

Abstract
Purpose
Ischemia-reperfusion (IR) injury is involved in the pathology of many retinal disorders since it contributes to the death of retinal neurons and the subsequent decline in vision. We determined the molecular patterns of some of the principal molecules involved in necroptosis and investigated whether IR retinal injury is associated with the extracellular signal-regulated kinase-1/2- receptor-interacting protein kinase 3 (ERK1/2-RIP3) pathway.
Methods
The cellular and subcellular localization of molecules involved in the cell death pathway, including RAGE, ERK1/2, FLIP, and RIP3, was determined with immunohistochemistry of cryosections of IR-injured retina from 2-month-old Long Evans rats. The total and phosphorylated protein levels were analyzed with quantitative western blots. ERK1/2 activity was inhibited by intravitreal injection of U0126, a highly selective inhibitor of mitogen-activated protein kinase 1/2 (MEK1/2).
Results
The IR-injured rat retinas expressed two RAGE isoforms with different intracellular localizations at early time points after injury. At that time point, frame inhibition of ERK activation decreased RIP3 accumulation and cell death. FLIP was detected in the IR-injured rat retinas at early time points after ischemia reperfusion.
Conclusions
We report that the necroptotic cell death mechanism is executed by an ERK1/2-RIP3 pathway in the retinal ganglion cells in early stages after IR injury. Inhibition of ERK1/2 activity increased retinal ganglion cell (RGC) survival indicating that targeting of this pathway within the initial 12 h after IR injury can be used to inhibit the necroptosis pathway. We also provide evidence that a novel RAGE isoform is expressed in the early stages in rat retinal RGCs.
Introduction
Ischemia-reperfusion (IR) injury has been associated with several retinal diseases, such as acute angle-closure glaucoma, central retinal artery occlusion (CRAO), and ophthalmic artery occlusion [1]. Although these blinding disorders might be linked with systemic disease such as atherosclerosis, they often occur spontaneously in the eyes of healthy patients and without a known cause [2]. Studies have shown that IR injury leads to neuronal cell degeneration [3-5]. In the retina, this degeneration has two phases. The first phase occurs within 24 h following IR injury, and the second phase occurs over the course of several days [5]. The mechanisms of retinal cell death are of interest because detailed knowledge may facilitate development of treatment.
Cell death can be executed by at least two well-established mechanisms, necrosis and apoptosis [6]. It has been found that necrosis, similar to apoptosis, can be “programmed,” and this form of necrotic death is known as necroptosis [7]. It has been established that these cell death processes are interconnected and share regulatory mechanisms [6]. However, they each contain key molecules that might be targeted to specifically prevent a particular mode of cell death that is prevalent in pathology-specific time windows. Earlier research has demonstrated that necrosis and apoptosis are critical factors in neuronal degeneration in brain and retinal injury [8-11]. However, current studies have shown that necroptosis contributes to retinal disorders such as retinal detachment [12], age-related macular degeneration (AMD) [13], and retinitis pigmentosa [14] and has been associated with retinal IR injury [15,16]. Although the underlying molecular mechanisms continue to emerge, it is now known that necroptosis is dependent on the kinase activity of receptor-interacting protein kinase 1 (RIP1 or RIPK1) and receptor-interacting protein kinase 3 (RIP3 or RIPK3) [17-20]. It has been recently demonstrated that pharmacological inhibition of RIP1 and RIP3 activity contributed to delayed cone cell death in pde6cw59 mutant zebrafish [21]. Both kinases have been detected in the ganglion cells in the IR-injured mouse retina [15], and the Nec1 inhibitor exhibited neuroprotective effects on these cells [15,16]. All these studies suggest involvement of RIP1 and RIP3 in regulation of necroptosis in the retina.
The initiation of necroptosis is associated with the release of damage-associated molecular patterns (DAMPs) that enhance innate inflammation and lead to tissue injury and cell death [22-26]. Once released, DAMPs evoke an inflammatory response through their binding to receptors, among which is the receptor for advanced glycation end products (RAGE) [25,27-29]. Distinct RAGE isoforms have been reported in various tissues, and expression of these isoforms has been associated with neuronal damage and inflammatory response in various diseases [29-32]. In addition, RAGE proteins are known to undergo different degrees of glycosylation and dimerization that change their three-dimensional configuration to influence ligand selectivity [33-42]. Interruption of the ligand or RAGE receptor activity of the retina decreased cell death [25]. However, temporal and molecular events in the signal transduction pathways downstream of RAGE activity have yet to be determined.
Occurrence of IR injury cannot be precisely predicted in the general human population. Therefore, the rescue of retinal neurons may be possible only after the precipitating event. Thus, it would be important to deliver the right therapeutics, targeting the most appropriate DAMP, within an appropriate time window after the initial injury. Since necroptosis contributes to retinal degeneration in early stages after IR, the goals of this study were to shed light on the signal transduction pathway associated with this cell death mechanism and to determine if manipulation of this pathway might have a neuroprotective effect on retinal neurons.
We demonstrate that between 12 and 24 h following IR-injury inhibition of ERK1/2 activity leads to downregulation of RIP3 and decreased RGC death. We report the expression of a novel RAGE isoform, which is significantly upregulated in RGCs in early stages after IR injury.
Methods
Animals
All animals were treated in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research, using protocols approved by the University of Louisville Institutional Animal Care and Use Committee. A total of 84 female Long Evans rats (250–300 g) were used in this study: 36 for the IR injury experiments, with six rats (three IR injured, three sham controls) used for each of the six time points for each of the two assessment types (western blot and immunohistochemistry; total of 72), and 12 for the IR injury experiments with pathway inhibition.
Ischemia-reperfusion injury
Ischemia was induced as previously described [43]. For the experimental group of animals, ischemia was induced by canulating the anterior chamber of the left eye with a 30-gauge needle attached to an elevated 1 L sterile saline bag. The intraocular pressure (IOP) was increased to 80 mmHg by elevating the saline bags and maintained at this pressure for 60 min. Retinas were then reperfused for various time periods (0 h, 12 h, 1 day, 3 days, 5 days, and 10 days) following the 1 h ischemic period. The eyes of the animals in the sham control group were canulated for the same amount of time without opening the outlet of the saline bags. At the specified times, the rats were euthanized with an overdose of pentobarbital. At the specified times, the rats were euthanized with an overdose of sodium pentobarbital, pharm grade in dosage 90 mg/kg. Route of administration was i.p. The retinas were collected and flash frozen in liquid nitrogen. For immunohistochemistry, the rats were perfused transcardially using 4% paraformaldehyde solution in PBS (1X; 155 mM NaCl, 1 mM KH2PO4, 3 mM Na2HPO4-7H2O, pH 7.4).. The eyeballs were embedded in Tissue-Tek OCT compound (Thermo Fisher Scientific, Bellefonte, PA) and stored at −80 °C before the tissue sections were cut.
