

Abstract
We previously reported that continuous 24-month costimulation blockade by abatacept significantly slows the decline of β-cell function after diagnosis of type 1 diabetes. In a mechanistic extension of that study, we evaluated peripheral blood immune cell subsets (CD4, CD8-naive, memory and activated subsets, myeloid and plasmacytoid dendritic cells, monocytes, B lymphocytes, CD4+CD25high regulatory T cells, and invariant NK T cells) by flow cytometry at baseline and 3, 6, 12, 24, and 30 months after treatment initiation to discover biomarkers of therapeutic effect. Using multivariable analysis and lagging of longitudinally measured variables, we made the novel observation in the placebo group that an increase in central memory (CM) CD4 T cells (CD4+CD45R0+CD62L+) during a preceding visit was significantly associated with C-peptide decline at the subsequent visit. These changes were significantly affected by abatacept treatment, which drove the peripheral contraction of CM CD4 T cells and the expansion of naive (CD45R0−CD62L+) CD4 T cells in association with a significantly slower rate of C-peptide decline. The findings show that the quantification of CM CD4 T cells can provide a surrogate immune marker for C-peptide decline after the diagnosis of type 1 diabetes and that costimulation blockade may exert its beneficial therapeutic effect via modulation of this subset.
Introduction
Type 1 diabetes results from autoimmune damage to pancreatic islet β-cells, a process that is widely believed to be mediated by the combined effects of the innate and adaptive immune systems (1). In recent decades, this knowledge has spawned numerous attempts to halt or limit immune-mediated β-cell destruction by using immunosuppressive (2,3) or antigen-based therapies (4,5). Some trials have shown important proof-of-concept that immune-based interventions can successfully delay the decline of functional β-cell mass, when assessed by the measurement of stimulated C-peptide release. A short course of nondepleting monoclonal antibody directed against CD3 on T cells (6,7) and depletion of B lymphocytes with a short-course of anti-CD20 monoclonal antibody (8) showed similar potency in delaying the decline of stimulated C-peptide release. More recently, the Type 1 Diabetes TrialNet Abatacept Study Group showed the benefit of continued administration of the costimulation blocking biologic agent CTLA-4-Ig (abatacept) (9). These are landmark studies, providing incremental advances in immune-based intervention strategies to prevent β-cell loss. Nevertheless, a clear understanding of the mechanisms of action of these agents on relevant immunological pathways is lacking.
This knowledge gap contributes to a bottleneck in the further development of type 1 diabetes interventions. It is difficult to build upon these successes and rationally design next-generation trials without some insight into the mechanism responsible for the achievement of therapeutic benefit. It has also been suggested (10) that future strategies for type 1 diabetes prevention might make use of combination approaches to achieve synergistic effects with more than one agent. This approach, in particular, would benefit from biomarkers of the individual component therapies to maximize and monitor success (11).
A further missing component in the translational pathway to successful type 1 diabetes prevention and intervention is a lack of biomarkers that reflect ongoing activity of the autoimmune process. Such measures could be deployed as surrogate end points for therapeutic interventions, as means of stratification for entry into clinical trials, and to provide an indication of the mechanism of action of a particular agent or combination. Importantly, the use of biomarkers as surrogate end points can limit patient exposures to potentially toxic drugs, expense, and time. To address these key knowledge gaps, it is important that opportunities for mechanistic studies and biomarker discovery are maximized, especially in the context of successful intervention studies and longitudinal sample collections in which data on β-cell function are collected.
An opportunity to address some of these issues arises in the context of the recent TrialNet study (9) of abatacept, a CTLA-4-Ig–soluble chimeric protein (extracellular domain of human CD152 and a fragment [hinge, CH2, and CH3 domains] of the Fc portion of human IgG1). Abatacept binds to CD80/86 on antigen-presenting cells and blocks their interaction with CD28 on T cells, a key second signal for T-cell activation (12,13). We hypothesized that abatacept treatment would interfere with T-cell activation and blunt the autoimmune destruction of β-cells, and that in the process there would be measurable effects on relevant immune cell populations such as CD4 and CD8 T-cell subsets, dendritic cells, and monocytes. Where these immune changes are disease critical, a direct relationship with the change in C-peptide levels might be uncovered. Here, we report these findings and provide evidence for a potent abatacept-mediated effect on selected T-cell populations that is associated with clinical efficacy and may also serve as the first immune biomarker of β-cell decline in the postdiagnosis period.
