

Highlights
-
•
Intravenous administration of VNAs can protect mice from developing rabies when the BBB permeability is enhanced.
-
•
MCP-1 enhances BBB permeability by down-regulation of TJ proteins in the brain microvasculature.
-
•
Enhancement of the BBB permeability correlates with the increase of VNA in and the clearance of RABV from the CNS.
Keywords: Rabies virus, MCP-1, BBB (blood–brain barrier), VNA (virus-neutralizing antibody), Occludin, Claudin-5
Abstract
Rabies virus (RABV) is a neurotropic virus that causes fatal disease in humans and animals. Currently there is no cure for rabies once clinical signs appear. It is believed that once RABV enters the central nervous system (CNS), virus neutralizing antibodies (VNAs) in the periphery cannot pass through the blood–brain barrier (BBB) and into the CNS. Furthermore, it has been hypothesized that VNAs produced in the CNS by invading B cells, rather than those produced in the periphery and then transported into the CNS, are important in clearing RABV from the CNS. In the present study, mouse serum containing VNA was administered intravenously into mice after infection with wild-type RABV. Our studies demonstrate that exogenous administration of VNAs is crucial in the clearance of RABV from the brain and prevent the development of rabies in both immunocompetent and immunocompromised mice as long as the BBB permeability remains enhanced. This present study therefore provides a foundation for the possibility of developing VNA therapy for clinical rabies in humans.
1. Introduction
Rabies virus (RABV) is a negative-sense and single-stranded RNA virus which belongs to the Lyssavirus genus of the Rhabdoviridae family (Rupprecht, 1996). Rhabdoviruses are enveloped with a typical bullet- or rod-shaped morphology and characterized by an extremely broad host spectrum ranging from plants to insects to mammals (Rupprecht, 1996). Rabies has been known as a deadly neurological disease of both humans and animals for centuries (Jackson, 2002) and remains a major threat to public health (Jackson, 2013b, Knobel et al., 2005, Martinez, 2000, Meslin et al., 1994). Each year rabies causes more than 55,000 human deaths around the world (Meslin et al., 1994). Canine rabies is responsible for more than 99% of the human cases in Asia and Africa (Cleaveland et al., 2006). In the United States, dog rabies has been largely brought under control through pet vaccination programs and the number of human cases has declined dramatically during the past 60 years (Hampson et al., 2009). Most of the human cases in the USA have been associated with RABV found in bats, particularly the silver-haired bats with no obvious recognized exposure history (Messenger et al., 2003, Morimoto et al., 1996). Therefore, there is a need to develop therapeutics for clinical rabies although rabies can be prevented in humans after exposure (usually after an animal bite) by post-exposure prophylaxis (PEP), which is comprised of wound cleansing, vaccination and administration of anti-rabies immunoglobulin. PEP is very effective if it is initiated promptly after exposure (CDC, 2010). It is widely accepted that there is no effective treatment for rabies infection which is almost always fatal once neurological symptoms develop (WHO, 1992, Wilde, 2007). Although human survivors have been reported recently after treatment with the Milwaukee Protocol or a modification thereof (MCW, 2009, Willoughby et al., 2005), its effectiveness has been questioned (Hemachudha et al., 2006, Jackson, 2013a, McDermid et al., 2008).
Rabies clinical signs, especially neurologic signs, are believed to be indicative of virus replication resulting in neuronal dysfunction or injury in the central nervous system (CNS), where peripheral immune effectors have limited access (Yousaf et al., 2012). Viral infection of the CNS poses unique challenges to the immune system with regards to controlling and eliminating the invading pathogens (Griffin, 2003). The presence of a blood–brain barrier (BBB) provides a physical and physiological separation of the CNS from the periphery and thus cells and molecules cannot easily enter the CNS (Ballabh et al., 2004, Hosking and Lane, 2010, Roy and Hooper, 2008). Although enhancement of BBB permeability and infiltration of inflammatory cells have often been associated with pathological changes in the CNS when infected by viruses (Hosking and Lane, 2010, Kim et al., 2009, Phares et al., 2006), transiently increased BBB permeability has been found to be helpful in clearing RABV from the CNS (Phares et al., 2007, Roy and Hooper, 2007). Induction of an autoimmune CNS inflammation (experimental allergic encephalomyelitis) (Roy and Hooper, 2007) or administration of attenuated RABV (CVS-F3) (Phares et al., 2006) as well as recombinant RABV expressing three copies of the glycoprotein (G) (TriGAS) (Faber et al., 2009) or immune stimulating molecules (for example, GM-CSF) (Wang et al., 2011, Wen et al., 2011) all resulted in enhancement of BBB permeability, increased production of virus neutralizing antibodies (VNAs), clearance RABV from the CNS, and prevention of rabies in the mouse model. Furthermore, clearance of attenuated RABV from the CNS correlates with infiltration of B cells into the CNS, expressing high levels of κ-light chain mRNA (Phares et al., 2006). Passively-transferred VNA via intraperitoneal route was insufficient to mediate the CNS clearance of attenuated RABV in B-cell deficient mice (Hooper et al., 2009). These observations led to the hypothesis that it is the VNA produced in situ (CNS) by invading B cells, rather than those produced in the periphery and then crossed into the CNS, that are important in clearing RABV from the CNS (Hooper et al., 2009). Nevertheless, it has been reported that enhancing BBB permeability with delivering sufficient VNA to the brain may provide an effective treatment after the CNS infection has been established (Liao et al., 2012). In the present study, intravenous administration of VNA was found to be crucial in clearing RABV from the brain and preventing animals from developing rabies in both immunocompetent and B-cell deficient mice as long as the BBB permeability is enhanced.
2. Materials and methods
2.1. Viruses, cells, serum, and animals
Street RABV (Dog Rabies Virus from Mexico, DRV-Mexico) (Dietzschold et al., 2000, Zhang and Fu, 2012) was propagated in suckling mouse brains. Mouse neuroblastoma cells (NA) were maintained in RPMI 1640 medium (Mediatech, Herndon, VA) supplemented with 10% fetal bovine serum (FBS) (Gibco, Grand Island, NY). Recombinant murine monocyte chemotactic protein-1 (MCP-1) was purchased from PreproTech (Rocky Hill, NJ). RABV-antibody positive serum was prepared from blood of ICR mice immunized with rRABV-GMCSF vaccines (Wen et al., 2011). RABV-negative serum was prepared from blood of naïve ICR mice. All sera were pooled and titrated for VNA. Four to six-week old female ICR mice were purchased from Harlan (Indianapolis, IN). Four to six-week old female C57BL/6J, B6.129S2-Ighmtm1Cgn/J (B cell deficient) were purchased from the Jackson Laboratory (Bar Harbor, ME). All mice were housed in temperature- and light-controlled quarters in the Animal Resources Facility, College of Veterinary Medicine, University of Georgia. This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All animal procedures were approved by the Institutional Animal Care and Use Committee, University of Georgia (animal welfare assurance number: A3085-01). All efforts were made to minimize animal suffering.
2.2. Brain and serum collection
At indicated time points, whole blood was collected from mice, allowed to clot overnight, and spun down at 10,000g for 10 min. Supernatant was isolated for serum antibody analysis. Then, mice were perfused with PBS at a rate of 2 ml/min for 10 min. Independent samples of mouse brains from each group were then homogenized and prepared as 10% (w/v) suspension in DMEM. Homogenized brain samples were centrifuged at 14,000 rpm for 20mins. Supernatant was harvested for virus titration, antibody detection and Western blot analysis.
2.3. Virus titrations
Virus titers were determined by direct fluorescent antibody assay (dFA) in NA cells. NA cells in 96-well plate were inoculated with serial 10-fold dilutions of virus preparation and incubated at 34 °C for 2 days. The culture supernatant was removed and the cells were fixed with 80% ice-cold acetone for 30 min. The cells were washed twice with PBS and then stained with FITC-conjugated anti-RABV N antibodies (Fujirebio, Malvern, PA). Antigen-positive foci were counted under a fluorescent microscope (Zeiss, Germany) and viral titers calculated as fluorescent focus units (FFU) per ml. All titrations were carried out in quadruplicate.