Inhibition of ERK in the IR-injured rat retina
The IR injury was induced as described previously [43]. ERK inhibitor U0126 (Santa Cruz, Dallas, TX, sc-222395) was dissolved in 5% dimethyl sulfoxide (DMSO) in PBS. Inhibitor solutions were delivered intravitreally to the eye with the aid of a Hamilton 10 μl syringe (701RN, Cat# 7635–01) at two time points following the ischemic period (Table 1). At 12 h post-IR, three rats per group were euthanized with an overdose of sodium pentobarbital, pharm grade in dosage 90 mg/kg. Route of administration was i.p. The retinas were collected and flash frozen in liquid nitrogen.
Table 1. Experimental design for ERK inhibition experiment.
| Groups | First injection | Second injection | Injected amount (each time) | Animal # |
|---|---|---|---|---|
| IR + Saline |
0 h post IR |
6 h post IR |
4 μl saline |
3 |
| IR + DMSO |
0 h post IR |
6 h post IR |
4 μl 5% DMSO |
3 |
| IR + 0.4 mM U0126 |
0 h post IR |
6 h post IR |
4 μl 0.4 mM U0126 |
3 |
| IR + 0.8 mM U0126 | 0 h post IR | 6 h post IR | 4 μl 0.8 mM U0126 | 3 |
Western blots
For total protein extraction, rat retinas were homogenized in RIPA buffer (Cell Signaling Technology, Danvers, MA) containing a 1% protease inhibitor cocktail using an ultrasonic homogenizer (BioLogics, Manassas, VA). Equal amounts (20 μg) of protein were loaded onto Bolt 4–12% gradient Bis-Tris Plus gels (Life Technologies, Grand Island, NY). The western blot membranes were blocked for 1 h in 5% nonfat milk containing 0.01% TWEEN-20 and incubated with the primary antibodies against (1:100, Santa Cruz, SC-8436), RAGE (1:1,000; Abcam, Cambridge, MA, Ab3611), RAGE (1:250, R&D, Mab1179), RIP1 (1:1,000; Cell Signaling, #3493), RIP3 (1:200, Santa Cruz, SC-135171), and FLICE-like inhibitory protein (FLIP)S/L (1:100, Santa Cruz, SC-5276) overnight at 4 °C. After three washes with PBS for 10 min each, the blots were incubated with the corresponding secondary antibodies, conjugated with horseradish peroxidase for 1 h at room temperature. Antibody binding was visualized using enhanced chemiluminescence (ECL, Thermo Scientific) and with the aid of a Western Workflow Complete System V3 (Bio-Rad, Hercules, CA).
Immunohistochemistry
Frozen tissue sections taken from whole eyes of rats in the different groups were cut at 10 μm with the aid of a cryostat (Microm HM 550, Thermo Scientific). After three washes in PBS for 10 min each, the sections were blocked for 1 h in 5% donkey serum containing 0.5% Triton X-100. The RAGE antibody produced by the R&D Systems (Toronto, Ontario, Canada), was raised against recombinant mouse protein in the rat. To prevent binding of the secondary anti-rat antibody to endogenous rat tissue immunoglobulin (IgG), the frozen sections were preincubated with unconjugated AffiniPure Fab fragments of donkey anti-rat IgG (H+L; Jackson ImmunoResearch Labs, West Grove, PA, Cat# 712–007–003) for 1 h. The sections were incubated overnight with primary antibodies against either RAGE (1:500, Abcam, Ab3611), RAGE (1:50, R&D, Mab1179, MN), poly ADP-ribose polymerase (PARP; 1:50, Abcam, Ab32138), or Brn3a (1:500, Santa Cruz, SC-31984) at 4 °C. Following three washes with PBS (5 min each), the sections were incubated with the corresponding secondary antibodies, conjugated with Alexa Fluor 488 or 568 and 4',6-diamidino-2-phenylindole (DAPI; 1 mg/ml, 1:1,000; Life Technologies, D1306) for 1 h at room temperature. Finally, the sections were washed in PBS and mounted on slides with Prolong Gold Antifade Reagent (Life Technologies). Negative controls were incubated with secondary antibodies only. The sections were viewed with the aid of an Eclipse Ti Confocal Microscope (Nikon Instruments, Melville, NY) with 40X objective magnification.
Retinal ganglion cell counts
Flatmounted retinas were imaged with confocal microscopy. For each retina, a total of 20 images were taken (five from each of the four retinal quadrants) using a 20X objective lens. Brn3a-positive neurons in the ganglion cell layer were counted semiautomatically using ImageJ software. Cell death in the IR-injured retinas was calculated as a percentage of the mean cell density in the untreated contralateral eyes.
Statistical analysis
The quantitative data are expressed as standard error of the mean (±SEM, n=9). Two-way ANOVA was used to compare temporal differences in the optical density values of the antibody-labeled bands in western blots for the various groups. Alpha was set to p<0.05.
Results
RAGE protein accumulation in the IR-injured rat retina
The abundance of the RAGE protein was determined using western blots and antibodies directed against different domains of the RAGE protein. One was raised against the folded form of the extracellular RAGE-domains (V, C1, and C2), denoted in this study as eRAGE, and the second one was raised against the C-terminal and cytoplasmic part of the RAGE protein, denoted in this study as cRAGE (Figure 1). Interestingly, each antibody revealed a different pattern of RAGE accumulation (Figure 2). eRAGE was increased over the controls, but only at the 12 h post-ischemic time. At this time point, three distinct bands with a proximal size of approximately 55, 70, and 110 kDa were evident in the western blots. The use of blocking peptide decreased the immunoreactivity of all three bands, confirming the specificity of the antibody (Figure 2B). The use of reducing agents (dTT or β-mercaptoethanol) before application of the antibody abolished the appearance of all three bands, which confirmed that the antibody recognizes only the folded structure of the RAGE protein, since disulfide bonds were obviously present in all three bands (Figure 2C). In contrast to this pattern, the second anti-RAGE antibody raised against the cytoplasmic C-terminal region of the protein revealed a single protein band with an approximate molecular size of 40 kDa, as expected (Figure 2D). No differences in the cRAGE protein levels were detected between the experimental and control animals among any of the post-ischemic time points.
Figure 1.

Schematic representation of RAGE protein domains. The position of the glycosylation sites, disulfide bonds, and recognition areas for the antibodies used in this experiment are shown. The monoclonal anti-RAGE antibody shown in pink recognizes the extracellular domains of full-length RAGE. The polyclonal anti-RAGE antibody, shown in blue, recognizes the cytoplasmic tail of full-length RAGE.
Figure 2.