Research Design and Methods
Study Design
This phase 2 clinical trial was registered with ClinicalTrials.gov (NCT00505375), and the primary outcome results have been published (9). A total of 112 type 1 diabetes patients, ages 6–45 years, who were autoantibody positive, had a diagnosis of type 1 diabetes for <100 days, and had stimulated C-peptide levels of ≥0.2 pmol/mL measured during a mixed-meal tolerance test, were enrolled in a study with parallel-group design. Patients were randomized in a 2:1 ratio, with 77 subjects receiving abatacept and 35 subjects receiving placebo. Abatacept (Orencia; Bristol-Myers Squibb) was administered on days 1, 14, and 28, and then every 28 days, with the last administration on day 700 (month 24; 27 total doses) as a 30-min intravenous infusion (10 mg/kg; maximum 1,000 mg/dose) in a 100-mL solution of 0.9% sodium chloride. Normal saline solution infusion was used as a placebo. Changes in β-cell function during the study were assessed by measurement of the area under the curve (AUC) from the 2-h oral glucose tolerance test–based measurement of C-peptide. The AUC values were divided by 120 min and were reported as the mean C-peptide levels (9). Longitudinal C-peptide data from 74 abatacept-treated subjects and 32 placebo-treated subjects with a minimal number missing were available for analysis.
Using a predetermined mechanistic study plan, fresh peripheral blood immune cell immunophenotyping was performed by five-color flow cytometry at baseline, and 3, 6, 12, 24, and 30 months during the trial. Eight panels of immune cell markers (Table 1), designed by the Immune Tolerance Network (ITN), were deployed according to their standard operating procedures. All antibodies and other flow cytometry assay reagents, except for phycoerythrin (PE)-conjugated anti-CD25 (2A3; BD Pharmingen) were obtained from Beckman Coulter. One hundred microliters of fresh blood collected in heparin blood tube were mixed with titrated antibody and incubated at room temperature in the dark for 15 min. At the end of the incubation, erythrocytes were lysed and washed twice with phosphate-buffered saline. Stained cells were analyzed on an FC-500 flow cytometer (Beckman Coulter). CD152 was stained intracellularly using the IntraPrep kit (Beckman Coulter) according to the manufacturer’s instructions. Isotype controls and cell viability check by 7-amino-actinomycin D were also applied to each sample. Absolute T-cell subpopulation counts were obtained by the integration of percentage subset data obtained on the flow cytometer with total lymphocyte counts obtained using Beckman Coulter AcT–derived peripheral complete blood counts. Longitudinal T-cell data from 60 abatacept-treated and 27 placebo-treated subjects with a minimal amount of missing data were available for analysis.
Table 1.
Flow cytometry staining panels used to monitor abatacept study
| ITN panel no. | Fluorochrome and cell markers |
Main subpopulations identifiable | ||||
|---|---|---|---|---|---|---|
| FITC | PE | APC | PE-Cy5.5 | PE-Cy7 | ||
| 7 | CD45RA | CD45R0 | CD62L | CD8 | CD4 | CD4, CD8-naive, and memory subsets |
| 54 | CD11c | CD80-BMS | DUMP | HLA-DR | CD123 | Myeloid and plasmacytoid dendritic cell subsets |
| 55 | CD11c | CD86-BMS | DUMP | HLA-DR | CD123 | Myeloid and plasmacytoid dendritic cell subsets |
| 57 | CD14 | CD80-BMS | CD19 | CD3 | CD86-BMS | Monocytes, B cells, T cells |
| 122 | CD62L | CD25 | CD8 | CD4 | CD45R0 | Tregs (CD62L+CD25highCD8−CD4+CD45R0+) |
| 123 | 6B11* | Vα24 | CD8 | CD4 | CD69 | Invariant NK T cells |
| 124 | CD62L | CD25 | CD8 | CD4 | CTLA-4/CD152 | Tregs (CD62L+CD25highCD8−CD4+CD152+) |
| 131 | CD2 | CD69 | CD8 | CD4 | CD45R0 | Activated (CD69+) CD4 and CD8 T cells |
APC, antigen-presenting cell; FITC, fluorescein isothiocyanate.
*Clone recognizes invariant CDR3 region of TCR Vα24-JαQ.
