

Abstract
Introduction
Earlier, we reported syntheses of ethyl-β-D-galactopyranosyl-(1,4′)-2′-deoxy-2′-[18F]fluoro-β-D-glucopyranoside (Et-[18F]FDL) and 1′-[18F]fluoroethyl-β-D-lactose ([18F]-FEL) for positron emission tomography (PET) of pancreatic carcinoma. Et-[18F]FDL requires a precursor, which involves 11 steps to synthesize and produces overall low yields. Synthesis of precursors for [18F]-FEL requires four steps, but those precursors produced low radiochemical yields. Here, we report new precursors and an improved synthesis of [18F]-FEL.
Method
Two precursors, 1′-(methanesulfonyl)ethyl-2′,3′,6′,2,3,4,6-hepta-O-acetyl-β-D-lactose 2a and 1′-(p-nitrophenyl-sulfonyl)ethyl-2′,3′,6′,2,3,4,6-hepta-O-acetyl-β-D-lactose 2b, were synthesized from lactose in four steps. Radiofluorination reactions were performed using K18F/kryptofix and the crude product [18F]-3 was purified by HPLC. Basic hydrolysis of [18F]-3 produced 1′-[18F]fluoroethyl-β-D-lactose [18F]-4, which was neutralized, diluted with saline, filtered on a 0.22-μm filter, and analyzed by radio-TLC.
Results
The average radiochemical yields of [18F]-4 (d. c.) from 2a and 2b were 21% (n = 6) and 65% (n = 6), respectively, with > 99% radiochemical purity and specific activity of 55.5 GBq/μmol. Synthesis time was 90–95 min from the end of bombardment.
Conclusion
An improved synthesis of [18F]FEL has been achieved in high yields, with high purity and specific activity. Precursor 2b with this method should be applicable for high yield automated production in a commercial synthesis module for clinical application.
Keywords: fluorine-18, lactose, HIP/PAP, pancreatic carcinoma, PET
Introduction
Early detection of pancreatic cancer with highly sensitive diagnostic imaging modality could save many thousands of patients by increasing resectability and survival rates. We have been developing novel probes for positron emission tomography (PET) in early detection of pancreatic cancer and hepatocellular carcinoma. In previous studies, we reported the synthesis of ethyl-β-D-galactopyranosyl-(1,4′)-2′-deoxy-2′-[18F]fluoro-β-D-glucopyranoside (Et-[18F]FDL),1 its PET imaging results on orthotopically implanted pancreatic cancer in nude mice,2 and the radiosynthesis of 1′-[18F]fluoroethyl-β-D-lactose ([18F]-FEL)3 for PET imaging of pancreatic carcinoma. These compounds, Et-[18F]FDL and [18F]-FEL, were developed based on the fact that D-lactose and its analogues bind with the hepatocarcinoma-intestine-pancreas/pancreatitis-associated protein (HIP/PAP),4–6 which is overexpressed (130-fold) in peritumoral pancreatic acinar cells compared with normal pancreas7,8 and in hepatocellular carcinomas.9–13 Our previous studies revealed that Et-[18F]FDL is an excellent PET imaging agent for detection of pancreatic cancer by targeting HIP/PAP in the peritumoral region.2 However, the synthesis of Et-[18F] FDL was performed using a precursor, which requires 11 steps to synthesize and produced overall low yields. Preliminary studies of pancreatic cancer in an orthotopic nude mouse model and chemically induced hepatocellular carcinoma in swine liver using [18F]-FEL showed that the efficacy of [18F]-FEL was similar to that of Et-[18F]FDL for PET imaging of these cancers [unpublished data]. Alternatively, the synthesis of precursors for [18F]-FEL required only four steps and produced high yields. However, those precursors produced low radiochemical yields (9–11%) of [18F]-FEL. Therefore, a convenient high-yield synthesis of [18F]-FEL is needed for routine production of the radiotracer and to pursue pre-clinical and clinical applications.