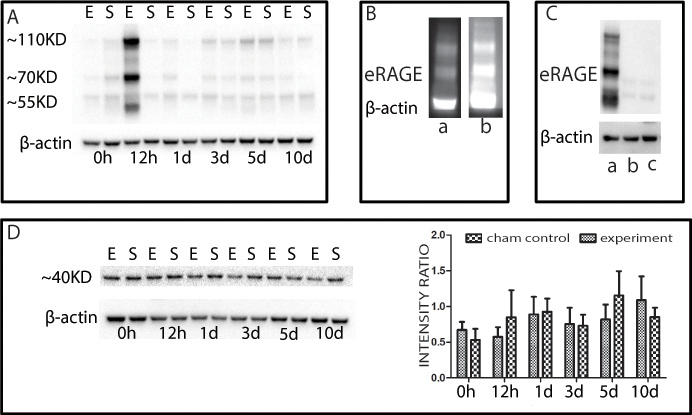
Western blots indicating the presence of the RAGE protein in the retina of the IR-injured rat. Six time points are presented on each blot. Every time point contained the experimental group (E) and the sham control group (S). A: The R&D-anti-RAGE antibody detected increased eRAGE accumulation at 12 h post ischemia. B: The peptide competition assay confirmed the specific band reactivity of the R&D-anti-RAGE antibody; lane-a western blot with R&D-anti-RAGE antibody preincubated with blocking peptide; lane-b western blot with a R&D-anti-RAGE antibody not preincubated with peptide. C: Western blots with R&D-anti-RAGE antibody performed under reducing and non-reducing conditions; lane-a non-reducing condition; lane-b reducing condition (β-mercaptoethanol); lane-c reducing condition (β-mercaptoethanol and dithiothreitol (DTT)). D: Abcam-anti RAGE-antibody shows the presence of cRAGE in every post-ischemic time point in the experimental and sham control animals. No significant difference in the accumulation of the cRAGE protein was detected among the examined post-ischemic time points.
Localization of the RAGE protein in retinal tissue sections
To determine the localization of the two RAGE isoforms, retina sections were assessed with immunohistochemistry. In agreement with the results from the western blots, eRAGE was observed only at the 12 h post-ischemic time point and not at all in the naïve retinas (Figure 3). The eRAGE immunolabeling was most prominent within cells in the retinal ganglion cell (RGC) layer. Colabeling the sections of 12 h post-ischemia retina for eRAGE and Brn3a—a marker for a subset of RGCs—revealed that all eRAGE-expressing cells also expressed Brn3a (Figure 4A–C), indicating that the RAGE protein is specifically localized within RGCs. In addition, eRAGE and PARP (reported to be a mediator of necrotic cell death [44,45]) were colocalized in cells within the RGC layer (Figure 4D–F).
Figure 3.
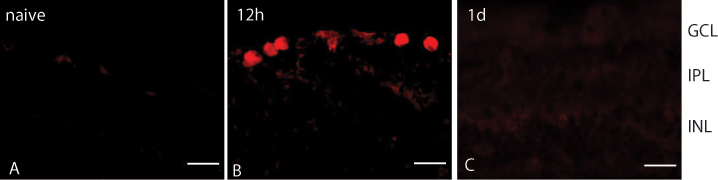
Immunohistochemical staining of the RAGE protein in the rat retina. A: The eRAGE protein is absent in the naïve (wild-type, WT) rat retina. B: The eRAGE protein accumulates in the retinal ganglion layer (RGC) layer of the retina at the 12 h post-ischemic time point. C: The eRAGE protein is not present in the retina at the 1 d post-ischemia-reperfusion period. The R&D anti-RAGE antibody (red) was used. Scale bars=20 μm.
Figure 4.

Colocalization of RAGE with Brn3a and PARP in the IR-injured rat retina at the12 h post-ischemia-reperfusion period. A: The transcription factor BRN3a (green) is present in a subset of cells in the retinal ganglion layer (RGC) layer at the12 h post-ischemia-reperfusion period. B: eRAGE (red) is localized to cells in the RGC layer in the ischemia reperfusion (IR)-injured rat retina at 12 h post-IR. C: Double immunolabeling with the eRAGE antibody (red) and the ganglion cell marker, BRN3a (green), at the 12 h reperfusion time shows they are colocalized. D: Necrotic cell marker, poly ADP-ribose polymerase (PARP) (green), is localized to cells in the RGC layer in the IR-injured rat retina at 12 h post-IR. E: eRAGE (red) is localized to cells in the RGC layer in the IR-injured rat retina at the 12 h post-ischemia-reperfusion period. F: Double immunolabeling with the RAGE antibody (red) and PARP at 12 h post-IR shows they are colocalized. Scale bars=20 μm.
In contrast, cRAGE was detected in the RGC layer and the inner nuclear layer (INL) of the retina in the naïve rats and the IR-injured rats at several post-ischemia-reperfusion time points (Figure 5). The observed lack of altered protein accumulation in the tissue sections is consistent with the results observed in the western blots (Figure 2D).
Figure 5.

Localization of the cRAGE protein in the retina from control, 12 h post-ischemia reperfusion (IR), 1 day post-IR, and 3 days post-IR, using the Abcam-anti-RAGE antibody. The cRAGE protein was detected mainly in the ganglion cell and inner nuclear layers. Scale bars=50 μm.
The intracellular distribution of each RAGE protein isoform appeared to be different. The antibody against the extracellular domain, eRAGE, demonstrated the protein in the cytoplasm and the nucleus, while the antibody against the cytoplasmic tail localized cRAGE only in the nucleus (Figure 6).
Figure 6.
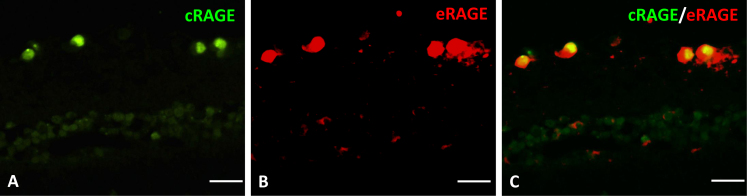
Localization of the RAGE protein in the retina using the two anti-RAGE antibodies at 12 h post-ischemia. A: The Abcam-anti-RAGE antibody (green) localized the cRAGE protein in the nucleus only. B: The R&D-anti-RAGE antibody (red) localized the eRAGE protein in the cytoplasm and the nucleus. C: eRAGE and cRAGE colocalized in cells of the retinal ganglion layer (RGC) layer at the 12 h post-ischemic time point. The colocalization shows the nucleus contains cRAGE and eRAGE. Scale bars=20 μm.
ERK in the IR-injured rat retina
Recent studies indicated that the activation of ERK plays a critical role in necroptosis in HT-22 cells [46]. Phosphorylated ERK protein (p-ERK) and total ERK protein (t-ERK) in the IR-injured rat retina were measured with western blots. The total amount of ERK protein did not change among the post-ischemia-reperfusion periods when compared with the naïve retina. In contrast, the amount of p-ERK increased at the 12 h post-ischemic time point and gradually returned to normal levels in later reperfusion periods (Figure 7).
Figure 7.

Western blots indicating accumulation of p-ERK and t-ERK in the IR-injured rat retina. A: There was little difference in total ERK (t-ERK) abundance at each time point in the experimental and sham control groups. However, the phosphorylated ERK protein (p-ERK) levels were increased at 12 h post-ischemia. The western blot is representative of the blots for each of the three animals. B: At 12 h post-ischemia reperfusion, the p-ERK accumulation was significantly increased relative to the sham controls and compared to all other time points. There was a gradual decrease in phosphorylated ERK with time after the 12 h ischemia reperfusion (IR) injury period. Data are presented as mean ± standard error of the mean (SEM), *p<0.05, ***p<0.001. E=experimental group; S=sham control group.