Statistics
Cell proportion data were log(natural) transformed prior to analysis. Changes in cell populations from baseline across time (equivalent to the log of the ratio of cell population proportion to the baseline proportion) and changes in the log of the ratio of CD4-naive to central memory (CM) T-cell proportions were analyzed with mixed-effects general linear models (GLMs) having subject as a random effect. When type 3 tests of treatment (i.e., the effect of treatment controlling for time) were found to be significant at the 5% level, treatment groups were then compared at each visit using least-squares means and the Bonferroni correction for multiple testing. Pearson correlations in the change from baseline between each of three pairs of CD4+ T-cell populations (regulatory T cells [Tregs] vs. CM T cells, Tregs vs. naive cells, and CM T cells vs. naive cells) at monthly time points were computed and considered statistically significant at P < 0.01 (Bonferroni corrected). Changes in C-peptide levels from baseline were analyzed with mixed-effects GLM as well. Parameter estimates from the GLM analysis were used to estimate the reduction in the decline of C-peptide level per unit of change in T-cell populations. These models considered lag-0 and lag-1 versions of the T-cell variables. All statistical analyses were conducted with SAS version 9.2 (SAS Institute, Cary, NC). For the C-peptide AUC, we analyzed the log-transformed ratio of the AUC value divided by baseline AUC. For T-cell populations, we analyzed the log of the ratio of the percentage of the T-cell population at each visit to baseline value. Hence, both C-peptide AUC and T-cell variables can be interpreted as the log (proportion of baseline), making it easy to translate results and inferences back to statements about change, expressed as the percentage of baseline values (proportion of baseline × 100%). Examination of normal quantile-quantile plots supported the assumption of normality in the distribution of the log-transformed data. In particular, because the log-transformed data appear be symmetrically distributed, inferences about mean log(proportion of baseline) back-translate to inferences about the median proportion of baseline. The mixed-effects GLM is appropriate to the analysis of data when, as in our case, missing data are minimal and are assumed to occur at random.
This study conformed to all applicable regulatory requirements and was conducted according to the principles of the Declaration of Helsinki. The protocol and consent documents were approved by appropriate independent ethics committees or institutional review boards. All participants (or parents) provided written informed consent; in addition to their parents providing consent, participants younger than 18 years of age signed a study assent form.
Results
Immune Cell Changes in Peripheral Blood
We analyzed eight sets of immune cell markers (Table 1) using a fresh whole-blood assay performed at baseline, during the 2 years of intervention (abatacept or placebo infusions) at 3, 6, 12, and 24 months, and at month 30, 6 months after the last infusion. In the placebo group, there were no significant changes from baseline at these time points for any of the subsets identified by the eight sets of markers. There was no significant change from baseline in total CD3, CD4, and CD8 T-cell populations, in CD152 subset, or in absolute lymphocyte count in either subject group. In the abatacept-treated group, there was no change from baseline in B-cell, dendritic cell, or invariant NK T-cell subsets, or in activated (CD69+; panel 131) CD4, CD8, and CD2 T cells.
However, in the abatacept-treated group, we noted significant treatment-induced changes in subsets defined by ITN panels 7, 122, and 124. Abatacept treatment significantly reduced the median percentage of circulating CD45R0+CD62L+ (CM) CD4+ T cells, and significantly increased the median percentage of naive (CD45R0−CD62L+) CD4 T cells at 6, 12, and 24 months of treatment (Figs. 1A–C and 2). Concomitantly, the ratio of naive to CM cells also increased significantly over the same time period. All of these changes return close to baseline values 6 months after treatment cessation (month 30). Of importance, these changes were found in both panels 7 and 122, both of which contained monoclonal antibodies for CD4, CD45R0, and CD62L, but with varied fluorochromes. We also detected a significant reduction from baseline at 6, 12, and 24 months in the median percentage of CD4+CD25high T cells, which represent a population that is highly enriched for thymus-derived Tregs (Fig. 1D). Again, these changes had returned to baseline at 30 months, and identical findings were made in both panels 122 and 124, which contained the same fluorochrome-conjugated monoclonal antibodies for CD4 and CD25.
Figure 1.

Percentage change from baseline of CD4 T-cell subsets identified as representing naive (A) and CM (B) cell populations, as well as the naive cell/CM cell ratio (C) and Treg populations (D). Closed circles represent abatacept-treated subjects, and open circles represent placebo-treated subjects; symbols represent the median, and error bars represent 95% CIs. P values and dashed lines indicate that the two groups differ significantly over the time points indicated.
Figure 2.
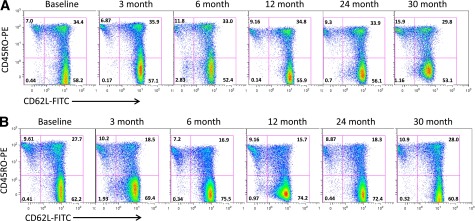
Representative flow cytomteric analyses of gated CD4 T cell–naive (CD45R0−CD62L+) and CM T cell (CD45R0+CD62L+) subpopulations at different time points during the study in which patients were receiving maintenance therapy (abatacept or placebo) when tested at 3, 6, 12, and 24 months, at which point treatment ceased. Numbers in quadrants are the percentages of each subset. A: Lysed whole-blood staining of a type 1 diabetes patient from the placebo group. There is no notable change in the percentage of CM cells (top right quadrant) or the percentage of naive cells (bottom right quadrant). B: A patient in the abatacept-treated arm of the study, in whom there is marked change in the proportion of circulating CM (reduced) and naive (increased) CD4 T cells, respectively. FITC, fluorescein isothiocyanate.