To improve the radiochemical yield of [18F]-FEL, we have designed and synthesized two new precursors and evaluated them for radiosynthesis of [18F]-FEL using K18F/kryptofix 2.2.2 in dry MeCN. In this article, we report a high-yield (>sixfold) radiosynthesis of [18F]-FEL using a new precursor, 1′-(p-nitrophenylsulfonylethyl)-2′,3′,6′,2,3,4,6-hepta-O-acetyl-β-D-lactose. This precursor should be suitable for routine production of high-yield [18F]-FEL, and the method may be adapted to a commercially available automated synthesis module for pre-clinical and clinical applications of the probe.
Experimental
Reagents and instrumentation
Reagents and solvents were purchased from Aldrich Chemical Co. (Milwaukee, WI, USA) and used without further purification. K18F/ kryptofix 2.2.2. was purchased from Cyclotope Inc. (Houston, TX, USA) as an aqueous solution. Silica gel solid-phase extraction cartridges (Sep-Pak, 900 mg) were purchased from Alltech Associates (Deerfield, IL, USA). Thin layer chromatography (TLC) was performed on precoated Kieselgel 60 F254 (Merck, Darmstadt, Germany) glass plates. Proton and 19F NMR spectra were recorded using tetramethylsilane as an internal reference or hexafluorobenzene as an external reference at The University of Texas MD Anderson Cancer Center using a Bruker 300 MHz spectrometer. High-resolution mass spectra (HRMS) were obtained on a Bruker BioTOF II mass spectrometer at the University of Minnesota using electrospray ionization.
High-performance liquid chromatography was performed using a 1100 series pump (Agilent Technologies, Stuttgart, Germany) with a built-in ultraviolet detector operated at 210 nm and a radioactivity detector with a single-channel analyzer (Bioscan, Washington, DC, USA). Crude product was purified on a semipreparative C18 reverse-phase column (Alltech, Econosil, 10 × 250 mm), and quality control analyses were performed on an analytical C18 column (Alltech, Econosil, 4.6 × 250 mm). An acetonitrile/water solvent system (45% MeCN/H2O) was used for purification of the radiolabeled product at a flow of 4 mL/min. Quality control analyses were performed using 55% MeCN/H2O solvents at a flow of 1 mL/min. Radio-TLC plates were scanned on an AR-2000 imaging scanner (Bioscan, Washington, DC, USA).
Preparation of 1′-(methanesulfonylethyl)-2′,3′,6′,2,3,4,6-hepta-O-acetyl-β-D-lactose: 2a
1′-(Hydroxyethyl)-2′,3′,6′,2, 3, 4,6-hepta-O-acetyl-β-D-lactose 13 (68.0 mg, 0.1 mmol) was dissolved in dry pyridine (3.0 mL) in a small flask and cooled to 0°C under argon. Methanesulfonyl chloride (12 μL, 1.5 equiv.) was added, and the reaction mixture was stirred for 10 min and then stirred for 1 h at room temperature. An aliquot was taken out, and pyridine was removed using a high-vacuum pump; the material was redissolved in CH2Cl2 and analyzed by TLC, which showed that no starting material remained. The rest of the reaction mixture was evaporated using a high-vacuum pump. The crude product was purified by flash chromatography on a silica gel column and eluted with 40% acetone in hexane. The solvent was evaporated to afford 2a (65 mg) in 86% yield as a white solid. 1H NMR (CDCl3) δ: 5.36 (d, J = 2.4 Hz, 1H), 5.22 (t, J = 9.3 Hz, 1H), 5.13 (2d, J = 7.8, 8.1 Hz, 1H), 4.9–4.89 (m, 2H), 4.56–4.49 (m, 3H), 4.36 (t, J = 4.5 Hz, 2H), 4.16–4.03 (m, 4H), 3.91–3.79 (m, 3H), 3.66–3.64 (m, 1H), 3.04 (s, 3H, Ms), 2.17 (s, 3H, acetate), 2.15 (s, 3H, acetate), 2.08 (s, 3H, acetate), 2.07 (s, 3H, acetate), 2.06 (s, 6H, acetate), 1.98 (s, 3H, acetate). HRMS: calculated for C29H42O21SNa (M + Na) 781.1832, found 781.1850.