Accumulation of the RIP3 protein in the IR rat retina
Previous studies have shown that increased RIP1 activity may lead to either apoptotic or necrotic cell death while the RIP1/RIP3 complex preferentially plays a critical role in the development of necroptosis [17,47]. Western blot assessment revealed that, compared with the naïve retinas, an increased amount of RIP3 protein was present at 12 h, as well as at days 1, 3, and 5 post-ischemia. Among the post-IR periods studied, the highest protein accumulation was observed at 12 h. After 12 h, the amount of RIP3 gradually returned to normal levels over a period of several days (Figure 8).
Figure 8.

Changes over time in RIP3 accumulation in the rat retina following ischemia. A: The amount of RIP3 protein was elevated at the 12 h post- ischemic time point in the animals from the experimental group when compared with the 0 h and later post-ischemic time points. The western blot is representative of blots for each of the three animals. After the increase at 12 h, the level of the RIP3 protein gradually returned to normal. B: The increase at the 12 h post-ischemic reperfusion time point in the animals from the experimental group was significant compared to the 0 h and later time points. Data are presented as mean ± standard error of the mean (SEM). ***p<0.001. E=experimental group; S=sham control group; ns=not significant.
Colocalization of RIP3 with eRAGE in the IR-injured rat retina
The RIP3 and eRAGE proteins were upregulated at 12 h post-IR, and eRAGE expression was restricted to the RGCs. We therefore examined possible colocalization of eRAGE with the necroptotic marker RIP3 in the RGCs. We first colabeled tissue sections with antibodies to RIP3 and the retinal ganglion cell marker Brn3a. All RIP3-positive cells expressed Brn3a as well (Figure 9A–C), indicating that the RIP3 protein is localized within retinal ganglion cells, much as we had observed for eRAGE. Sections colabeled with antibodies to RIP3 and eRAGE revealed that both proteins were expressed predominantly in the same RGCs at 12 h post-ischemia in the IR-injured rat retina (Figure 9D–F). The majority of the eRAGE-expressing cells were RIP3-positive.
Figure 9.

Colocalization of RIP3 and eRAGE with Brn3a-labeled retinal ganglion cells in the IR-injured rat retina at the 12 h post-ischemic reperfusion time point. A: The transcription factor BRN3a (green) is present in the ganglion cells of the retinal ganglion cell (RGC) layer. B: RIP3 (red) is localized to cells in the RGC layer in the ischemia reperfusion (IR)-injured rat retina. C: Double immunolabeling shows Brn3a (green) and RIP3 (red) are colocalized. D: eRAGE (red) is localized to cells in the RGC layer in the IR-injured retina. E: RIP3 (green) is localized to cells in the RGC layer in the IR-injured retina F: Double immunolabeling shows eRAGE (red) and RIP3 (green) are colocalized in the IR-injured retina. G: BRN3a (green) in the healthy rat retina (control). H: RIP3 (red) in the healthy rat retina (control). I: Double immunolabeling with Brn3a (green) and RIP3 (red) in the healthy rat retina (control). Scale bars=20 μm.
Presence of FLIP in the IR-injured rat retina
Previous studies have shown that the RIP1/RIP3 complex plays a critical role in the development of not only necroptosis but also apoptosis. The activity of a caspase-8 like protein, FLIPS/L, is a postulated control point influencing the assembly of the RIP1/RIP3 complex [48] and thus affecting the relative contribution of each apoptosis and necroptosis pathway to cell death. The protein levels of FLIPS/L were evaluated at six post-IR points using western blots. FLIPS/L was detected only at the 12 h post-ischemic time point in the animals from the experimental group. The presence of FLIPS/L could not be detected in the experimental retinas at the other time points or the controls at any time (Figure 10).
Figure 10.
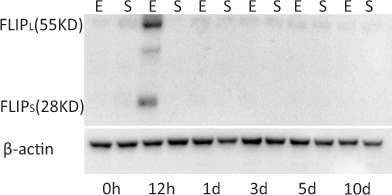
Western blots indicating the presence of two FLIP isoforms, FLIPL and FLIPS, in the retina of the IR-injured rat. The FLIPS/L protein was strongly expressed only at the 12 h post-ischemic time point. Although weak FLIPL bands are visible, the FLIPS protein was detected only at the 12 h post- ischemic time point. E=experimental group; S=sham control group.
Inhibition of ERK activation increased RGC survival in the IR-injured rat retina
The amount of p-ERK as well as the level of the RIP3 protein were increased at the 12 h post-IR time point in the retina and gradually returned to normal levels in later reperfusion periods (Figure 7 and Figure 8). To determine if there is a possible association between the kinase activity of ERK and the accumulation of the RIP3 protein in the retina, ERK activation was inhibited with the highly selective mitogen-activated protein kinase (MEK) inhibitor U0126. Accumulation of p-ERK was significantly decreased in the presence of 0.8 mM U0126 (Figure 11A). We then measured the level of RIP3 in IR-injured rats at the 12 h time point. The IR-injured animals from the two control groups were dissimilar in that the RIP3 was elevated in the DMSO control treatment group relative to the saline control group (Figure 11B). This is likely because DMSO can induce retinal cell death in vivo and in vitro [49]. Relative to the DMSO control group, the 0.4 mM U0126 treatment group showed a decreased amount of RIP3, but this decrease was not apparent when compared with the saline control group. However, the 0.8 mM U0126 significantly reduced the amount of RIP3 protein in the IR-injured retina when compared to all other groups (Figure 11B). As a comparison, we also measured the levels of the RIP1 protein in the IR-injured rats at the 12 h time point, which was different from RIP3 in that the control IR-injured animals, treated with either saline or solvent, showed no difference in RIP1 accumulation compared to the animals treated with 0.4 or 0.8 mM U0126 (Figure 11C).
Figure 11.

Inhibition of ERK activation using MEK inhibitor U0126 led to decreased accumulation of RIP3 but not of RIP1 in the IR-injured rat retina. A: The amount of phosphorylated ERK protein (p-ERK) at 12 h post-ischemia reperfusion (IR) decreased in the animals treated with 0.4 or 0.8 mM U0126 relative to the solvent- (5% DMSO) or saline-treated animals. The decreased amount of p-ERK protein in the animals treated with 0.8 mM U0126 was significant compared to the groups treated with saline, solvent (5% DMSO), or 0.4 mM U0126. B: The amount of accumulated RIP3 protein at 12 h post-IR decreased in the animals treated with 0.4 or 0.8 mM U0126 when compared with the solvent- (5% DMSO) or saline-treated animals. The decreased amount of RIP3 in the animals treated with 0.8 mM U0126 was significant compared to the groups treated with saline, solvent (5% DMSO), or 0.4 mM U0126. C: There was no difference in the accumulation of the RIP1 protein at 12 h post-IR in the animals treated with 0.8 mM U0126 compared to the groups treated with saline, solvent (5% DMSO), or 0.4 mM U0126. Data are presented as mean ± standard error of the mean (SEM). *p<0.05, ***p<0.001.
