC, Dietmaier W, Hofstädter F, Burgart LJ, Blaszyk H. Hereditary nonpolyposis colorectal cancer syndrome in a patient with urothelial carcinoma of the upper urothelial tract. Arch Pathol Lab Med. 2003;127:E60–E63. doi: 10.5858/2003-127-e60-HNCCSI. [DOI] [PubMed] [Google Scholar]
- 10.Peltomäki P, Vasen HF. Mutations predisposing to hereditary nonpolyposis colorectal cancer: database and results of a collaborative study. The International Collaborative Group on Hereditary Nonpolyposis Colorectal Cancer. Gastroenterology. 1997;113:1146–1158. doi: 10.1053/gast.1997.v113.pm9322509. [DOI] [PubMed] [Google Scholar]
- 11.Peltomäki P, Vasen H. Mutations associated with HNPCC predisposition -- Update of ICG-HNPCC/INSiGHT mutation database. Dis Markers. 2004;20:269–276. doi: 10.1155/2004/305058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anwar S, Hall C, White J, Deakin M, Farrell W, Elder JB. Hereditary non-polyposis colorectal cancer: an updated review. Eur J Surg Oncol. 2000;26:635–645. doi: 10.1053/ejso.2000.0974. [DOI] [PubMed] [Google Scholar]
- 13.Järvinen HJ, Aarnio M, Mustonen H, Aktan-Collan K, Aaltonen LA, Peltomäki P, De La Chapelle A, Mecklin JP. Controlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterology. 2000;118:829–834. doi: 10.1016/s0016-5085(00)70168-5. [DOI] [PubMed] [Google Scholar]
- 14.Brand RM, Jones DD, Lynch HT, Brand RE, Watson P, Ashwathnayaran R, Roy HK. Risk of colon cancer in hereditary non-polyposis colorectal cancer patients as predicted by fuzzy modeling: Influence of smoking. World J Gastroenterol. 2006;12:4485–4491. doi: 10.3748/wjg.v12.i28.4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Domanska K, Nilbert M, Soller M, Silfverberg B, Carlsson C. Discrepancies between estimated and perceived risk of cancer among individuals with hereditary nonpolyposis colorectal cancer. Genet Test. 2007;11:183–186. doi: 10.1089/gte.2007.9999. [DOI] [PubMed] [Google Scholar]
- 16.Jakubowska A, Górski B, Kurzawski G, Debniak T, Hadaczek P, Cybulski C, Kladny J, Oszurek O, Scott RJ, Lubinski J. Optimization of experimental conditions for RNA-based sequencing of MLH1 and MSH2 genes. Hum Mutat. 2001;17:52–60. doi: 10.1002/1098-1004(2001)17:1<52::AID-HUMU6>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 17.Cai Q, Sun MH, Lu HF, Zhang TM, Mo SJ, Xu Y, Cai SJ, Zhu XZ, Shi DR. Clinicopathological and molecular genetic analysis of 4 typical Chinese HNPCC families. World J Gastroenterol. 2001;7:805–810. doi: 10.3748/wjg.v7.i6.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gebert J, Sun M, Ridder R, Hinz U, Lehnert T, Möller P, Schackert HK, Herfarth C, von Knebel Doeberitz M. Molecular profiling of sporadic colorectal tumors by microsatellite analysis. Int J Oncol. 2000;16:169–179. [PubMed] [Google Scholar]
- 19.Hendriks Y, Franken P, Dierssen JW, De Leeuw W, Wijnen J, Dreef E, Tops C, Breuning M, Bröcker-Vriends A, Vasen H, et al. Conventional and tissue microarray immunohistochemical expression analysis of mismatch repair in hereditary colorectal tumors. Am J Pathol. 2003;162:469–477. doi: 10.1016/S0002-9440(10)63841-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wahlberg SS, Schmeits J, Thomas G, Loda M, Garber J, Syngal S, Kolodner RD, Fox E. Evaluation of microsatellite instability and immunohistochemistry for the prediction of germ-line MSH2 and MLH1 mutations in hereditary nonpolyposis colon cancer families. Cancer Res. 2002;62:3485–3492. [PubMed] [Google Scholar]