Preparation of 1′-(p-nitrophenylsulfonylethyl)-2′,3′,6′,2,3,4,6-hepta-O-acetyl-β-D-lactose: 2b
1′-(Hydroxyethyl)-2′,3′,6′,2,3,4,6-hepta-O-acetyl-β-D-lactose 1 (68.0 mg, 0.1 mmol) was dissolved in dry pyridine (3.0 mL) in a small flask and cooled to 0°C under argon. p-Nitrophenylsulfonyl chloride (55 mg, 2.5 equiv.) was dissolved in dry CH2Cl2 (1.0 mL) and added slowly to the reaction mixture and stirred for 1 h at 0°C. TLC showed that no starting material remained. The reaction mixture was evaporated using a high-vacuum pump, and the crude product was purified by flash chromatography on a silica gel column and eluted with 40% acetone in hexane. The solvent was evaporated to afford 2b (55 mg) in 64% yield as a white solid. 1H NMR (CDCl3) δ: 8.44 (d, J = 8.8 Hz, 2H, aromatic), 8.1 (d, J = 8.9 Hz, 2H, aromatic), 5.37 (d, J = 2.4 Hz, 1H), 5.20–5.089 (m, 2H), 4.98 (dd, J = 10.5 Hz, 3.6 Hz, 1H), 4.77 (dd, J = 7.8 Hz, 7.8 Hz, 1H), 4.51–4.46 (m, 3H), 4.29–4.25 (m, 2H), 4.16–4.06 (m, 3H), 4.04–3.39 (m, 1H), 3.92–3.88 (m 1H), 3.85–3.72 (m, 2H), 3.64–3.57 (m, 1H), 2.17 (s, 3H, acetate), 2.14 (s, 3H, acetate), 2.08 (s, 3H, acetate), 2.07 (s, 3H, acetate), 2.06 (s, 3H, acetate), 2.05 (s, 3H, acetate), 1.98 (s, 3H, acetate). HRMS: calculated for C34H43NO23SNa (M + Na) 888.1839, found 888.1873.
Preparation of 1′-fluoroethyl-2′,3′,6′,2,3,4,6-hepta-O-acetyl-β-D-lactose: 3
Compound 3 was prepared from 2a and 2b by fluorination with Bu4NF; a representative preparation from 2a is described here. Compound 2a (10 mg, 13.2 μmol) was dissolved in MeCN (1.0 mL) in a V-vial. To this solution, 26 μL of Bu4NF in tetrahydrofuran (1 M solution, 2 equiv.) was added, and the reaction mixture was heated at 90°C for 20 min, when TLC showed no starting material remained. The reaction mixture was cooled at room temperature, the solvent was evaporated, and the crude product was chromatographed on a small silica gel column using 40% acetone/hexane. The solvent was evaporated, and 6.0 mg of the product 3 was obtained in 65% yield. Precursor 2b produced 85% yield of 3 with all spectral data identical as reported earlier.3
Preparation of 1′-fluoroethyl-β-D-lactose: 4
Compound 4 was prepared as previously reported3 and fully characterized by spectroscopic methods, and the spectral data were consistent with those previously reported in the literature.3
Radiosynthesis of 1′-[18F]fluoroethyl-2′,3′,6′,2,3,4,6-hepta-O-acetyl-β-D-lactose: [18F]-3
This compound was also prepared as previously reported.3 A brief description using precursor 2b is presented here. The aqueous solution of K[18F]/kryptofix 2.2.2 was evaporated with acetonitrile (1.0 mL) by an azeotropic removal of water at 90 °C under a stream of argon. A solution of 2b (4–5 mg) in dry acetonitrile (0.4 mL) was added to the dry K18F/ kryptofix 2.2.2. The reaction mixture was heated at 80 °C for 20 min. The crude reaction mixture was passed through a silica Sep-Pak cartridge and eluted with 10% MeOH/CH2Cl2 (2.5 mL). The solvent was evaporated, and the residue was dissolved in 60% acetonitrile/water (1.0 mL) and purified by HPLC. The product [18F]-3 was collected between 16.5 and 18.0 min, and an aliquot was analyzed on an analytical HPLC to verify its identity and purity by coinjection with the nonradioactive authentic sample 3.