Next, we examined the retinal ganglion cell survival post ischemia-reperfusion injury in the rat retina using Brn3a labeling and compared it to the group treated with the ERK inhibitor and the DMSO control treatment group. As expected, RGC survival was significantly decreased in the ischemia-reperfusion injured eye compared to the contralateral eye. Treatment with the ERK inhibitor increased RGC survival by approximately 20% in the injured retinas relative to the non-treated (IR) and DMSO-treated groups (Figure 12).
Figure 12.
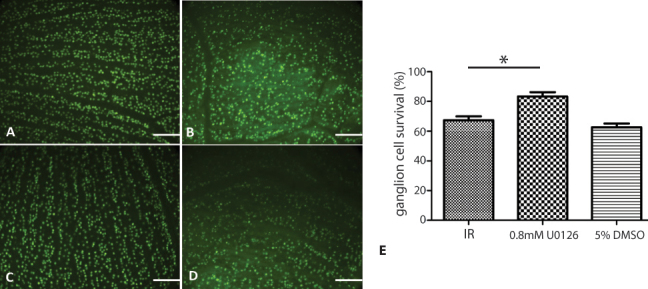
Retinal ganglion cell survival at 24 h post ischemia-reperfusion injury in the rat retina. The retinal ganglion cells (RGCs) were labeled with Brn3a. The cell counts are relative to the contralateral eye. Three animals per group were analyzed. A: Naïve rat retina. B: Ischemia reperfusion (IR)-injured rat retina. C: IR-injured rat retina treated with 0.8 mM U0126. D: IR-injured rat retina treated with 5% dimethyl sulfoxide (DMSO). E: Percentage of ganglion cell survival in the IR-injured rat retina. Scale bars=10 μm. *p<0.01.
Discussion
In this study, we examined the response of the adult rat retina to IR injury and confirmed that necroptotic cell death occurs in the early stages of the reperfusion period. We identified some of the principal players involved in the necroptotic signal transduction pathway and demonstrated that inhibition of this pathway leads to increased retinal ganglion cell survival.
RAGE isoforms are differentially localized in the retina
We identified two RAGE isoforms in the retina. Although both isoforms colocalized in the same RGCs, we detected differences in the isoforms’ intracellular localizations. The eRAGE isoform was detected in the cytoplasm and the nucleus of the RGCs, while the cRAGE isoform existed only in the nucleus. It seems likely that these different intracellular localizations might contribute to distinct functions of each RAGE isoform. Another possibility is that both isoforms might be expressed in different subpopulations of RGCs. In this study, we used Brn3a as a ganglion cell marker, which labels one particular RGC subpopulation [50].
We propose that eRAGE is a novel isoform, with a different C-terminal cytoplasmic domain than the cRAGE isoform. The cytoplasmic C-terminus of the RAGE protein contains motifs that are essential for transducing extracellular to intracellular signaling through binding to different partners [51-53]. The isoforms that lack a cytoplasmic domain are soluble and block signal transduction [33]. Studies have shown that the soluble RAGE isoform could act as a decoy receptor that antagonizes full-length RAGE by competing for ligand binding [51,54]. The full length of our novel eRAGE is indicated by the size of the detected bands (approximately 55, 70, and 100 kDa) while soluble isoforms are approximately 20 kDa [51]. Three protein bands were detected in the lung homogenate using the same anti-RAGE antibody as used in our study [55]. The sizes for two of the reported RAGE protein bands correspond to the sizes obtained in our blots. In that study, matrix-assisted laser desorption/ionization time of flight/mass spectrometry (MALDI-TOF/MS) supported their identification as a RAGE protein [55]. The third band obtained in our western blots (approximately 100 kDa) might result from dimerization of the low protein band (approximately 55 kDa). Homodimerization changes the three-dimensional configuration and might be essential for RAGE-mediated signal transduction [34].
Reasons for the difference in size between the eRAGE (approximately 55 kDa) and cRAGE (approximately 40 kDa) isoforms could be the posttranslational modification. It is possible that the continuously expressed cRAGE isoform does not undergo glycosylation and its size corresponds to the only known RAGE variant in the rat. In contrast, the novel eRAGE isoform possibly undergoes a different degree of glycosylation and dimerization. The two N-glycosylation sites in the V domain of RAGE (Figure 1) could affect the three-dimensional configuration of the protein, which is reported to influence ligand selection [33-35,42]. For example, studies have shown that glycosylation of RAGE mediated the interaction between RAGE and HMGB1 in mouse embryonic cortical neurons [42]. It has also been shown that RAGE could form oligomers on cell membranes to increase the extracellular V-domains for higher ligand binding affinity [34,36].
In the RAGE-deficient IR mouse retinal model, there is increased survival of cells in the ganglion cell layer [25]. Other studies have reported that the use of recombinant soluble RAGE provides cell protection from various types of injuries [56-58]. Interestingly, Tae et al. showed that the glycosylation of soluble RAGE impacts its bioactivity and appears to amplify its therapeutic efficacy [57]. Targeting the glycosylated transmembrane eRAGE receptor with putative antagonists (e.g., glycosylated soluble RAGE) within the 12 h post-IR injury period might be used to rescue neurons from their prospective fate.
ERK1/2 and RIP3 in the IR-injured retina
Our results showed that the phosphorylation of ERK was significantly increased at the early post-IR injury period, confirming the association of ERK in neuronal cell death as previously reported [2,59]. In the tumor necrosis factor-α tumor-necrosis factor receptor-1 (TNF-α TNFR1) pathway, formation of the RIP1/RIP3 complex, called the necrosome, is necessary for execution of the necroptosis program [17,60]. Although RIP1 has been associated with necroptosis and apoptosis, RIP3-driven cell death is linked predominantly to necroptosis [47,60]. We have shown that RIP1 and RIP3 are present in the retina at the 12 h post-ischemic time point and that RIP3 accumulation significantly increased. These data and recently published studies [15,16] confirm that in early time periods following IR injury, one of the mechanisms driving retinal cell death is necroptosis.
Decreasing the level of ERK phosphorylation in the early post-IR periods led to markedly decreased accumulation of RIP3 in the ischemic rat retina, while RIP1 accumulation was not changed. These results imply involvement of ERK in the critical steps leading to the execution of necroptosis at early time points following retinal IR injury. This interesting result also indicates the possibility that ERK activation is upstream of RIP3 in this cell death pathway. These data showed that 4 μl of the 800 μM U0126 injection, which yields an estimated vitreal concentration of approximately 50 μM, is needed to diffuse into the retina and indicates that local concentrations in the retina exceed the calculated IC50 value of 15 μM reported for NIH3T3 cells [61]. However, we were inhibiting tissue in vivo, and it is not known how well the retina is permeated or how much of the inhibitor gets into the cells.