2.4. Quantitative real-time PCR (qRT-PCR)
A real-time (RT) SYBR green PCR assay was carried out in an Mx3000P apparatus (Stratagene, La Jolla, CA) to quantify the expression of viral genomic RNA (copy number) as well as mRNA of tight junction proteins (occludin and claudin-5) and κ-light chain. Total RNA was extracted from brain homogenates using the Qiagen RNeasy kit (Qiagen, Redwood, CA) and treated with DNase (Qiagen). The reverse transcriptase and DNA polymerase were utilized from a one-step Brilliant II SYBR green qRT-PCR master mix kit (Stratagene). Each reaction was carried out in duplicate with approximately 100 ng of DNase-treated RNA and 5 nM each primer pairs described previously (Armstrong et al., 2012, Braniste et al., 2009, Faber et al., 2002, Phares et al., 2006). Amplification was carried out at 50 °C for 2 min and 95 °C for 10 min, followed by 40 cycles in two steps: 95 °C for 15 s and 60 °C for 1 min. For absolute quantification of viral genomic RNA, a standard curve was generated by using a serially diluted RNA in vitro transcribed from a plasmid expressing RABV G, and the copy numbers of viral genomic RNA were normalized to 1 g of total RNA. For measurement of the mRNA for tight junction proteins and κ-light chain, the copy numbers were normalized to those of the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Levels of gene expression in a test sample are presented as the fold increase over that detected in sham-infected controls.
2.5. Rapid fluorescent focus inhibition test (RFFIT)
VNA measurement was carried out by the RFFIT as previously described (Favoretto et al., 1993). 50 μl of serial fivefold dilutions of serum were prepared in Lab-Tek Chamber slides (Nalge Nunc International, Rochester, NY). 50% Fluorescing Foci dose (Fifty FFD50) of challenge virus standard (CVS-11) was added to each chamber and incubated for 90 min at 37 °C. NA cells (105 cells) were added into each chamber and the slides were incubated at 37 °C for 20 h. The cells then were fixed with ice-cold 80% acetone and stained with FITC-conjugated anti-RABV N antibodies for 1 h at 37 °C. Twenty fields in each chamber were assessed under a fluorescent microscope. The 50% endpoint titers were calculated according to the Reed–Muench formula (Reed and Muench, 1938). Then the values were compared with that of a reference serum (obtained from the National Institute for Biological Standards and Control, Herts, UK) and normalized to international units (IU/ml).
2.6. Total IgG ELISA assay
ELISA was carried out by using Mouse IgG Titer ELISA Kit (General Bioscience, Brisbane, CA) according to the manufacturer’s instructions. The optical density was measured at 450 nm using a spectrophotometer (BioTek Instruments, VT). Determination of the total IgG concentration was performed by linear regression analysis using software KC4 Signature Ver. 3.4 (Bio-Tek Instruments). A standard curve was prepared using known concentrations of mouse IgG provided in the ELISA Kit (General Bioscience). The IgG concentration of each sample and control was calculated from each corresponding reference standard curve using a 4-parameter logistic regression equation of the KC4 program. The results were expressed as μg/ml for both brain homogenates and serum samples.
2.7. Measurement of BBB permeability
BBB permeability was determined by measuring Sodium Fluorescein (NaF) uptake as previously described (Kuang et al., 2009, Phares et al., 2006, Phares et al., 2007). 100 μl of 100 mg/ml of NaF, used as a tracer, was injected into the tail vein of each mouse. Peripheral blood was collected after 10 min and PBS-perfused brains were then harvested. Serum recovered was mixed with equal volume of 10% trichloroacetic acid (TCA) and centrifuged for 10 min. The supernatant was collected after centrifugation and made up to 150 μl by mixing with 5 M NaOH and 7.5% TCA. Homogenized brain samples in cold 7.5% TCA were centrifuged for 10 min at 10,000g to remove debris. The supernatant was made up to 150 μl by adding 5 M NaOH. The fluorescence of serum and brain homogenate samples was measured using a spectrophotometer (BioTek Instruments) with excitation at 485 nm and emission at 530 nm. NaF taken up into brain tissues is expressed as the micrograms of fluorescence per mg of cerebrum or cerebellum divided by the micrograms of fluorescence per μl of serum to normalize the uptake amounts of marker from peripheral blood at the time of brain tissue collection (Phares et al., 2007, Trout et al., 1986). Data are expressed as a fold change in the amount of tracer in tissues by comparison with the values obtained for tissues from negative control.
2.8. Western blot analysis
The brain homogenates were subjected to 8–16% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) (Thermo Scientific, Rockford, IL). Separated proteins were electroblotted onto nitrocellulose membranes and incubated with primary antibodies overnight. After extensive washing with PBS, the blots were incubated with secondary antibodies. Proteins were detected by SuperSignal West Pico chemiluminescence (Thermo Scientific). Band signals corresponding to immunoreactive proteins were captured and chemiluminescence intensities were analyzed using ChemiDoc 4000 MP documentation System (Biorad, CA).
2.9. Statistical analyses
Graphpad Prism 5 v. 5.01(1992–2009 Graphpad Software Inc., La Jolla, CA, USA) was used for statistical analysis. Survival curves were estimated through Kaplan–Meier method and statistical significance of survival rates between experimental groups was determined by log rank test. Student’s t-test or one-way ANOVA was used to evaluate the significant difference between experimental group and control or experimental groups, respectively. Statistical significance was set at p < 0.05.
3. Results
3.1. Intravenous administration of VNA can prevent immunocompetent mice from developing rabies if BBB permeability is enhanced
Our previous studies revealed that intracerebral administration of rRABV-GMCSF or a combination of inactivated rRABV and MCP-1 can prevent mice from developing rabies by stimulating the production of VNA, enhancing the BBB permeability, and clearing RABV from the CNS (Wang et al., 2011). To investigate if intravenous administration of VNA together with MCP-1 can protect mice from RABV infection, 4–6 week-old ICR mice were infected intramuscularly (biceps femoris muscle of left hindlimb) with 10 IMLD50 (50% mouse intramuscular lethal dose) of a street RABV (DRV-Mexico) as described (Wang et al., 2011). At 5 days post infection (dpi), mice were treated intravenously from the tail vein with 150 μl of RABV-negative serum or RABV-antibody positive serum (VNA titer: 8 IU). To enhance the BBB permeability, mice were treated intracerebrally with 25 μg recombinant murine MCP-1 (suspended in 40 μl of sterile ddH2O), which is known to transiently enhance the BBB permeability (Stamatovic et al., 2005). Mice were observed daily for 3 weeks for the development of rabies and the results are shown in Fig. 1 . All infected mice treated with medium only died by day 14 p.i. Only 10% of the mice survived after treatment with the combination of MCP-1 and RABV-negative serum or with the positive serum alone without MCP-1. These results indicate that treatment with MCP-1 alone does not increase the survival rate after RABV infection in the absence of passively-transferred VNAs. Without MCP-1, VNA administered intravenously are unable to pass into the brain to clear RABV. Combined treatment with RABV-antibody positive serum and MCP-1 increased the survival rate to 60% (p < 0.05).
Fig. 1.