Preparation of 1′-[18F]fluoroethyl-β-D-lactose: [18F]-4
The product [18F]-3 was dried under reduced pressure, dissolved in CH2Cl2, and transferred to a V-vial; then, the solvent was evaporated. The residue was dissolved in methanol (0.4 mL), 0.5 M NaOMe/MeOH solution (0.1 mL) was added, and the mixture was heated at 80 °C for 7 min, when HPLC showed complete hydrolysis of [18F]-3. The solvent was evaporated, and the residue, [18F]-4, was neutralized with HCl, diluted with saline, and filtered through a 0.22-μm Millipore filter. Compound [18F]-4 was analyzed by TLC as previously reported,3 which showed >99% pure product with the same Rf value as the authentic compound.
Results and discussion
Scheme 1 describes the non-radioactive synthesis of 1′-fluoroethyl-β-D-lactose 4 and radiosynthesis of [18F]-FEL [18F]-4. Compound 1 was prepared from lactose using methods described previously3,14 and characterized by 1H NMR spectroscopy and MS, which were consistent with those previously reported in the literature.3,14 Compound 2a was obtained in 86% yield; its identity was fully characterized by 1H NMR spectroscopy and HRMS. The 1H NMR spectrum showed a new peak (s) at 3.04 ppm with an integration of 3H, suggesting the presence of the mesylate and the HRMS showed M + Na ion at m/z 781.1850. Compound 2b was obtained in 64% yield, and this compound was also characterized by 1H NMR spectroscopy and HRMS. The NMR spectrum showed new characteristic peaks in the aromatic region with an integration of 2H each, and the HRMS showed M + Na ion at m/z 888.1873. Compound 2b was highly reactive as opposed to 2a; for example, when pyridine was evaporated from the crude reaction mixture at an elevated temperature (50 °C), the product was converted to a new compound, which was characterized by 1H NMR and MS as the 1′-chloroethyl-2′,3′,6′,2,3,4,6-hepta-O-acetyl-β-D-lactose. The chloride ion generated from the reaction of nosyl chloride acted as a nucleophile to produce the new derivative. About 80% of the product was converted to the 1′-chloroethyl derivative at 50°C. However, the problem was resolved by keeping the reaction mixture at 0°C and evaporating pyridine at a lower temperature (7–10 °C). With this precautionary measure, the yield could be improved to 64%.
Scheme 1.
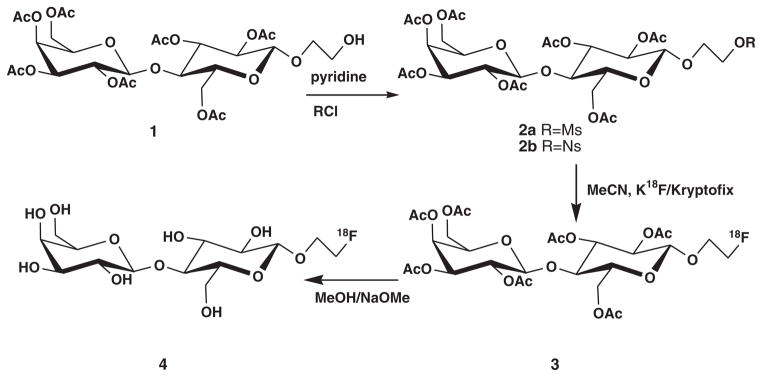
Scheme for synthesis and radiosynthesis of 1′-[18F]fluoroethyl-β-D-lactose [18F]-4.
The reaction of 2a with Bu4NF at 90°C and 100°C produced 3 in 65% yields. The 1H NMR spectrum of 3, as previously reported,3 was quite complex in the region where the CH2-F protons resonate. The 19F NMR spectrum (coupled), however, was clear, showing a peak at −224.22 ppm as a multiplet, which supports the presence of CH2-F. Both 1H NMR and 19F NMR spectra of 3 were consistent, as previously reported.3 The reaction of 2b with Bu4NF at 80 °C and 90°C produced 3 in 85% yields. NMR spectra of 3 from 2a and 2b were identical, and 3 from both precursors had the same retention time (tR) in the HPLC chromatograms.