Similar findings to those described above for CM, naive, and Treg CD4 T cells were made when absolute counts of these subpopulations were analyzed, but these failed to reach statistical significance. The variances of the absolute T-cell subset counts were high (SD range 5.6–92 million counts).
CD4 T-Cell Subsets and Metabolic Changes
We then explored whether any of these changes in T-cell subsets were associated with metabolic changes using mixed linear modeling for longitudinal data. Our model-fitting strategy started with a multivariable analysis of treatment, with each of the T-cell populations (CD4+ T cells of CM, naive cells, and Treg subsets) included in the same model, time (expressed in months), and interaction of treatment with T-cell change. Only treatment and time were found to be significantly associated with metabolic change.
To explore possible interactions further, we then used a common statistical technique of “lagging” longitudinally measured explanatory variables (i.e., using the value of the T-cell change seen at the prior visit) and discovered that changes in certain cell populations during the preceding visit were significantly associated with C-peptide change after controlling for the other variables in the model. That is, we again found significant associations with treatment (type 3 P value = 0.0033) and time (P < 0.001), but, in addition, we also found that changes at the prior visit in CM (P = 0.0025) and naive (P = 0.0059) cell subsets were associated with the change in C-peptide levels seen at the current visit. Moreover, we found that the association between prior values of CM T cells and a subsequent change in C-peptide levels was affected by treatment (P = 0.0126).
Estimating the Effect of CD4+CD45R0+CD62L+ Change on Subsequent Change in C-Peptide Levels
Based on these findings, we then fit models having only the lagged versions of each of the T-cell changes separately, along with treatment and time. These models confirmed statistically significant associations between metabolic changes and previous changes in CM (type 3 P value = 0.0097) and naive (P = 0.0327) cell populations, and the ratio of these two populations (P = 0.0006), but not Tregs (P = 0.0817). In addition, the analysis found again that treatment significantly altered the association between prior values of CM T cells and the naive cell/CM cell ratio and the subsequent change in C-peptide levels (P = 0.0004 and 0.0010, respectively, based on Wald tests).
To illustrate, Fig. 3 plots the 2-year log change from baseline in C-peptide level versus the 1-year log change in CM cells. Superimposed are regression lines from our fitted model. We observe that 1-year increases in CM cells are associated with an increased loss in C-peptide levels at 2 years and that abatacept reduces the 2-year rate of C-peptide loss compared with placebo (P = 0.0004) toward zero. Most striking, however, is the finding that the estimated rate of C-peptide loss related to the 1-year change in CM cells in the abatacept group is not significantly different from zero (0.0418 [95% CI −0.0025 to 0.0865]; P = 0.0659). These findings therefore suggest that abatacept attenuates metabolic loss related to the change in CM cells in addition to reducing CM cell populations. Similar findings held for the naive cell/CM cell ratio.
Figure 3.

Log change in C-peptide 2 years after baseline vs. log change in the percentage of CD4+ CM T cells at 1 year. Filled circles represent abatacept-treated subjects, and open circles represent placebo-treated subjects. Lines represent regression estimates from the fitted statistical model: abatacept, solid line; placebo, dashed line.
The estimated effects of previous changes in CM cells on the subsequent change in C-peptide AUC are presented in Table 2. The unadjusted columns are estimates from the fitted mixed linear model described previously. Since we have shown here that treatment affects CM cell populations, and have previously shown that treatment affects C-peptide AUC (9), there is a potential for treatment to act as a confounder influencing the statistical estimates. To evaluate this potential, we adjusted the change in both the CM cell population and the change in C-peptide level for the effect of treatment using a regression model. The residuals from the model were then reanalyzed using the same mixed model as was used for the unadjusted data. The adjusted columns in Table 2 are, therefore, estimates that have been adjusted for the potential confounding influence of treatment. Since the unadjusted and adjusted estimates for CM cells and the effect of treatment on the influence of CM cells on the change in C-peptide levels are identical, we conclude that our findings have not been confounded with treatment.
Table 2.