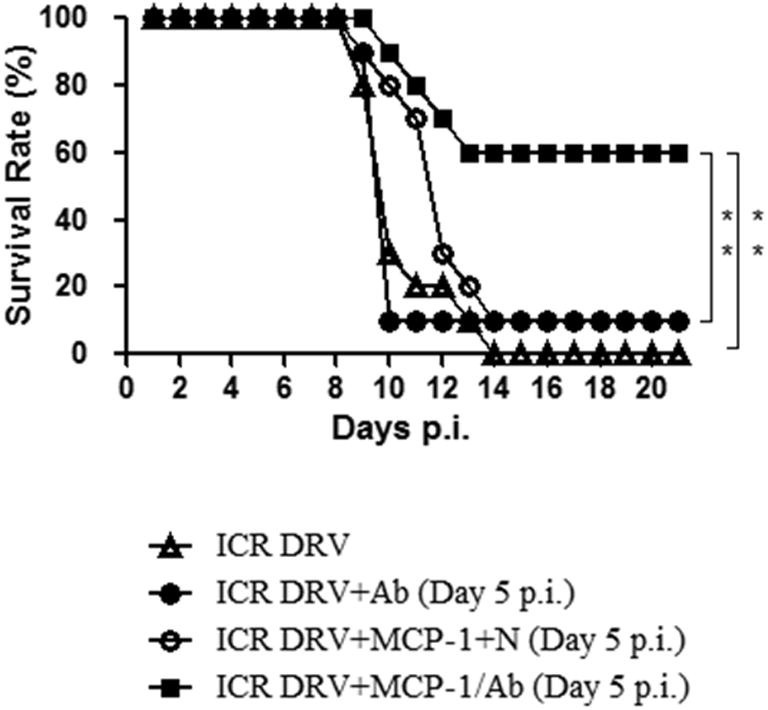
Protective efficacy of immune-competent mice treated with VNA with or without MCP-1 after infection with DRV. ICR mice (groups of 10) at the age of 4–6 weeks were infected i.m. with 10 IMLD50 DRV-Mexico and then treated intravenously with RABV-negative serum (N) or RABV-antibody positive serum (Ab) with or without recombinant murine MCP-1 (25 μg/mouse, i.c.) at 5 dpi. Infected and treated mice were observed daily for 21 days and survivorship was recorded and analyzed. Asterisks indicate significant differences (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001) between the indicated experimental groups.
Brains from mice survived (3 weeks after treatment) from or succumb (at the time of death) to rabies were harvested for virus titration and detection of antibody κ-light chain by RT-PCR (Phares et al., 2006). The results are depicted in Fig. 2 . High virus titers (>104 FFU/ml) were detected in the brains of mice succumbing to rabies and no virus was detected in the survived ones (Fig. 2A). Similar results were also obtained when the viral genomic RNA was detected (Fig. 2B). High VNA titers were detected in the serum of surviving mice (Fig. 2C). The κ-light chain mRNA level in the brain was significantly higher in the surviving mice than in the dead ones (Fig. 2D). These results indicate that intravenous administration of VNA is crucial in the clearance of RABV from the brain when the BBB permeability is enhanced. However, detection of κ-light chain in the brain suggests that plasma cells entering into the CNS could have produced VNA in situ to provide protection.
Fig. 2.

Virus titers and virus copy number in brains, serum VNA and the expression of κ-light chain mRNA in brains of immune-competent mice treated with VNA with or without MCP-1 after infection with DRV-Mexico. ICR mice (groups of 10) at the age of 4–6 weeks were infected i.m. with 10 IMLD50 DRV-Mexico and then treated with RABV-antibody negative (N) or positive serum (Ab) with or without recombinant murine MCP-1 at 5 dpi. Sera and brains were harvested once mice reached the criteria of euthanasia by developing paralysis in both hind legs (Dead) or survived through day 21 (Survived). Virus titers (A) and viral genomic RNA copy number (B) in brains and serum VNA (C) as well as the expression of κ-light chain mRNA in brains (D) were determined. Data from groups of n ⩾ 3 mice are presented as mean values ± SEM, except for group f and g with only one C57BL/6 mouse survived. Asterisks indicate significant differences (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001) between the indicated experimental groups. ND: not done.
3.2. Passively transferred antibodies are able to reach the brain once BBB permeability is enhanced
It has been shown that a single intracerebral dose of MCP-1(25 μg) can induce FITC-albumin leakage to the mouse brain (Stamatovic et al., 2005). To ensure that intravenously administered RABV VNAs are able to reach the brain after BBB permeability is enhanced by MCP-1, ICR mice were given RABV-antibody positive serum intravenously with or without MCP-1 and the antibody levels were analyzed 24 h later in both the brain and the serum. The results are depicted in Fig. 3 . The level of VNAs was found to be similar in the serum of mice with or without MCP-1 treatment (Fig. 3B) while the VNA levels in the brains of mice treated with RABV-antibody positive serum in conjunction with MCP-1 were significantly higher (∼0.3 IU) than in mice treated with VNA-positive serum without MCP-1 (<0.1 IU) (Fig. 3A). Similar findings were also observed when total IgG levels were measured by ELISA (Fig. 3C and D). These results demonstrate that passively administered VNAs are able to pass through BBB when BBB permeability is enhanced with MCP-1.
Fig. 3.
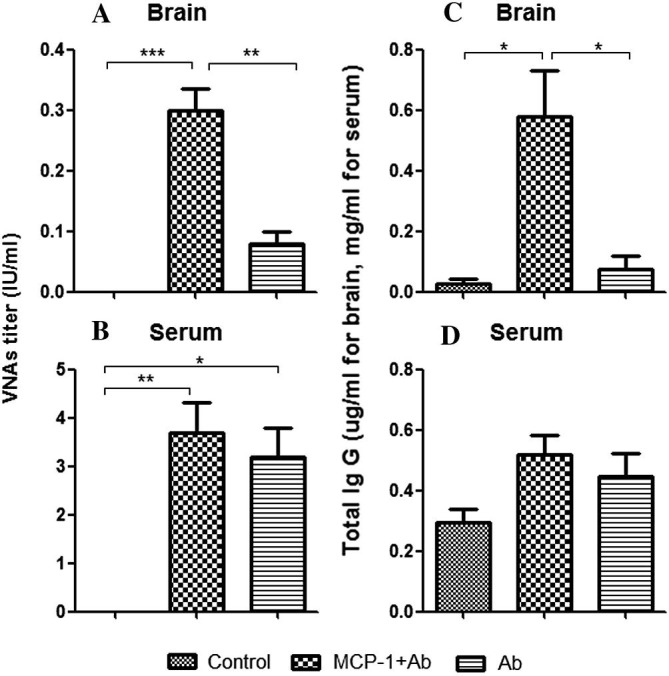
Serum VNA and total IgG in brains and sera of immune-competent mice treated with VNA and/or MCP-1. ICR mice (groups of 3) at the age of 4–6 weeks were treated with RABV-antibody positive serum (Ab) with or without recombinant murine MCP-1. Mice were euthanized 24 h after the treatment and both sera and brains were harvested. VNA in brains (A), serum VNA (B), total IgG in brains (C) and serum total IgG (D) were determined. Data are presented as mean values ± SEM. Asterisks indicate significant differences (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001) between the indicated experimental groups.
3.3. Enhancement of BBB permeability peaked at 36 h after administration of MCP-1 that correlates with the down-regulation of tight junction protein expression
It has been shown that MCP-1 enhanced the BBB permeability 6–12 h after injection and the effect waned off 24–48 h (Stamatovic et al., 2005). To investigate if infection with wt RABV would change the dynamics of BBB permeability enhancement by MCP-1, mice were infected with DRV-Mexico and then treated with MCP-1 and RABV-antibody positive or negative serum at 5 dpi. NaF uptake was measured in mice at 12, 24, 36, and 48 h after treatment with MCP-1. As shown in Fig. 4 , infection with DRV-Mexico alone did not change the BBB permeability when compared to the controls. The enhancement of BBB permeability was detected at 12 h, reached a peak at 36 h, and declined at 48 h after MCP-1 injection with the greater effect observed in the cerebrum than in the cerebellum (Fig. 4A and B). These studies indicate that infection with RABV does not change the dynamics of BBB permeability enhancement by MCP-1.
Fig. 4.