Previous studies have shown that RIP kinases act upstream of ERK [60] and that RIP3 acts upstream to phosphorylate RIP1 [62]. The phosphorylation of RIP1 and RIP3 can stabilize their interaction within the pronecrotic complex, evokes downstream reactive oxygen species (ROS) production, and leads to necroptosis [62]. Other studies have suggested that RIP1 is recruited to RIP3 followed by mutual phosphorylation of RIP1 and RIP3 [63,64]. Based on our current results with the inhibitor U0126, ERK phosphorylation occurs upstream of RIP3 and directly or indirectly leads to increased RIP3 accumulation to initiate necroptosis.
Switch between necroptosis and apoptosis
The notion that necroptosis is initiated early in the IR-injury model is supported by previous studies [15,16], but also by the presence of cellular FLICE-like inhibitory protein (cFLIP). This protein interferes with apoptosis by competing with caspase-8 [48,65,66]. The presence of the two isoforms, c-FLIPL and c-FLIPS, at 12 h post-IR, indicates a possible switch from the mechanism of apoptosis in favor of necroptosis at the early IR injury time point of 12 h.
Summary
In summary, the present study demonstrates that in early stages after IR injury RGC death occurs through the ERK1/2-RIP3 dependent pathway. Directly or indirectly, ERK contributes to increased RIP3 accumulation to induce the assembly of a necrosome, which in turn activates downstream signaling leading to necroptosis. The occurrence of ERK phosphorylation as an upstream event of RIP3 activation differs from the TNF-α-TNFR1-induced cell death pathway, where involvement of ERK is not reported. Our data suggest that the necroptotic cell death process peaks at 12 h post-ischemia and may continue to be active for limited time afterwards. Therapeutic targeting of the ERK1/2-RIP3 pathway at the appropriate time might prevent the triggering of necroptosis and contribute to extended survival of retinal neurons. In addition, our study suggests the existence of a novel RAGE isoform in the ischemic rat retina that is specifically expressed in RGCs at 12 h post-IR injury. Further studies might explore the possibility of eRAGE-evoked ERK phosphorylation.
Acknowledgments
This work was supported by NEI R01EY017594, the National Institute of General Medical Sciences (8 P20 GM103436–13). Sheng Gao was supported by the Chinese Government (201,206,240,121). The confocal microscope used in this study is part of a core facility supported by NIH/NIGMS P30 GM103507. The authors thank Dr. R. Benton, Dr. J. Petruska and Dr. S. Florea for the critical reading of this manuscript.
References
- 1.Osborne NN, Casson RJ, Wood JP, Chidlow G, Graham M, Melena J. Retinal ischemia: mechanisms of damage and potential therapeutic strategies. Prog Retin Eye Res. 2004;23:91–147. doi: 10.1016/j.preteyeres.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 2.Yokota H, Narayanan SP, Zhang W, Liu H, Rojas M, Xu Z, Lemtalsi T, Nagaoka T, Yoshida A, Brooks SE, Caldwell RW, Caldwell RB. Neuroprotection from retinal ischemia/reperfusion injury by NOX2 NADPH oxidase deletion. Invest Ophthalmol Vis Sci. 2011;52:8123–31. doi: 10.1167/iovs.11-8318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anderson DR, Davis EB. Sensitivities of ocular tissues to acute pressure-induced ischemia. Arch Ophthalmol. 1975;93:267–74. doi: 10.1001/archopht.1975.01010020277006. [DOI] [PubMed] [Google Scholar]
- 4.Nucci C, Tartaglione R, Rombola L, Morrone LA, Fazzi E, Bagetta G. Neurochemical evidence to implicate elevated glutamate in the mechanisms of high intraocular pressure (IOP)-induced retinal ganglion cell death in rat. Neurotoxicology. 2005;26:935–41. doi: 10.1016/j.neuro.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 5.Fujita R, Ueda M, Fujiwara K, Ueda H. Prothymosin-alpha plays a defensive role in retinal ischemia through necrosis and apoptosis inhibition. Cell Death Differ. 2009;16:349–58. doi: 10.1038/cdd.2008.159. [DOI] [PubMed] [Google Scholar]
- 6.Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol. 2014;15:135–47. doi: 10.1038/nrm3737. [DOI] [PubMed] [Google Scholar]
- 7.Takahashi N, Duprez L, Grootjans S, Cauwels A, Nerinckx W, DuHadaway JB, Goossens V, Roelandt R, Van Hauwermeiren F, Libert C, Declercq W, Callewaert N, Prendergast GC, Degterev A, Yuan J, Vandenabeele P. Necrostatin-1 analogues: critical issues on the specificity, activity and in vivo use in experimental disease models. Cell Death Dis. 2012;3:e437. doi: 10.1038/cddis.2012.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang GQ, Hao Y, Wong F. Apoptosis: final common pathway of photoreceptor death in rd, rds, and rhodopsin mutant mice. Neuron. 1993;11:595–605. doi: 10.1016/0896-6273(93)90072-y. [DOI] [PubMed] [Google Scholar]
- 9.Sancho-Pelluz J, Arango-Gonzalez B, Kustermann S, Romero FJ, van Veen T, Zrenner E, Ekstrom P, Paquet-Durand F. Photoreceptor cell death mechanisms in inherited retinal degeneration. Mol Neurobiol. 2008;38:253–69. doi: 10.1007/s12035-008-8045-9. [DOI] [PubMed] [Google Scholar]
- 10.Guo L, Moss SE, Alexander RA, Ali RR, Fitzke FW, Cordeiro MF. Retinal ganglion cell apoptosis in glaucoma is related to intraocular pressure and IOP-induced effects on extracellular matrix. Invest Ophthalmol Vis Sci. 2005;46:175–82. doi: 10.1167/iovs.04-0832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bonfoco E, Krainc D, Ankarcrona M, Nicotera P, Lipton SA. Apoptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with N-methyl-D-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc Natl Acad Sci USA. 1995;92:7162–6. doi: 10.1073/pnas.92.16.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trichonas G, Murakami Y, Thanos A, Morizane Y, Kayama M, Debouck CM, Hisatomi T, Miller JW, Vavvas DG. Receptor interacting protein kinases mediate retinal detachment-induced photoreceptor necrosis and compensate for inhibition of apoptosis. Proc Natl Acad Sci USA. 2010;107:21695–700. doi: 10.1073/pnas.1009179107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hanus J, Zhang H, Wang Z, Liu Q, Zhou Q, Wang S. Induction of necrotic cell death by oxidative stress in retinal pigment epithelial cells. Cell Death Dis. 2013;4:e965. doi: 10.1038/cddis.2013.478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sato K, Li S, Gordon WC, He J, Liou GI, Hill JM, Travis GH, Bazan NG, Jin M. Receptor interacting protein kinase-mediated necrosis contributes to cone and rod photoreceptor degeneration in the retina lacking interphotoreceptor retinoid-binding protein. J Neurosci. 