Estimates of the effect of previous change in CM cells on the subsequent change in C-peptide levels with and without adjustment for potential treatment confounding
| Effect | Unadjusted |
Adjusted |
|||
|---|---|---|---|---|---|
| Estimate | SE | Pr > |t| | Estimate | Pr > |t| | |
| Intercept | −0.07780 | 0.01710 | <0.0001 | 0.08146 | <0.0001 |
| Treatment | 0.07356 | 0.02051 | 0.0005 | 0.001889 | 0.9257 |
| Month | −0.00642 | 0.000475 | <0.0001 | −0.00642 | <0.0001 |
| CM cells | −0.1019 | 0.03177 | 0.0018 | −0.1019 | 0.0018 |
| CM cell × treatment interaction | 0.1438 | 0.03893 | 0.0004 | 0.1438 | 0.0004 |
Unadjusted estimates refer to the intercept or slope of the linear relationships between the change in C-peptide levels and treatment with abatacept, time, and previously measured change in CM cell population. CM cell × treatment interaction estimates the difference in slopes between treatment groups (treated slope − placebo group). Since treatment could affect both C-peptide level and the number of CM cells, estimates were then adjusted for this potential confounding and are presented in the “Adjusted” portion of the table.
From Table 2, we observe that abatacept therapy is associated with a reduction in the loss of C-peptide across time seen in the placebo group (difference in mean reduction = 0.07356, P = 0.0005), and thus our model agrees with clinical findings (9). The model also estimates that unit increases in the log CM percentage change from baseline are associated with a subsequent decrease in C-peptide levels by −0.1019 (P = 0.0018).
Translating these findings to a more concrete setting, consider a placebo-treated individual who experiences a 10% increase in CM cells from baseline at 1 year during treatment. The model then predicts a reduction in C-peptide level at 2 years of 21% (95% CI 17–23%). On the other hand, should (despite treatment) an abatacept-treated subject also experience a 10% increase in CM cells, the model then projects their loss in C-peptide at 24 months to be much smaller (14% [95% CI 12–17%]). For example, a 10% increase in CM cells from baseline equates to a 0.095 increase in the log scale (ln[1.10] = 0.095). Using the unadjusted coefficients from Table 2, the model then predicts a change in log C-peptide level at 2 years of −0.07780 − 0.00642 × 24 (months) − 0.1019 × 0.095 = −0.241. This change in log equates to a 21% reduction in C-peptide level at 2 years (i.e., [1 − exp(−0.241)] × 100%). The associated 95% CI of 17–23% was computed on the log data using model-based estimation error and transformed into the percentage change using similar computations. Similar computations were used for the example of the abatacept-treated subject.
Validation and Implications of the Model for Therapeutic Studies
After performing the above analyses and modeling, follow-up results from the abatacept study became available providing C-peptide data after the 30-month period, but no further flow cytometry analyses were available. Since this time point is beyond the range of the data that we used to build the model predicting the 2-year change in C-peptide from 1-year change in CM cells, we used these follow-up data as a semi-independent validation data set (although it was not used in constructing the prediction model, the same subjects are used).
We tested the model prediction of the change seen in C-peptide data at 3 years from the change seen in CM cells at 2 years. The “baseline” for measuring a 2-year change in C-peptide level was, therefore, the 1-year value. In addition, the 1-year change in CM cells would therefore need to be the change seen at 2 years (using the baseline of 1 year). Table 3 summarizes the results from this preliminary validation study. “Prediction error” is the difference between our prediction and the observed C-peptide level change at 3 years (predicted − observed in the log scale). Table 3 reports the median prediction error expressed as a percentage of observed and associated 95% confidence limits.
Table 3.
Analysis of prediction error
| Treatment | n | Median prediction error (%) | 95% CI on median prediction error (%) |
|---|---|---|---|
| Placebo | 16 | 1.6 | −7.8 to 10.2 |
| Abatacept | 38 | 8.5 | 1.7 to 15.8 |
Prediction error refers to the difference between the 3-year change in C-peptide levels predicted by our model and the change actually observed in an individual.
In the placebo group, our predictions were quite accurate with the median error being within 1.6% of the actual C-peptide level change at 3 years. This is less than one half of the SD of the prediction error in the original model (which was 9%). The results in the placebo group therefore provide preliminary validation of the model. However, the small sample size (n = 16 placebo-treated subjects) leads to wide CIs, which include values greater than ±9%. The point estimate of the abatacept-treated group is surprising as it validates the model despite the fact that during the period of our prediction a major event occurred (i.e., treatment was suspended). Moreover, we believe that this event may lead to increased variability in the prediction error in the abatacept-treated group and, hence, wide confidence bounds. It should be emphasized that, because of the limited availability of subjects with a 30-month follow-up at the time of the writing of this article, the validation study is based on approximately one half of the total number that will, ultimately, be available.
In sum, with the above caveats in mind, this analysis provides an initial validation of our model. Yet, since we have applied it to data from the same subjects used to create the prediction model, it might still be biased and points to the need for further validation studies.