BBB permeability in cerebrum and cerebellum as well as the expression of tight junction proteins (occludin and claudin-5) in brains of immune-competent mice treated with MCP-1 with or without VNA after infection with DRV. ICR mice (groups of 3) at the age of 4–6 weeks were infected i.m. with 10 IMLD50 DRV-Mexico and then treated with recombinant murine MCP-1 and RABV-antibody negative (N) or positive serum (Ab) at 5 dpi. BBB permeability in cerebrum and cerebellum, (A) and (B), respectively, and mRNA and quantitative (Western blotting) expression of occludin (C and E) and claudin-5 (D and F) were determined at 12, 24, 36, 48 h after the treatment. Data from groups are presented as mean values ± SEM. Asterisks indicate significant differences (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001) between the control and experimental groups.
MCP-1 has been reported to attract leukocytes and to increase BBB permeability by redistributing tight junction proteins and reorganizing actin cytoskeleton (Stamatovic et al., 2003). The expression of tight junction proteins (occludin and claudin-5) was measured by RT-PCR and Western blotting as described (Chai et al., 2014). An inverse relationship was found between the BBB permeability and the expression of tight junction proteins (Fig. 4C and D). When the NaF intake increased in the brain of mice after treatment with MCP-1 (12–36 h), the expression of tight junction proteins decreased (12–36 h). When the BBB permeability started to decline at 48 h, the expression of tight junction proteins increased. These observations were confirmed by quantitative Western blotting (Fig. 4E and F). These results suggest that MCP-1 enhances the BBB permeability possibly through down-regulation of tight junction proteins in the brain vasculature.
3.4. Treatment with VNA and MCP-1 is crucial for the clearance of RABV from the brain
To determine if combined treatment with MCP-1 and RABV-antibody positive serum is crucial in clearing RABV from the brain, mice were infected with DRV-Mexico. At 5 dpi, mice were treated with MCP-1 with or without RABV-antibody positive serum. Animals were sacrificed at 12, 24, 36, 48 h after treatment. Animals were also sacrificed at the time when DRV-infected mice without any treatment develop hindlimb paralysis (reached the euthanasia criteria) at 10 dpi (indicated as D10) or survived at 15 dpi (indicated as D15) to ensure the clearance of RABV from the brain in an established infection. Brain and serum samples were collected from sacrificed mice for detection of VNAs and total IgG. Virus copy number and mRNA level of κ-light chain were measured in the brain samples. The results are summarized in Fig. 5 . All mice treated with MCP-1 without RABV-antibody positive serum reached euthanasia criteria before D15. Therefore, no samples were collected for this group (DRV+ MCP-1+N/D15).
Fig. 5.

VNA and total IgG in brains and sera, viral copy number and the expression of κ-light chain mRNA in brains of immunocompetent mice treated with MCP-1 with or without VNA after infection with DRV. ICR mice (groups of 3) at the age of 4–6 weeks were infected i.m. with 10 IMLD50 DRV-Mexico and then treated with recombinant murine MCP-1 (25 μg/mouse, i.c.) and RABV-antibody negative (N) or positive serum (Ab) at 5 dpi. Mice were euthanized at 12, 24, 36, 48 h after the treatment) as well as at 10 dpi (indicated as D10) or survived at 15 dpi (indicated as D15). There is another group of one ICR mouse with only DRV infection and sacrificed at 10 dpi (DRV/D10). Both sera and brains from all groups were harvested for determination of VNA in brains (A) and in sera (B), total IgG in brains (C) and sera (D), viral genomic RNA copy number in brains (E) and the expression of κ-light chain mRNA in brains (F). Data are presented as mean values ± SEM. Asterisks indicate significant differences (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001) between the corresponding time points of MCP-1 with N or Ab groups. ND: not done.
In mice treated with MCP-1 without RABV-antibody positive serum, no detectable levels of VNAs were found in the brain (Fig. 5A) or in the serum (Fig. 5B) during the observation period, suggesting that wt RABV (DRV-Mexico) is incapable of inducing VNAs. VNAs were detectable in the brains shortly (12 h) after the combined treatment (MCP-1 and RABV-antibody positive serum). The level of VNAs in the brain reached a peak at 36 h and then declined (Fig. 5A), corresponding to the enhancement of BBB permeability (Fig. 4). In the serum, VNA levels remained constant after administration (Fig. 5B). VNA levels in both the brain and serum continued to be significant at 10 and 15 dpi in mice treated with MCP-1 and RABV-antibody positive serum while VNAs remained undetectable in mice treated with MCP-1 but without RABV-antibody positive serum (Fig. 5A). These mice developed severe signs of rabies. On the other hand, no significant differences were observed in IgG levels in the brain and serum between DRV-infected mice treated with MCP-1 with or without RABV-antibody positive serum (Fig. 5C and D). IgG levels in the brains peaked immediately 12 h after administration which correlates with the timing of BBB permeability being enhanced by MCP-1 (Fig. 5C). Together, these results suggest that wt RABV fails to elicit VNA responses and passively-transferred RABV-antibody positive serum along with MCP-1 is needed for mice to maintain a protective VNA level in the brain.
As shown in Fig. 5E, virus copy numbers increased steadily in the brain of mice infected with DRV-Mexico and treated with MCP-1 plus RABV-negative serum. The highest virus copy number was observed on 10 dpi when infected mice developed severe clinical signs. In contrast, viral genomic RNA gradually decreased in the brains of mice infected with DRV-Mexico and treated with MCP-1 and RABV-antibody positive serum. Similar results were also observed for virus titers (data not shown). The decrease in virus copy numbers and titers corresponds to the increase of VNA in the brain. At 10 dpi, virus copy number in the brain of mice treated with RABV-negative serum was 1000 times more than in mice treated with RABV-antibody positive serum. Similar virus copy numbers were detected in the brains of mice infected with DVR-Mexico and treated with MCP-1 alone and those without any treatment at 10 dpi. By 15 dpi (10 days after treatment), virus was no longer detectable in the surviving mice treated with MCP-1 and RABV-antibody positive serum. These findings indicated that MCP-1 alone is not capable of clearing RABV from the brain. Instead, intravenous administration of VNA, in addition to MCP-1, is crucial in clearing RABV from the brain.
The copy numbers of κ-light chain as detected by RT-PCR were similar in the brains of mice treated with MCP-1 plus RABV-antibody positive or negative serum (Fig. 5F). The level was very low in the beginning stage of infection and reached high levels by 10 dpi. However, the copy numbers of κ-light chain were significantly higher in mice with MCP-1 treatment than without. These data may indicate that plasma cells induced by infection with DRV-Mexico can cross the BBB after treatment with MCP-1.
3.5. VNA in the absence of antibody-producing cells can prevent mice from developing rabies after BBB is compromised in B-deficient mice
To exclude the possibility that VNA-producing plasma cells entering into the CNS are absolutely required to clear RABV from the CNS, the experiment as summarized in Fig. 1 was repeated in B-cell deficient mice. B6.129S2-Ighmtm1Cgn/J (B-cell deficient) and C57BL/6J (background) mice were inoculated intramuscularly with 10 IMLD50 DRV-Mexico. Half of the mice were left untreated and the other half treated intravenously with RABV-antibody positive serum in conjunction with MCP-1 at 5 dpi. Without treatment (MCP-1 or VNA), B-cell deficient and C57BL/6J mice all died by 12–14 dpi (Fig. 6 ). Eighty percent of the C57BL/6J mice treated with RABV-antibody positive serum and MCP-1 survived (Fig. 6) and κ-light chain mRNA was detected in the CNS of surviving mice (Fig. 7 D). Only 25% of the B6.129S2-Ighmtm1Cgn/J mice survived when treated at day 5 after infection (Fig. 6) despite the fact that VNA titers were still high in the serum (Fig. 7C). This might be due to the fact that VNA level declined 48 h after injection of MCP-1 (Fig. 5). Thus additional dose of MCP-1 was given at day 7 after infection, which increased significantly (p < 0.05) the survival rate from 25% (MCP-1 given only at 5 dpi) to 78% (MCP-1 given at both 5 and 7 dpi) in B6.129S2-Ighmtm1Cgn/J mice (Fig. 6). Yet, no κ-light chain was detected in the CNS of these mice (Fig. 7D). Virus was detected only in the brains of mice succumbed to rabies, but not in those that survived (Fig. 7A and B). From these results, it is clear that antibody-producing plasma cells are not absolutely required to clear RABV if sufficient VNAs are allowed to enter the CNS.