After deacetylation of peracetyl-1′-fluoroethyl-β-lactose 3 by reaction with 0.5 M sodium methoxide, 1′-fluoroethyl-β-lactose 4 was produced in quantitative yield.
For synthesis of [18F]-3 and eventually [18F]-4, both precursors 2a and 2b were radiofluorinated with K18F/kryptofix 2.2.2. in dry MeCN (Scheme 1) at different temperatures. Compound 2a was radiofluorinated at 90°C and 100°C, and 2b was radiofluorinated at 80°C and 90°C. Because the higher temperatures did not render any improvement in yields, we maintained the lower temperatures; thus, 2a was reacted at 90°C and 2b reacted at 80°C for 20 min. Passing [18F]-3 through a silica gel cartridge helped to remove unreacted fluoride that may have co-eluted with the desired product during HPLC purification. The precursor 2a produced [18F]-3 in 15–27% yields at 90°C after HPLC purification, and the precursor 2b produced [18F]-3 in 58–69% yields at 80°C. Figure 1 represents the HPLC chromatogram for the purification of [18F]-3 from the nosylate precursor. The nosylate produced much a cleaner product, and the recovery from the HPLC column was about 75%, much higher than that previously reported.3 Analysis of [18F]-3 by HPLC on an analytical column showed a single radioactive peak co-eluted with an authentic nonradioactive standard (Figure 2). The nosylate precursor 2b appeared to be much better and produced high yields. Thus, 2b at 80°C for 20 min seems to be ideal for the routine production of [18F]-fluoroethyl lactose. One experiment was performed at 70°C, which also produced high yields, thereby suggesting that this temperature may be applicable; however, further experiments should be performed.
Figure 1.

HPLC purification of 1′-[18F]fluoroethyl-2′,3′,6′,2,3,4,6-hepta-O-acetyl-β-D-lactose, [18F]-3: semipreparative C18 Column; 45% MeCN and 55% H2O; flow 4.0 mL/min.
Figure 2.

HPLC chromatogram of 1′-[18F]fluoroethyl-2′,3′,6′,2,3,4,6-hepta-O-acetyl-β-D-lactose, [18F]-3, co-injected with standard 1′-fluoroethyl-2′,3′,6′,2,3,4,6-hepta-O-acetyl-β-D-lactose 3: Analytical C18 Column; 55% MeCN and 45% H2O; flow 1.0 mL/min.
Hydrolysis of [18F]-3 with NaOMe produced the final product, [18F]-4, in quantitative yield. Hydrolysis was monitored by HPLC, which showed that the hydrolysis was complete in 7 min. After the solvent was evaporated from [18F]-4, the product required neutralization with 1 M HCl solution followed by dilution with saline and finally filtration through a 0.22-μm Millipore filter. Radio-TLC of [18F]-4 showed a single product with an Rf value of 0.82, consistent with that of the non-radioactive compound (Figure 3). In general, about 10–12% of the product was lost during evaporation of the HPLC solvent from [18F]-3, transfer, hydrolysis, and filtration of [18F]-4. The radiochemical purity was >99% with specific activity of 55.5 GBq/μmol at the end of synthesis. The synthesis time was 90–95 min from the end of bombardment.
Figure 3.

Radio-TLC of 1′-[18F]fluoroethyl-β-D-lactose [18F]-4: developing solvent; MeOH:H2O (95:5).
Mesylate at 90°C for 30 min produced 21% yield; on the other hand, nosylate at 80°C produced 65% yield. At 90°C, nosylate did not produce any higher yield. Lower temperatures are preferred for any chemical reaction to eliminate the formation of any byproduct, and in this synthesis, nosylate at 80°C should be used for routine production. Thus, nosylate produced the highest radiochemical yield at 65%, whereas mesylate produced 21% yield, which is lower than that of nosylate but still higher than those obtained from bromo- and tosylate precursors, 9% and 11%, respectively.3
Conclusion
We have achieved an improved synthesis of the lactose analogue, [18F]-FEL, in high yields, with high purity and high specific activity. The product is suitable for PET imaging of early pancreatic carcinomas. Nosylate precursor 2b with this method should be applicable for automated production in a commercial synthesis module for clinical application.