2013;33:17458–68. doi: 10.1523/JNEUROSCI.1380-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dvoriantchikova G, Degterev A, Ivanov D. Retinal ganglion cell (RGC) programmed necrosis contributes to ischemia-reperfusion-induced retinal damage. Exp Eye Res. 2014;123:1–7. doi: 10.1016/j.exer.2014.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosenbaum DM, Degterev A, David J, Rosenbaum PS, Roth S, Grotta JC, Cuny GD, Yuan J, Savitz SI. Necroptosis, a novel form of caspase-independent cell death, contributes to neuronal damage in a retinal ischemia-reperfusion injury model. J Neurosci Res. 2010;88:1569–76. doi: 10.1002/jnr.22314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity. 2013;38:209–23. doi: 10.1016/j.immuni.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 18.Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FKM. Phosphorylation-Driven Assembly of the RIP1–RIP3 Complex Regulates Programmed Necrosis and Virus-Induced Inflammation. Cell. 2009;137:1112–23. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–14. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- 20.Long JS, Ryan KM. New frontiers in promoting tumour cell death: targeting apoptosis, necroptosis and autophagy. Oncogene. 2012;31:5045–60. doi: 10.1038/onc.2012.7. [DOI] [PubMed] [Google Scholar]
- 21.Viringipurampeer IA, Shan X, Gregory-Evans K, Zhang JP, Mohammadi Z, Gregory-Evans CY. Rip3 knockdown rescues photoreceptor cell death in blind pde6c zebrafish. Cell Death Differ. 2014;21:665–75. doi: 10.1038/cdd.2013.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murakami Y, Matsumoto H, Roh M, Giani A, Kataoka K, Morizane Y, Kayama M, Thanos A, Nakatake S, Notomi S, Hisatomi T, Ikeda Y, Ishibashi T, Connor KM, Miller JW, Vavvas DG. Programmed necrosis, not apoptosis, is a key mediator of cell loss and DAMP-mediated inflammation in dsRNA-induced retinal degeneration. Cell Death Differ. 2014;21:270–7. doi: 10.1038/cdd.2013.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pouwels SD, Heijink IH, Ten Hacken NH, Vandenabeele P, Krysko DV, Nawijn MC, van Oosterhout AJ. DAMPs activating innate and adaptive immune responses in COPD. Mucosal Immunol. 2014;7:215–26. doi: 10.1038/mi.2013.77. [DOI] [PubMed] [Google Scholar]
- 24.Muhammad S, Barakat W, Stoyanov S, Murikinati S, Yang H, Tracey KJ, Bendszus M, Rossetti G, Nawroth PP, Bierhaus A, Schwaninger M. The HMGB1 receptor RAGE mediates ischemic brain damage. J Neurosci. 2008;28:12023–31. doi: 10.1523/JNEUROSCI.2435-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dvoriantchikova G, Hernandez E, Grant J, Santos AR, Yang H, Ivanov D. The high-mobility group box-1 nuclear factor mediates retinal injury after ischemia reperfusion. Invest Ophthalmol Vis Sci. 2011;52:7187–94. doi: 10.1167/iovs.11-7793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zitvogel L, Kepp O, Kroemer G. Decoding cell death signals in inflammation and immunity. Cell. 2010;140:798–804. doi: 10.1016/j.cell.2010.02.015. [DOI] [PubMed] [Google Scholar]
- 27.Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–5. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 28.van Beijnum JR, Buurman WA, Griffioen AW. Convergence and amplification of toll-like receptor (TLR) and receptor for advanced glycation end products (RAGE) signaling pathways via high mobility group B1 (HMGB1). Angiogenesis. 2008;11:91–9. doi: 10.1007/s10456-008-9093-5. [DOI] [PubMed] [Google Scholar]
- 29.Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ. HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol. 2010;28:367–88. doi: 10.1146/annurev.immunol.021908.132603. [DOI] [PubMed] [Google Scholar]
- 30.Dvoriantchikova G, Hernandez E, Grant J, Santos ARC, Yang H, Ivanov D. The High-Mobility Group Box-1 Nuclear Factor Mediates Retinal Injury after Ischemia Reperfusion. Invest Ophthalmol Vis Sci. 2011;52:7187–94. doi: 10.1167/iovs.11-7793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hassid BG, Nair MN, Ducruet AF, Otten ML, Komotar RJ, Pinsky DJ, Schmidt AM, Yan SF, Connolly ES. Neuronal RAGE expression modulates severity of injury following transient focal cerebral ischemia. J Clin Neurosci. 2009;16:302–6. doi: 10.1016/j.jocn.2007.12.011. [DOI] [PubMed] [Google Scholar]
- 32.Kalea AZ, Reiniger N, Yang H, Arriero M, Schmidt AM, Hudson BI. Alternative splicing of the murine receptor for advanced glycation end-products (RAGE) gene. FASEB J. 2009;23:1766–74. doi: 10.1096/fj.08-117739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koch M, Chitayat S, Dattilo BM, Schiefner A, Diez J, Chazin WJ, Fritz G. Structural basis for ligand recognition and activation of RAGE. Structure. 2010;18:1342–52. doi: 10.1016/j.str.2010.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zong H, Madden A, Ward M, Mooney MH, Elliott CT, Stitt AW. Homodimerization is essential for the receptor for advanced glycation end products (RAGE)-mediated signal transduction. J Biol Chem. 2010;285:23137–46. doi: 10.1074/jbc.M110.133827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marinakis E, Bagkos G, Piperi C, Roussou P, Diamanti-Kandarakis E. Critical role of RAGE in lung physiology and tumorigenesis: a potential target of therapeutic intervention? Clin Chem Lab Med. 2014;52:189–200. doi: 10.1515/cclm-2013-0578. [DOI] [PubMed] [Google Scholar]
- 36.Sparvero LJ, Asafu-Adjei D, Kang R, Tang DL, Amin N, Im J, Rutledge R, Lin B, Amoscato AA, Zeh HJ, Lotze MT. RAGE (Receptor for Advanced Glycation Endproducts), RAGE Ligands, and their role in Cancer and Inflammation. J Transl Med. 2009;7:7. doi: 10.1186/1479-5876-7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huskens D, Princen K, Schreiber M, Schols D. The role of N-glycosylation sites on the CXCR4 receptor for CXCL-12 binding and signaling and X4 HIV-1 viral infectivity. Virology. 2007;363:280–7. doi: 10.1016/j.virol.2007.01.031. [DOI] [PubMed] [Google Scholar]
- 38.Arey BJ. The Role of Glycosylation in Receptor Signaling. In: Petrescu DS, ed.: InTech, 2012. [Google Scholar]
- 39.Yan X, Han J, Zhang Z, Wang J, Cheng Q, Gao K, Ni Y, Wang Y. Lung cancer A549 cells migrate directionally in DC electric fields with polarized and activated EGFRs. Bioelectromagnetics. 2009;30:29–35. doi: 10.1002/bem.20436. [DOI] [PubMed] [Google Scholar]