Discussion
The application of therapeutic strategies that target molecular pathways to ameliorate complex, chronic inflammatory autoimmune conditions such as type 1 diabetes has entered a key phase, in which the tangible progress in efficacy that is seen with several agents needs to be allied with a greater understanding of their modes of action. Better understanding would have several benefits. First, not all patients see clinical efficacy from a specific agent, suggesting that stratification and targeting of selected subgroups, effectively a personalized health care approach, may be required. Second, single-agent effects are often not sustained, making decisions regarding treatment duration, maintenance, and periodicity complex. And third, where combination therapies are being considered, as is the case for type 1 diabetes, there is a need for objective means to guide the administration of the individual component drugs. The discovery of biomarkers relevant to the molecular pathways through which immune-modulating drugs act is a first step in addressing these issues, and represents the main aim of our study.
Using polychromatic flow cytometry, we searched for changes in immune cell populations and their major subsets following the infusion of abatacept. While several cell types did not change in frequency (CD3, CD4, and CD8 T cells; B cells; dendritic cells; or invariant NK T cells), there were marked and significant alterations in CD4 T-cell subsets defined by CD45R0 and CD62L. In broad terms, the CD45R0+CD62L+ population is enriched for CD4 T cells with the characteristics of a CM phenotype, and this population is reduced in the circulation by abatacept treatment. Concomitantly, the circulating CD45R0−CD62L+ population, which is highly enriched for naive CD4 T cells, increases following therapy. These changes revert after the cessation of treatment, implying a direct drug effect. The CM CD4 T-cell population is marked by the acquisition of CD45R0, which occurs after priming against specific antigens in the lymph node, and the expression of CD62L, an important cell adhesion molecule. CD62L facilitates entry into secondary lymphoid tissues via high endothelial venules and thus enables antigen-experienced CD4 T cells to recirculate between blood, tissues, and lymph nodes as part of an immune surveillance process. Upon activation in the lymph node, CM CD4 T cells secrete interleukin-2 and have high proliferative capacity, enabling them to participate rapidly in adaptive immune responses. Activation of these cells results in differentiation toward a more effector phenotype with secretion of cytokines such as interferon-γ, resulting in efficient responses to viruses and other pathogens (14). Analysis of cell subset concentrations in our study indicates that, for the most part, the treatment-induced changes we observed result from a reduction in the absolute numbers of CM cells in the circulation. This could be because of drug effects that promote migration out of the circulation or, alternatively, enhance retention within the lymph node or tissues. Increased retention within the lymph node seems the most likely of these options (the CD62L−CD45R0+ effector memory, which does not circulate via lymph nodes was not affected by treatment), but a mechanism is not immediately obvious since the role of CD80/CD86 molecules in cell migration has been little studied. We have previously shown that abatacept inhibits the transmigration of CD45R0+ CD4 T cells across CD86+ microvascular endothelial cells in vitro (15), indicating that it is capable of modulating the migration behavior of memory cells. Future studies conducted in vivo in preclinical models will be required to address the migration fate of CM CD4 T cells in the presence of CD80/86 blockade. Such studies should also help to elucidate whether the redistribution of CM CD4 T cells is associated with the beneficial therapeutic effect of abatacept, for example, by restricting circulatory access of pathogenic, autoreactive T helper type 1 and T helper type 17 cells to the islets of Langerhans.
We are not aware that changes in CM or naive CD4 T cells following abatacept treatment have been previously reported as a biomarker of immunological efficacy. Scarsi et al. have shown changes in populations of CD28-expressing CD8 T cells in abatacept-treated rheumatoid arthritis (16) and that the baseline number of circulating CD28-negative T cells predicts remission (17). These and our findings may be related, but CD28 was not incorporated into our current panel, and future studies will be needed to examine whether these observations align. Abatacept also induced a decline in the number of circulating CD4+CD25high cells, and we observed a nonsignificant trend for this change to associate with C-peptide level decline. A recent clinical trial (18) using abatacept in the treatment of patients with rheumatoid arthritis detected a decrease in Treg numbers with a simultaneous increase in their function. Future studies using more specific Treg markers, such as CD127low and FOXP3, and studies of Treg function should be able to establish whether Tregs are a useful additional biomarker of costimulation blockade.
In a second phase of our analyses, we examined the relationship of cellular changes to metabolic function, and made the novel observation that an increase in the number of circulating CM CD4 T cells and a decrease in CD4-naive cell/CM cell ratio were significantly associated with a subsequent rise in the rate of C-peptide decline. Moreover, abatacept treatment significantly altered this association between prior values of CM cells and CD4-naive cell/CM cell ratio and the subsequent change in C-peptide levels. In effect, these data indicate that an increase in the proportion of CM CD4 T cells and a decrease in CD4 naive cell/CM cell ratio act as a surrogate for C-peptide level decline. Costimulation blockade reverts these immunological changes and the associated metabolic effects. This provides a very strong indication that the acquisition of CD45R0+CD62L+ CD4 T cells in the circulation is a key process in the damaging autoreactivity that underlies type 1 diabetes, and it is a clear sign that abatacept controls this process to achieve therapeutic benefit. All samples were collected in a double-blinded fashion. Although we could not test all patients at each time point, a potential limitation on detecting less prominent T-cell subset changes, our statistical methods pooled all available data when testing for differences between treatment groups and when analyzing the effect of time, and thus made the most efficient use of the available data.