Acknowledgments
This work was supported by the NCI CCSG Core Grant CA 106672 and 1R01DA030333–01; NIH/NIDA.
Footnotes
Conflict of Interest
The authors did not report any conflict of interest.
References
- 1.Ying Y, Ghosh P, Guo L, Pal A, Mukhapadhyay U, Peng Z, Yeh HH, Bertolini S, Flores LG, Young D, Volgin A, Soghomonyan S, Bornmann W, Logsdon C, Alauddin MM, Gelovani JG. Mol Imaging Biol. 2011;13:536–546. doi: 10.1007/s11307-010-0334-9. [DOI] [PubMed] [Google Scholar]
- 2.Flores L, Bertolini S, Yeh HH, Young D, Mukhapadhyay U, Pal A, Ying Y, Volgin A, Shavrin A, Soghomonyan S, Tong W, Bornmann WG, Alauddin MM, Logsdon C, Gelovani JG. PLoS One. 2009;4(11):e7977. doi: 10.1371/journal.pone.0007977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Turkman N, Pal A, Tong W, Gelovani JG, Alauddin MM. J Label Compd Radiopharm. 2011;54:233–238. [Google Scholar]
- 4.Drickamer K. Curr Opin Struct Biol. 1999;9:585–590. doi: 10.1016/s0959-440x(99)00009-3. [DOI] [PubMed] [Google Scholar]
- 5.Rosty C, Christa L, Kuzdzal CS, Balwin WM, Zahurak ML, Carnot F, Cham DW, Canto M, Lillemoe KD, Cameron JL, Yeo CJ, Hruban RH, Goggins M. Cancer Res. 2002;62:1868–1875. [PubMed] [Google Scholar]
- 6.Iovanna JL, Dagorn JC. Biochim Biophys Acta. 2005;1723:8–18. doi: 10.1016/j.bbagen.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 7.Fukushima N, Koopmann J, Sato N, Prasad N, Carvalho R, Leach SD, Hruban RH, Goggins M. Mod Pathol. 2005;18:779–787. doi: 10.1038/modpathol.3800337. [DOI] [PubMed] [Google Scholar]
- 8.Demaugre F, Philippe Y, Sar S, Pileire B, Christa L, Lasserre C, Brechot C. Eur J Biochem. 2004;271:3812–3820. doi: 10.1111/j.1432-1033.2004.04302.x. [DOI] [PubMed] [Google Scholar]
- 9.Christa L, Felin M, Morali O, Simon MT, Lasserre C, Brechot C, Seve AP. FEBS Lett. 1994;337:114–118. doi: 10.1016/0014-5793(94)80640-3. [DOI] [PubMed] [Google Scholar]
- 10.Lasserre C, Christa L, Simmon MT, Vernier P, Brechot C. Cancer Res. 1992;52:5089–5095. [PubMed] [Google Scholar]
- 11.Lasserre C, Colnot C, Brechot C, Poirier F. Am J Pathol. 1999;154:1601–1610. doi: 10.1016/S0002-9440(10)65413-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cervello M, Giannitrapani L, La Rosa M, Notarbartolo M, D’Alessandro N, Virruso L, Iovanna JL, Montalto G. Ann N Y Acad Sci. 2002;963:53–58. doi: 10.1111/j.1749-6632.2002.tb04094.x. [DOI] [PubMed] [Google Scholar]
- 13.Cavard C, Terris B, Grimber G, Christa L, Audard V, Radenen-Bussiere B, Simon MT, Renard CA, Buendia MA, Perret C. Oncogene. 2006;25:599–608. doi: 10.1038/sj.onc.1208860. [DOI] [PubMed] [Google Scholar]
- 14.Šardzík R, Noble GT, Weissenborn MJ, Martin A, Webb SJ, Flitsch SL. Beilstein J Org Chem. 2010;6:699–703. doi: 10.3762/bjoc.6.81. [DOI] [PMC free article] [PubMed] [Google Scholar]