- 40.Laberge M. Intrinsic protein electric fields: basic non-covalent interactions and relationship to protein-induced Stark effects. Biochim Biophys Acta. 1998;1386:305–30. doi: 10.1016/s0167-4838(98)00100-9. [DOI] [PubMed] [Google Scholar]
- 41.Solá RJ, Griebenow K. Effects of glycosylation on the stability of protein pharmaceuticals. J Pharm Sci. 2009;98:1223–45. doi: 10.1002/jps.21504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Srikrishna G, Huttunen HJ, Johansson L, Weigle B, Yamaguchi Y, Rauvala H, Freeze HH. N -Glycans on the receptor for advanced glycation end products influence amphoterin binding and neurite outgrowth. J Neurochem. 2002;80:998–1008. doi: 10.1046/j.0022-3042.2002.00796.x. [DOI] [PubMed] [Google Scholar]
- 43.Zhang Z, Qin X, Tong N, Zhao X, Gong Y, Shi Y, Wu X. Valproic acid-mediated neuroprotection in retinal ischemia injury via histone deacetylase inhibition and transcriptional activation. Exp Eye Res. 2012;94:98–108. doi: 10.1016/j.exer.2011.11.013. [DOI] [PubMed] [Google Scholar]
- 44.Ha HC, Snyder SH. Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc Natl Acad Sci USA. 1999;96:13978–82. doi: 10.1073/pnas.96.24.13978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu Y, Huang S, Liu ZG, Han JH. Poly(ADP-ribose) polymerase-1 signaling to mitochondria in necrotic cell death requires RIP1/TRAF2-mediated JNK1 activation. J Biol Chem. 2006;281:8788–95. doi: 10.1074/jbc.M508135200. [DOI] [PubMed] [Google Scholar]
- 46.Zhang M, Li J, Geng R, Ge W, Zhou Y, Zhang C, Cheng Y, Geng D. The inhibition of ERK activation mediates the protection of necrostatin-1 on glutamate toxicity in HT-22 cells. Neurotox Res. 2013;24:64–70. doi: 10.1007/s12640-012-9361-4. [DOI] [PubMed] [Google Scholar]
- 47.Moriwaki K, Chan FK. RIP3: a molecular switch for necrosis and inflammation. Genes Dev. 2013;27:1640–9. doi: 10.1101/gad.223321.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gong J, Kumar SA, Graham G, Kumar AP. FLIP: Molecular Switch Between Apoptosis and Necroptosis. Mol Carcinog. 2014;53:675–85. doi: 10.1002/mc.22027. [DOI] [PubMed] [Google Scholar]
- 49.Galvao J, Davis B, Tilley M, Normando E, Duchen MR, Cordeiro MF. Unexpected low-dose toxicity of the universal solvent DMSO. FASEB J. 2014;28:1317–30. doi: 10.1096/fj.13-235440. [DOI] [PubMed] [Google Scholar]
- 50.Shi M, Kumar SR, Motajo O, Kretschmer F, Mu X, Badea TC. Genetic interactions between Brn3 transcription factors in retinal ganglion cell type specification. PLoS ONE. 2013;8:E76347. doi: 10.1371/journal.pone.0076347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rai V, Maldonado AY, Burz DS, Reverdatto S, Yan SF, Schmidt AM, Shekhtman A. Signal transduction in receptor for advanced glycation end products (RAGE): solution structure of C-terminal rage (ctRAGE) and its binding to mDia1. J Biol Chem. 2012;287:5133–44. doi: 10.1074/jbc.M111.277731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sakaguchi M, Murata H, Yamamoto K, Ono T, Sakaguchi Y, Motoyama A, Hibino T, Kataoka K, Huh NH. TIRAP, an adaptor protein for TLR2/4, transduces a signal from RAGE phosphorylated upon ligand binding. PLoS ONE. 2011;6:e23132. doi: 10.1371/journal.pone.0023132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ishihara K, Tsutsumi K, Kawane S, Nakajima M, Kasaoka T. The receptor for advanced glycation end-products (RAGE) directly binds to ERK by a D-domain-like docking site. FEBS Lett. 2003;550:107–13. doi: 10.1016/s0014-5793(03)00846-9. [DOI] [PubMed] [Google Scholar]
- 54.Borsi V, Cerofolini L, Fragai M, Luchinat C. NMR characterization of the C-terminal tail of full-length RAGE in a membrane mimicking environment. J Biomol NMR. 2012;54:285–90. doi: 10.1007/s10858-012-9671-0. [DOI] [PubMed] [Google Scholar]
- 55.Mizumoto S, Takahashi J, Sugahara K. Receptor for Advanced Glycation End Products (RAGE) Functions as Receptor for Specific Sulfated Glycosaminoglycans, and Anti-RAGE Antibody or Sulfated Glycosaminoglycans Delivered in Vivo Inhibit Pulmonary Metastasis of Tumor Cells. J Biol Chem. 2012;287:18985–94. doi: 10.1074/jbc.M111.313437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bowman MA, Schmidt AM. The next generation of RAGE modulators: implications for soluble RAGE therapies in vascular inflammation. J Mol Med (Berl) 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tae HJ, Kim JM, Park S, Tomiya N, Li G, Wei W, Petrashevskaya N, Ahmet I, Pang J, Cruschwitz S, Riebe RA, Zhang Y, Morrell CH, Browe D, Lee YC, Xiao RP, Talan MI, Lakatta EG, Lin L. The N-glycoform of sRAGE is the key determinant for its therapeutic efficacy to attenuate injury-elicited arterial inflammation and neointimal growth. J Mol Med (Berl) 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ha CH, Kim S, Chung J, An SH, Park S, Choi D, Kwon K. Inhibitory effect of soluble RAGE in disturbed flow-induced atherogenesis. Int J Mol Med. 2013;32:373–80. doi: 10.3892/ijmm.2013.1393. [DOI] [PubMed] [Google Scholar]
- 59.Gesslein B, Hakansson G, Carpio R, Gustafsson L, Perez MT, Malmsjo M. Mitogen-activated protein kinases in the porcine retinal arteries and neuroretina following retinal ischemia-reperfusion. Mol Vis. 2010;16:392–407. [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang D, Lin J, Han J. Receptor-interacting protein (RIP) kinase family. Cell Mol Immunol. 2010;7:243–9. doi: 10.1038/cmi.2010.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.WesterAssay I–C. Complete Sample Protocol for Measuring IC50 of Inhibitor U0126 in NIH3T3 Responding to Acidic Fibroblast Growth Factor (aFGF-1). 2012 [Google Scholar]
- 62.Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK. Phosphorylation-driven assembly of the RIP1–RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–23. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100–11. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 64.Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, Han J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332–6. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 65.Safa AR, Pollok KE. Targeting the Anti-Apoptotic Protein c-FLIP for Cancer Therapy. Cancers. 2011;3:1639–71. doi: 10.3390/cancers3021639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shirley S, Micheau O. Targeting c-FLIP in cancer. Cancer Lett. 2013;332:141–50. doi: 10.1016/j.canlet.2010.10.009. [DOI] [PubMed] [Google Scholar]