In summary, to our knowledge these data describe both the first immunological biomarker of C-peptide level decline in patients in whom type 1 diabetes has recently developed and clearly demonstrate cellular immune changes by which the drug may induce the slowing of this process in a type 1 diabetes intervention study. Other drugs such as rituximab and monoclonal anti-CD3 antibody have also been shown to delay C-peptide level decline in treated patients (6,8). Studies in these prior trials have discovered T-cell biomarkers that associate with clinical responses, such as enhanced reactivity to diabetes antigens in rituximab-treated clinical responders (19) and/or an increase in tetramer-positive CD8 T cells that is associated with treatment (20).
Future studies will be required to confirm these findings, elucidate the precise subsets that change within the CM CD4 T-cell populations, and unravel the potential mechanisms involved. In the future, the early measurement of the rate of change of CM CD4+ T cells could help to identify those subjects with a more aggressive disease course, and/or those suitable for costimulation blockade and other biologic therapies, or those relapsing under treatment. In clinical trials, measurements of these naive and CM CD4+ T-cell populations can help to improve monitoring of the efficacy of the interventions. Moreover, such immune markers could also have an impact in improving the prediction of progression to overt type 1 diabetes in high-risk, multiple islet cell, autoantibody-positive subjects.
Article Information
Funding. The sponsor of the trial was the Type 1 Diabetes TrialNet Study Group. The Type 1 Diabetes TrialNet Study Group is a clinical trials network funded by the National Institutes of Health through the National Institute of Diabetes and Digestive and Kidney Diseases, the National Institute of Allergy and Infectious Diseases, and The Eunice Kennedy Shriver National Institute of Child Health and Human Development through cooperative agreements U01-DK-061010, U01-DK-061016, U01-DK-061034, U01-DK-061036, U01-DK-061040, U01-DK-061041, U01-DK-061042, U01-DK-061055, U01-DK-061058, U01-DK-084565, U01-DK-085453, U01-DK-085461, U01-DK-085463, U01-DK-085466, U01-DK-085499, U01-DK-085505, and U01-DK-085509 and contract HHSN267200800019C; the National Center for Research Resources through Clinical Translational Science Awards UL1-RR-024131, UL1-RR-024139, UL1-RR-024153, UL1-RR-024975, UL1-RR-024982, UL1-RR-025744, UL1-RR-025761, UL1-RR-025780, UL1-RR-029890, and UL1-RR-031986 and General Clinical Research Center Award M01-RR-00400; the Juvenile Diabetes Research Foundation International; and the American Diabetes Association.
Duality of Interest. Bristol-Myers Squibb provided abatacept. The LifeScan Division of Johnson & Johnson provided blood glucose monitoring meters and strips to participants. M.P. has received funding via the U.K. Department of Health National Institute for Health Research Biomedical Research Centre award to Guy's and St Thomas' National Health Service Foundation Trust in partnership with King's College London. No other potential conflicts of interest relevant to this article were reported.
Author Contributions. T.O. conceived the idea, designed the study, analyzed the data, and wrote the manuscript. C.A.B. performed the statistical analysis and wrote the manuscript. P.X. assisted with the statistical analysis. K.M. and Q.J. contributed to the flow cytometry analysis. J.D. performed the flow cytometry analysis and reviewed the manuscript. S.M. assisted in project and data management. P.G. contributed to discussions. L.S. supervised the project, contributed to the study design, analyzed the data, and wrote the manuscript. M.P. analyzed the data and wrote the manuscript. T.O. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Prior Presentation. Parts of this study were presented in poster form at FOCIS 2012, the annual meeting of the Federation of Clinical Immunology Societies, Vancouver, BC, Canada, 20–23 June 2012.
Footnotes
Clinical trial reg. no. NCT00505375, clinicaltrials.gov.
The contents of this article are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health, Juvenile Diabetes Research Foundation International, or American Diabetes Association.