- 21.Liu SR, Zhao B, Wang ZJ, Wan YL, Huang YT. Clinical features and mismatch repair gene mutation screening in Chinese patients with hereditary nonpolyposis colorectal carcinoma. World J Gastroenterol. 2004;10:2647–2651. doi: 10.3748/wjg.v10.i18.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aarnio M, Sankila R, Pukkala E, Salovaara R, Aaltonen LA, de la Chapelle A, Peltomäki P, Mecklin JP, Järvinen HJ. Cancer risk in mutation carriers of DNA-mismatch-repair genes. Int J Cancer. 1999;81:214–218. doi: 10.1002/(sici)1097-0215(19990412)81:2<214::aid-ijc8>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 23.Park YJ, Shin KH, Park JG. Risk of gastric cancer in hereditary nonpolyposis colorectal cancer in Korea. Clin Cancer Res. 2000;6:2994–2998. [PubMed] [Google Scholar]
- 24.Lynch HT, Taylor RJ, Lynch JF, Knezetic JA, Barrows A, Fodde R, Wijnen J, Wagner A. Multiple primary cancer, including transitional cell carcinoma of the upper uroepithelial tract in a multigeneration HNPCC family: molecular genetic, diagnostic, and management implications. Am J Gastroenterol. 2003;98:664–670. doi: 10.1111/j.1572-0241.2003.07329.x. [DOI] [PubMed] [Google Scholar]
- 25.Watson P, Lin KM, Rodriguez-Bigas MA, Smyrk T, Lemon S, Shashidharan M, Franklin B, Karr B, Thorson A, Lynch HT. Colorectal carcinoma survival among hereditary nonpolyposis colorectal carcinoma family members. Cancer. 1998;83:259–266. [PubMed] [Google Scholar]
- 26.Elsakov P, Kurtinaitis J. Survival from colorectal carcinoma in HNPCC families as compared to the general population in Lithuania--initial results. Fam Cancer. 2006;5:369–371. doi: 10.1007/s10689-006-0007-7. [DOI] [PubMed] [Google Scholar]
- 27.Crijnen TE, Janssen-Heijnen ML, Gelderblom H, Morreau J, Nooij MA, Kenter GG, Vasen HF. Survival of patients with ovarian cancer due to a mismatch repair defect. Fam Cancer. 2005;4:301–305. doi: 10.1007/s10689-005-6573-2. [DOI] [PubMed] [Google Scholar]
- 28.Clark AJ, Barnetson R, Farrington SM, Dunlop MG. Prognosis in DNA mismatch repair deficient colorectal cancer: are all MSI tumours equivalent? Fam Cancer. 2004;3:85–91. doi: 10.1023/B:FAME.0000039915.94550.cc. [DOI] [PubMed] [Google Scholar]
- 29.Boks DE, Trujillo AP, Voogd AC, Morreau H, Kenter GG, Vasen HF. Survival analysis of endometrial carcinoma associated with hereditary nonpolyposis colorectal cancer. Int J Cancer. 2002;102:198–200. doi: 10.1002/ijc.10667. [DOI] [PubMed] [Google Scholar]
- 30.You JF, Hsieh LL, Changchien CR, Chen JS, Chen JR, Chiang JM, Yeh CY, Hsieh PS, Fan CW, Liu CT, et al. Inverse effects of mucin on survival of matched hereditary nonpolyposis colorectal cancer and sporadic colorectal cancer patients. Clin Cancer Res. 2006;12:4244–4250. doi: 10.1158/1078-0432.CCR-06-0202. [DOI] [PubMed] [Google Scholar]
- 31.Kouraklis G, Misiakos EP. Hereditary nonpolyposis colorectal cancer (Lynch syndrome): criteria for identification and management. Dig Dis Sci. 2005;50:336–344. doi: 10.1007/s10620-005-1607-z. [DOI] [PubMed] [Google Scholar]
- 32.Alazzouzi H, Domingo E, González S, Blanco I, Armengol M, Espín E, Plaja A, Schwartz S, Capella G, Schwartz S Jr. Low levels of microsatellite instability characterize MLH1 and MSH2 HNPCC carriers before tumor diagnosis. Hum Mol Genet. 2005;14:235–239. doi: 10.1093/hmg/ddi021. [DOI] [PubMed] [Google Scholar]
- 33.Caldés T, Godino J, Sanchez A, Corbacho C, De la Hoya M, Lopez Asenjo J, Saez C, Sanz J, Benito M, Ramon Y Cajal S, et al. Immunohistochemistry and microsatellite instability testing for selecting MLH1, MSH2 and MSH6 mutation carriers in hereditary non-polyposis colorectal cancer. Oncol Rep. 2004;12:621–629. [PubMed] [Google Scholar]