Fig. 6.
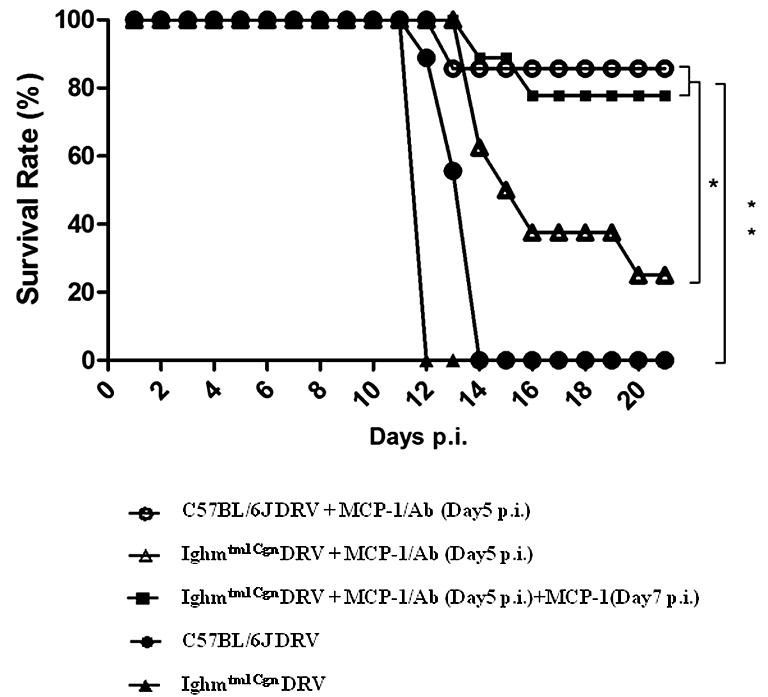
Protective efficacy in B-cell deficient mice treated with VNA and MCP-1after infection with DRV. C57BL/6J and Ighmtm1Cgn (B-cell deficient) mice (groups of 8) at the age of 4–6 weeks were infected i.m. with 10 IMLD50 DRV-Mexico and then treated with RABV- antibody positive serum (Ab) in conjunction with MCP-1 at 5 dpi or at both 5 and 7 dpi. Infected and treated mice were observed daily for 21 days and survivorship was recorded and analyzed. Asterisks indicate significant differences (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001) between the indicated experimental groups.
Fig. 7.

Virus titers and virus copy number in brains, serum VNA and the expression of κ-light chain mRNA in brains of B-cell deficient mice treated with RABV-antibody positive serum and MCP-1 after infection with DRV. C57BL/6J and Ighmtm1Cgn (B-cell deficient) mice (groups of 8) at the age of 4–6 weeks were infected i.m. with 10 IMLD50 DRV and then treated with RABV-antibody positive serum (Ab) in conjunction with MCP-1 at 5 dpi or at both 5 and 7 dpi. Infected and treated mice were observed daily for 21 days. Both sera and brains were harvested once mice reached the criteria of euthanasia by developing paralysis in both hind legs (Dead) or survived through day 21 (Survived) for the determination of virus titers (A) and viral genomic RNA copy number (B) in brains, serum VNA (C) and κ-light chain mRNA in brains (D). Data from groups of n ⩾ 3 mice are presented as mean values ± SEM, except for group e with only one C57BL/6J mouse dead. Asterisks indicate significant differences (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001) between the indicated experimental groups. ND: not done.
4. Discussion
Our previous studies indicated that intracerebral administration of rRABV-GMCSF could stimulate the production of VNA, enhance the BBB permeability, and clear RABV from the brain, therefore, preventing mice from developing rabies as late as 5 days after infection with a wt RABV. However, administration of inactivated rRABV-GMCSF did not improve the survival rate despite the fact that VNA was produced in the periphery. Yet enhancing the BBB permeability by MCP-1 could improve the survivor rate of mice treated with inactivated rRABV-GMCSF (Wang et al., 2011). In the present study, we demonstrated that exogenously delivered VNA is crucial in clearing an established infection by wt RABV in the brain and preventing the development of rabies in immunocompetent and immunodeficient mice as long as the BBB permeability remains enhanced.
Despite natural infection of mice being rare, mouse models have been used in many rabies studies given their short incubation period. Unlike humans where RABVs may take weeks to reach the CNS from the site of exposure and cause clinical illness, the spread of RABVs to the CNS in mice is rapid with virus generally being detectable in CNS tissues within 48 h of infection (Hooper, 2005). Clinical signs, particularly neurologic signs, are believed to indicate neuronal injury/dysfunction related to virus replication in CNS neurons. Hindlimb paralysis, a major neurological sign of rabies infection in mice, may not be the best endpoint for wt rabies infection in mice, especially given that infection of spinal neurons was believed to be the cause of hindlimb paralysis in infections with laboratory rabies strain (CVS-11). However, our previous studies demonstrated that the highest RABV titer was detected in the brains, at 9 dpi, right before RABV-infected mice develop hindlimb paralysis (Wang et al., 2011). Therefore, it was used as the humane endpoint in present this study to demonstrate the clearance of RABV from the CNS by passively transferred antibodies and also to minimize the suffering of the infected mice. BBB is a physical and physiological barrier formed by the endothelial tight junctions, impeding the influx of most compounds from the peripheral circulation to enter the CNS (Abbott et al., 2010, Ballabh et al., 2004). Thus, BBB plays an important role in maintaining the homeostasis in the CNS (Neuwelt, 2004) and enhancement of BBB permeability has often been associated with CNS diseases (Spindler and Hsu, 2012). Some viruses, such as lymphocytic choriomeningitis virus and attenuated RABV, that infect the CNS can enhance the BBB permeability resulting in infiltration of inflammatory cells and pathological changes (Kim et al., 2009, Phares et al., 2006). In wt RABV infection, however, BBB permeability is not enhanced since RABV bypasses the BBB and enter the CNS via axonal transport directly from the site of peripheral inoculation (Hooper et al., 2009, Roy and Hooper, 2008). Enhancement of BBB permeability is thus required for immune effectors to enter into the CNS to clear RABV and prevent the occurrence of rabies (Phares et al., 2006, Roy et al., 2007). To enhance the BBB permeability for RABV clearance from the CNS, lab-attenuated RABV (Phares et al., 2006) and recombinant RABV expressing three copies of G (Faber et al., 2009) or GM-CSF (Wang et al., 2011, Wen et al., 2011) have been directly injected into the brain and were found to stimulate VNA production and enhance the BBB permeability, resulting in the clearance of RABV from the CNS and prevention of the development of rabies in the mouse model after infection with wt RABV.