References
- 1.Herold KC, Vignali DA, Cooke A, Bluestone JA. Type 1 diabetes: translating mechanistic observations into effective clinical outcomes. Nat Rev Immunol 2013;13:243–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stiller CR, Dupré J, Gent M, et al. Effects of cyclosporine immunosuppression in insulin-dependent diabetes mellitus of recent onset. Science 1984;223:1362–1367 [DOI] [PubMed] [Google Scholar]
- 3.Harrison LC, Colman PG, Dean B, Baxter R, Martin FI. Increase in remission rate in newly diagnosed type I diabetic subjects treated with azathioprine. Diabetes 1985;34:1306–1308 [DOI] [PubMed] [Google Scholar]
- 4.Orban T, Farkas K, Jalahej H, et al. Autoantigen-specific regulatory T cells induced in patients with type 1 diabetes mellitus by insulin B-chain immunotherapy. J Autoimmun 2010;34:408–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wherrett DK, Bundy B, Becker DJ, et al. Type 1 Diabetes TrialNet GAD Study Group Antigen-based therapy with glutamic acid decarboxylase (GAD) vaccine in patients with recent-onset type 1 diabetes: a randomised double-blind trial. Lancet 2011;378:319–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herold KC, Hagopian W, Auger JA, et al. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med 2002;346:1692–1698 [DOI] [PubMed] [Google Scholar]
- 7.Keymeulen B, Vandemeulebroucke E, Ziegler AG, et al. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med 2005;352:2598–2608 [DOI] [PubMed] [Google Scholar]
- 8.Pescovitz MD, Greenbaum CJ, Krause-Steinrauf H, et al. Type 1 Diabetes TrialNet Anti-CD20 Study Group Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N Engl J Med 2009;361:2143–2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Orban T, Bundy B, Becker DJ, et al. Type 1 Diabetes TrialNet Abatacept Study Group Co-stimulation modulation with abatacept in patients with recent-onset type 1 diabetes: a randomised, double-blind, placebo-controlled trial. Lancet 2011;378:412–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matthews JB, Staeva TP, Bernstein PL, Peakman M, von Herrath M, ITN-JDRF Type 1 Diabetes Combination Therapy Assessment Group Developing combination immunotherapies for type 1 diabetes: recommendations from the ITN-JDRF Type 1 Diabetes Combination Therapy Assessment Group. Clin Exp Immunol 2010;160:176–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roep BO, Peakman M. Surrogate end points in the design of immunotherapy trials: emerging lessons from type 1 diabetes. Nat Rev Immunol 2010;10:145–152 [DOI] [PubMed] [Google Scholar]
- 12.Bluestone JA, St Clair EW, Turka LA. CTLA4Ig: bridging the basic immunology with clinical application. Immunity 2006;24:233–238 [DOI] [PubMed] [Google Scholar]
- 13.Marelli-Berg FM, Okkenhaug K, Mirenda V. A two-signal model for T cell trafficking. Trends Immunol 2007;28:267–273 [DOI] [PubMed] [Google Scholar]
- 14.Farber DL, Yudanin NA, Restifo NP. Human memory T cells: generation, compartmentalization and homeostasis. Nat Rev Immunol 2014;14:24–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lozanoska-Ochser B, Klein NJ, Huang GC, Alvarez RA, Peakman M. Expression of CD86 on human islet endothelial cells facilitates T cell adhesion and migration. J Immunol 2008;181:6109–6116 [DOI] [PubMed] [Google Scholar]
- 16.Scarsi M, Ziglioli T, Airò P. Decreased circulating CD28-negative T cells in patients with rheumatoid arthritis treated with abatacept are correlated with clinical response. J Rheumatol 2010;37:911–916 [DOI] [PubMed] [Google Scholar]
- 17.Scarsi M, Ziglioli T, Airò P. Baseline numbers of circulating CD28-negative T cells may predict clinical response to abatacept in patients with rheumatoid arthritis. J Rheumatol 2011;38:2105–2111 [DOI] [PubMed] [Google Scholar]
- 18.Álvarez-Quiroga C, Abud-Mendoza C, Doníz-Padilla L, et al. CTLA-4-Ig therapy diminishes the frequency but enhances the function of Treg cells in patients with rheumatoid arthritis. J Clin Immunol 2011;31:588–595 [DOI] [PubMed] [Google Scholar]
- 19.Herold KC, Pescovitz MD, McGee P, et al. Type 1 Diabetes TrialNet Anti-CD20 Study Group Increased T cell proliferative responses to islet antigens identify clinical responders to anti-CD20 monoclonal antibody (rituximab) therapy in type 1 diabetes. J Immunol 2011;187:1998–2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cernea S, Herold KC. Monitoring of antigen-specific CD8 T cells in patients with type 1 diabetes treated with antiCD3 monoclonal antibodies. Clin Immunol 2010;134:121–129 [DOI] [PMC free article] [PubMed] [Google Scholar]