MCP-1 has been reported to transiently enhance the BBB permeability in mice without causing obvious side effects (Stamatovic et al., 2005) and has been used to enhance the BBB permeability for RABV clearance in our previous studies (Wang et al., 2011). In the present study, the role of MCP-1 in enhancing the BBB permeability was investigated, including the dynamics of BBB permeability change, the level of RABV VNA in the brain, and the down-regulation of tight junction proteins in the brain vasculature. A single intracerebral dose of MCP-1 enhanced the BBB permeability significantly over the control mice. The enhancement reached a peak by 36 h, an observation similar to that previously reported (Stamatovic et al., 2005). Administration of MCP-1 resulted in the reduction of TJ protein (occludin and claudin-5) expression in our studies. It has been known that enhancement of BBB permeability correlates with the reduction of TJ protein expression in the brain vasculature (Chai et al., 2014, Kirk et al., 2003, Kniesel and Wolburg, 2000, Liao et al., 2012). Our results further support the functional role for the cytoplasmic anchorage of TJ particles for brain endothelial barrier function. VNA levels in the brains were also measured to evaluate whether the compromised BBB allows antibodies to leak into the brain. The possible interference of circulating antibodies, particularly VNAs, in brain vasculature was minimized by PBS perfusion before sample collection as previously described (Yu et al., 2011). It is always possible that residual antibodies which did not pass the BBB may not be completely eliminated by perfusion. However, VNA in the brain was found to be significantly higher in mice treated with the combination of VNA and MCP-1 than in mice treated with VNA alone, which indicates that significantly more VNA passed through the BBB with MCP-1 treatment since the VNA level in the serum was similar in these animals. Therefore, enhancement of BBB permeability allowed intravenously administered VNA to pass through the BBB and into the brain which inhibited RABV replication, leading to eventual clearance of RABV from the brain and prevention of the development of rabies. Without MCP-1, intravenously administered VNA cannot pass through the BBB to clear RABV in the brain and prevent the development of rabies. Enhancement of BBB permeability alone, however, cannot prevent the development of rabies in mice infected with wt RABV as has been demonstrated clearly in previous (Wang et al., 2011) and this study. Although MCP-1 is capable of recruiting leukocytes into the CNS (Yadav et al., 2010) and intracerebral injection of MCP-1 may induce a localized inflammation secondary to the mechanical stimuli, MCP-1 (i.c.) alone or a combination of MCP-1 (i.c.) and negative serum (i.v.) is insufficient to clear wt RABV from the brain and prevent the development of rabies. These studies indicate that MCP-1 alone or mechanical stimuli of intracerebral injection does not lead to the clearance of RABV from the brain or increase survival.
VNA has been suggested to be produced in the brains of mice infected with attenuated RABV (CVS-F3) which may thus contribute to RABV clearance (Hooper et al., 2009). However, it has been known for a long time that natural infection with wt RABV does not induce a strong antiviral immune responses (Baltazard and Ghodssi, 1954). Hemachudha (1994) reported that less than 30% of the human rabies patients developed VNA at the time of death. In this present study, no VNA was detected in mice infected with DRV-Mexico and all mice died without any intervention. This finding further suggests that although non-VNA could be made in the brain, it does not contribute to survival during wt rabies infections. The mechanism by which wt RABV does not induce the production of VNA is not entirely clear at the moment and further studies are warranted.
It has been hypothesized that it is the VNA produced in situ (CNS) by invading B cells, not those produced in the periphery and then transported into the CNS, are important in clearing RABV from the CNS (Hooper et al., 2009). Passively transferred VNA via intraperitoneal route failed to clear attenuated RABV from the brain in B-cell deficient mice. However, our studies summarized here demonstrate a different result in the wt RABV-infected mouse model, which is likely more relevant to clinical rabies in humans. The intravenously administered VNA is important in clearing wt RABV from the brain and preventing the development of rabies not only in immunocompetent mice, but also in B-cell deficient mice as long as the BBB permeability remains enhanced. Thus production of VNA in situ (brain) by plasma cells is not absolutely required for wt RABV clearance from the brain. Similar results have also been reported for coronavirus-induced encephalomyelitis. Although recruitment of virus-specific antibody-secreting cells (ASC) mediated by CXCR3 has been found helpful in controlling coronavirus-induced encephalomyelitis (Marques et al., 2011, Phares et al., 2013), viral recrudescence can be prevented in B-cell-deficient and IgM−/− mice by intraperitoneal administrations of JHMV-specific neutralizing antibodies (Lin et al., 1999, Ramakrishna et al., 2003). Thus these findings further demonstrate that production of VNA in situ (brain) by antibody-secreting cells (ASC) is not absolutely required for clearance of viruses from the brain.
It was found in our study that MCP-1 has to be administered twice (at 5 and 7 dpi) in B-cell deficient mice in order to achieve a similar rate of protection as found in immunocompetent mice after one injection. We attribute this to the transient nature of MCP-1 in enhancing the BBB permeability (Stamatovic et al., 2005) and the low level of VNA present in the brain (∼0.3 IU) despite the fact that VNA level in the serum remains high (3–4 IU). Thus continued enhancement of BBB permeability is required for maintaining sufficient VNA level in the brain to clear RABV. It was speculated that B cell deficient mice could mediate non-antibody dependent antiviral mechanisms and thus play a role in the clearance of RABV from the brain. However, all mice died without being treated with RABV antibody-positive serum and more mice survived when treated with MCP-1 twice than those treated only once, further indicating that VNA passing into the brain from the periphery played a decisive role in clearing RABV from the brain in the B-cell deficient mice.
Although traditional inactivated RABV vaccines have been used for pre- and post-exposure prophylaxis in humans with high safety and efficacy, they are not useful for delayed treatment and can even accelerate the rabies (CDC, 2010, Rupprecht et al., 2002, Sampath et al., 2005). Other vaccines, such as, live-attenuated RABV vaccine and recombinant vaccine expressing RABV G, are limited to use only in wildlife animals (Brochier et al., 1991, Hanlon et al., 1998, Schumacher et al., 1993). Recent studies suggested that live-attenuated vaccines expressing multiple copies of the G or immune-stimulating molecules not only have the potential to replace the traditional inactivated RABV vaccines, but also can clear RABV from the CNS if administered intracerebrally (Faber et al., 2009, Wang et al., 2011). However, intracerebral administration of live-attenuated or recombinant RABV still possesses safety concerns. Our results demonstrated that VNA can be administered by intravenous route and is crucial for RABV clearance from the brain before the appearance of clinical signs. It would be interesting to initiate the treatment after the appearance of clinical signs. However, there are still obstacles to overcome. For example, virus burden will increase and changes in neuronal injury/dysfunction may not be reversible at late stage of RABV infection. Thus further studies are warranted. Nevertheless, the findings presented in this study provide a foundation for developing VNA therapy for human clinical rabies.
Acknowledgement
This work was supported partially by Public Health Service Grants AI-051560 and AI-093369 from the National Institute of Allergy and Infectious Diseases.
References
- Abbott N.J., Patabendige A.A., Dolman D.E., Yusof S.R., Begley D.J. Structure and function of the blood–brain barrier. Neurobiol. Dis. 2010;37:13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- Armstrong S.M., Wang C., Tigdi J., Si X., Dumpit C., Charles S., Gamage A., Moraes T.J., Lee W.L. Influenza infects lung microvascular endothelium leading to microvascular leak: role of apoptosis and claudin-5. PLoS One. 2012;7:e47323. doi: 10.1371/journal.pone.0047323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballabh P., Braun A., Nedergaard M. The blood–brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol. Dis. 2004;16:1–13. doi: 10.1016/j.nbd.2003.12.016. [DOI] [PubMed] [Google Scholar]
- Baltazard M., Ghodssi M. Prevention of human rabies: treatment of persons bitten by rabid wolves in Iran. Bull. World Health Organ. 1954;10:797–803. [PMC free article] [PubMed] [Google Scholar]
- Braniste V., Leveque M., Buisson-Brenac C., Bueno L., Fioramonti J., Houdeau E. Oestradiol decreases colonic permeability through oestrogen receptor beta-mediated up-regulation of occludin and junctional adhesion molecule-A in epithelial cells. J. Physiol. 2009;587:3317–3328. doi: 10.1113/jphysiol.2009.169300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brochier B., Kieny M.P., Costy F., Coppens P., Bauduin B., Lecocq J.P., Languet B., Chappuis G., Desmettre P., Afiademanyo K. Large-scale eradication of rabies using recombinant vaccinia-rabies vaccine. Nature. 1991;354:520–522. doi: 10.1038/354520a0. [DOI] [PubMed] [Google Scholar]
- CDC, 2010. Use of a Reduced (4-Dose) Vaccine Schedule for Postexposure Prophylaxis to Prevent Human Rabies. MMWR 59. [PubMed]
- Chai Q., He W.Q., Zhou M., Lu H., Fu Z.F. Enhancement of blood–brain barrier permeability and reduction of tight junction protein expression are modulated by chemokines/cytokines induced by rabies virus infection. J. Virol. 2014;88:4698–4710. doi: 10.1128/JVI.03149-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleaveland S., Kaare M., Knobel D., Laurenson M.K. Canine vaccination – providing broader benefits for disease control. Vet. Microbiol. 2006;117:43–50. doi: 10.1016/j.vetmic.2006.04.009. [DOI] [PubMed] [Google Scholar]
- Dietzschold B., Morimoto K., Hooper D.C., Smith J.S., Rupprecht C.E., Koprowski H. Genotypic and phenotypic diversity of rabies virus variants involved in human rabies: implications for postexposure prophylaxis. J. Hum. Virol. 2000;3:50–57. [PubMed] [Google Scholar]
- Faber M., Pulmanausahakul R., Hodawadekar S.S., Spitsin S., McGettigan J.P., Schnell M.J., Dietzschold B. Overexpression of the rabies virus glycoprotein results in enhancement of apoptosis and antiviral immune response. J. Virol. 2002;76:3374–3381. doi: 10.1128/JVI.76.7.3374-3381.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber M., Li J., Kean R.B., Hooper D.C., Alugupalli K.R., Dietzschold B. Effective preexposure and postexposure prophylaxis of rabies with a highly attenuated recombinant rabies virus. Proc. Natl. Acad. Sci. USA. 2009;106:11300–11305. doi: 10.1073/pnas.0905640106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favoretto S.R., Carrieri M.L., Tino M.S., Zanetti C.R., Pereira O.A. Simplified fluorescent inhibition microtest for the titration of rabies neutralizing antibodies. Rev. Inst. Med. Trop. Sao Paulo. 1993;35:171–175. doi: 10.1590/s0036-46651993000200009. [DOI] [PubMed] [Google Scholar]
- Griffin D.E. Immune responses to RNA-virus infections of the CNS. Nat. Rev. Immunol. 2003;3:493–502. doi: 10.1038/nri1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampson K., Dushoff J., Cleaveland S., Haydon D.T., Kaare M., Packer C., Dobson A. Transmission dynamics and prospects for the elimination of canine rabies. PLoS Biol. 2009;7:e53. doi: 10.1371/journal.pbio.1000053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanlon C.A., Niezgoda M., Hamir A.N., Schumacher C., Koprowski H., Rupprecht C.E. First North American field release of a vaccinia-rabies glycoprotein recombinant virus. J. Wildl. Dis. 1998;34:228–239. doi: 10.7589/0090-3558-34.2.228. [DOI] [PubMed] [Google Scholar]
- Hemachudha T. Human rabies: clinical aspects, pathogenesis, and potential therapy. Curr. top. microbio. 1994;187:121–143. doi: 10.1007/978-3-642-78490-3_7. [DOI] [PubMed] [Google Scholar]
- Hemachudha T., Sunsaneewitayakul B., Desudchit T., Suankratay C., Sittipunt C., Wacharapluesadee S., Khawplod P., Wilde H., Jackson A.C. Failure of therapeutic coma and ketamine for therapy of human rabies. J. Neurovirol. 2006;12:407–409. doi: 10.1080/13550280600902295. [DOI] [PubMed] [Google Scholar]
- Hooper D.C. The role of immune responses in the pathogenesis of rabies. J. Neurovirol. 2005;11:88–92. doi: 10.1080/13550280590900418. [DOI] [PubMed] [Google Scholar]
- Hooper D.C., Phares T.W., Fabis M.J., Roy A. The production of antibody by invading B cells is required for the clearance of rabies virus from the central nervous system. PLoS Negl. Trop. Dis. 2009;3:e535. doi: 10.1371/journal.pntd.0000535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosking M.P., Lane T.E. The role of chemokines during viral infection of the CNS. PLoS Pathog. 2010;6:e1000937. doi: 10.1371/journal.ppat.1000937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson A.C. Rabies pathogenesis. J. Neurovirol. 2002;8:267–269. doi: 10.1080/13550280290100725. [DOI] [PubMed] [Google Scholar]
- Jackson A.C. Current and future approaches to the therapy of human rabies. Antiviral Res. 2013;99:61–67. doi: 10.1016/j.antiviral.2013.01.003. [DOI] [PubMed] [Google Scholar]
- Jackson A.C. third ed. Academic Press; 2013. Rabies: Scientific Basis of the Disease and its Management. [Google Scholar]
- Kim J.V., Kang S.S., Dustin M.L., McGavern D.B. Myelomonocytic cell recruitment causes fatal CNS vascular injury during acute viral meningitis. Nature. 2009;457:191–195. doi: 10.1038/nature07591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirk J., Plumb J., Mirakhur M., McQuaid S. Tight junctional abnormality in multiple sclerosis white matter affects all calibres of vessel and is associated with blood–brain barrier leakage and active demyelination. J. Pathol. 2003;201:319–327. doi: 10.1002/path.1434. [DOI] [PubMed] [Google Scholar]
- Kniesel U., Wolburg H. Tight junctions of the blood–brain barrier. Cell. Mol. Neurobiol. 2000;20:57–76. doi: 10.1023/A:1006995910836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knobel D.L., Cleaveland S., Coleman P.G., Fevre E.M., Meltzer M.I., Miranda M.E., Shaw A., Zinsstag J., Meslin F.X. Re-evaluating the burden of rabies in Africa and Asia. Bull. World Health Organ. 2005;83:360–368. [PMC free article] [PubMed] [Google Scholar]
- Kuang Y., Lackay S.N., Zhao L., Fu Z.F. Role of chemokines in the enhancement of BBB permeability and inflammatory infiltration after rabies virus infection. Virus Res. 2009;144:18–26. doi: 10.1016/j.virusres.2009.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao P.H., Yang H.H., Chou P.T., Wang M.H., Chu P.C., Liu H.L., Chen L.K. Sufficient virus-neutralizing antibody in the central nerve system improves the survival of rabid rats. J. Biomed. Sci. 2012;19:61. doi: 10.1186/1423-0127-19-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin M.T., Hinton D.R., Marten N.W., Bergmann C.C., Stohlman S.A. Antibody prevents virus reactivation within the central nervous system. J. Immunol. 1999;162:7358–7368. [PubMed] [Google Scholar]
- Marques C.P., Kapil P., Hinton D.R., Hindinger C., Nutt S.L., Ransohoff R.M., Phares T.W., Stohlman S.A., Bergmann C.C. CXCR3-dependent plasma blast migration to the central nervous system during viral encephalomyelitis. J. Virol. 2011;85:6136–6147. doi: 10.1128/JVI.00202-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez L. Global infectious disease surveillance. Int. J. Infect. Dis. 2000;4:222–228. doi: 10.1016/s1201-9712(00)90114-0. [DOI] [PubMed] [Google Scholar]
- McDermid R.C., Saxinger L., Lee B., Johnstone J., Gibney R.T., Johnson M., Bagshaw S.M. Human rabies encephalitis following bat exposure: failure of therapeutic coma. CMAJ. 2008;178:557–561. doi: 10.1503/cmaj.071326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCW, 2009. The Milwaukee Rabies Protocol, Version 3.1. <http://www.mcw.edu/FileLibrary/Groups/Pediatrics/InfectiousDiseases/Milwaukee_rabies_protocol_V3_1.pdf>.
- Meslin F.X., Fishbein D.B., Matter H.C. Rationale and prospects for rabies elimination in developing countries. Curr. Top. Microbiol. Immunol. 1994;187:1–26. doi: 10.1007/978-3-642-78490-3_1. [DOI] [PubMed] [Google Scholar]
- Messenger S.L., Smith J.S., Orciari L.A., Yager P.A., Rupprecht C.E. Emerging pattern of rabies deaths and increased viral infectivity. Emerg. Infect. Dis. 2003;9:151–154. doi: 10.3201/eid0902.020083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto K., Patel M., Corisdeo S., Hooper D.C., Fu Z.F., Rupprecht C.E., Koprowski H., Dietzschold B. Characterization of a unique variant of bat rabies virus responsible for newly emerging human cases in North America. Proc. Natl. Acad. Sci. USA. 1996;93:5653–5658. doi: 10.1073/pnas.93.11.5653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuwelt E.A. Mechanisms of disease: the blood–brain barrier. Neurosurgery. 2004;54:131–140. doi: 10.1227/01.neu.0000097715.11966.8e. discussion 141-132. [DOI] [PubMed] [Google Scholar]
- Phares T.W., Kean R.B., Mikheeva T., Hooper D.C. Regional differences in blood–brain barrier permeability changes and inflammation in the apathogenic clearance of virus from the central nervous system. J. Immunol. 2006;176:7666–7675. doi: 10.4049/jimmunol.176.12.7666. [DOI] [PubMed] [Google Scholar]
- Phares T.W., Fabis M.J., Brimer C.M., Kean R.B., Hooper D.C. A peroxynitrite-dependent pathway is responsible for blood–brain barrier permeability changes during a central nervous system inflammatory response: TNF-alpha is neither necessary nor sufficient. J. Immunol. 2007;178:7334–7343. doi: 10.4049/jimmunol.178.11.7334. [DOI] [PubMed] [Google Scholar]
- Phares T.W., Stohlman S.A., Bergmann C.C. Intrathecal humoral immunity to encephalitic RNA viruses. Viruses. 2013;5:732–752. doi: 10.3390/v5020732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishna C., Bergmann C.C., Atkinson R., Stohlman S.A. Control of central nervous system viral persistence by neutralizing antibody. J. Virol. 2003;77:4670–4678. doi: 10.1128/JVI.77.8.4670-4678.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed L.J., Muench H. A simple method of estimating fifty per cent endpoints. Am. J. Epidemiol. 1938;27:493–497. [Google Scholar]
- Roy A., Hooper D.C. Lethal silver-haired bat rabies virus infection can be prevented by opening the blood–brain barrier. J. Virol. 2007;81:7993–7998. doi: 10.1128/JVI.00710-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A., Hooper D.C. Immune evasion by rabies viruses through the maintenance of blood–brain barrier integrity. J. Neurovirol. 2008;14:401–411. doi: 10.1080/13550280802235924. [DOI] [PubMed] [Google Scholar]
- Roy A., Phares T.W., Koprowski H., Hooper D.C. Failure to open the blood–brain barrier and deliver immune effectors to central nervous system tissues leads to the lethal outcome of silver-haired bat rabies virus infection. J. Virol. 2007;81:1110–1118. doi: 10.1128/JVI.01964-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupprecht C.E. Rhabdoviruses: rabies virus. In: Baron S., editor. Medical Microbiology. fourth ed. University of Texas Medical Branch at Galveston; 1996. pp. 751–760. [PubMed] [Google Scholar]
- Rupprecht C.E., Hanlon C.A., Hemachudha T. Rabies re-examined. Lancet Infect. Dis. 2002;2:327–343. doi: 10.1016/s1473-3099(02)00287-6. [DOI] [PubMed] [Google Scholar]
- Sampath G., Parikh S., Sangram P., Briggs D.J. Rabies post-exposure prophylaxis in malnourished children exposed to suspect rabid animals. Vaccine. 2005;23:1102–1105. doi: 10.1016/j.vaccine.2004.08.036. [DOI] [PubMed] [Google Scholar]
- Schumacher C.L., Coulon P., Lafay F., Benejean J., Aubert M.F., Barrat J., Aubert A., Flamand A. SAG-2 oral rabies vaccine. Onderstepoort J. Vet. Res. 1993;60:459–462. [PubMed] [Google Scholar]
- Spindler K.R., Hsu T.H. Viral disruption of the blood–brain barrier. Trends Microbiol. 2012;20:282–290. doi: 10.1016/j.tim.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatovic S.M., Keep R.F., Kunkel S.L., Andjelkovic A.V. Potential role of MCP-1 in endothelial cell tight junction ‘opening’: signaling via Rho and Rho kinase. J. Cell Sci. 2003;116:4615–4628. doi: 10.1242/jcs.00755. [DOI] [PubMed] [Google Scholar]
- Stamatovic S.M., Shakui P., Keep R.F., Moore B.B., Kunkel S.L., Van Rooijen N., Andjelkovic A.V. Monocyte chemoattractant protein-1 regulation of blood–brain barrier permeability. J. Cereb. Blood Flow Metab. 2005;25:593–606. doi: 10.1038/sj.jcbfm.9600055. [DOI] [PubMed] [Google Scholar]
- Trout J.J., Koenig H., Goldstone A.D., Lu C.Y. Blood–brain barrier breakdown by cold injury. Polyamine signals mediate acute stimulation of endocytosis, vesicular transport, and microvillus formation in rat cerebral capillaries. Lab. Invest. 1986;55:622–631. [PubMed] [Google Scholar]
- Wang H., Zhang G., Wen Y., Yang S., Xia X., Fu Z.F. Intracerebral administration of recombinant rabies virus expressing GM-CSF prevents the development of rabies after infection with street virus. PLoS One. 2011;6:e25414. doi: 10.1371/journal.pone.0025414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Y., Wang H., Wu H., Yang F., Tripp R.A., Hogan R.J., Fu Z.F. Rabies virus expressing dendritic cell-activating molecules enhances the innate and adaptive immune response to vaccination. J. Virol. 2011;85:1634–1644. doi: 10.1128/JVI.01552-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO, 1992. WHO Expert Committee on rabies. In: World Health Organization Technical Report Series 824, pp. 1–84. [PubMed]
- Wilde H. Failures of post-exposure rabies prophylaxis. Vaccine. 2007;25:7605–7609. doi: 10.1016/j.vaccine.2007.08.054. [DOI] [PubMed] [Google Scholar]
- Willoughby R.E., Jr., Tieves K.S., Hoffman G.M., Ghanayem N.S., Amlie-Lefond C.M., Schwabe M.J., Chusid M.J., Rupprecht C.E. Survival after treatment of rabies with induction of coma. N. Engl. J. Med. 2005;352:2508–2514. doi: 10.1056/NEJMoa050382. [DOI] [PubMed] [Google Scholar]
- Yadav A., Saini V., Arora S. MCP-1: chemoattractant with a role beyond immunity: a review. Clin. Chim. Acta. 2010;411:1570–1579. doi: 10.1016/j.cca.2010.07.006. [DOI] [PubMed] [Google Scholar]
- Yousaf M.Z., Qasim M., Zia S., Khan M., Ashfaq U.A., Khan S. Rabies molecular virology, diagnosis, prevention and treatment. Virol. J. 2012;9:50. doi: 10.1186/1743-422X-9-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y.J., Zhang Y., Kenrick M., Hoyte K., Luk W., Lu Y., Atwal J., Elliott J.M., Prabhu S., Watts R.J., Dennis M.S. Boosting brain uptake of a therapeutic antibody by reducing its affinity for a transcytosis target. Sci. Transl. Med. 2011;3:84ra44. doi: 10.1126/scitranslmed.3002230. [DOI] [PubMed] [Google Scholar]
- Zhang G., Fu Z.F. Complete genome sequence of a street rabies virus from Mexico. J. Virol. 2012;86:10892–10893. doi: 10.1128/JVI.01778-12. [DOI] [PMC free article] [PubMed] [Google Scholar]