

GNE myopathy is an adult-onset muscle disease caused by genetic defects in sialic acid biosynthesis. In a mouse model of the disorder, Yonekawa et al. reveal that sialic acid supplementation reduces muscle atrophy in aged symptomatic animals. The sialic acid glycoconjugate 6'-sialyllactose has greater therapeutic efficacy than free sialic acid.
Keywords: amyloid inclusion, distal myopathy with rimmed vacuoles (DMRV)/hereditary inclusion body myopathy (hIBM), GNE myopathy, hyposialylation, sialyllactose
Abstract
Patients with GNE myopathy, a progressive and debilitating disease caused by a genetic defect in sialic acid biosynthesis, rely on supportive care and eventually become wheelchair-bound. To elucidate whether GNE myopathy is treatable at a progressive stage of the disease, we examined the efficacy of sialic acid supplementation on symptomatic old GNE myopathy mice that have ongoing, active muscle degeneration. We examined the therapeutic effect of a less metabolized sialic acid compound (6’-sialyllactose) or free sialic acid (N-acetylneuraminic acid) by oral, continuous administration to 50-week-old GNE myopathy mice for 30 weeks. To evaluate effects on their motor performance in living mice, spontaneous locomotion activity on a running wheel was measured chronologically at 50, 65, 72 and 80 weeks of age. The size, force production, and pathology of isolated gastrocnemius muscle were analysed at the end point. Sialic acid level in skeletal muscle was also measured. Spontaneous locomotion activity was recovered in 6’-sialyllactose-treated mice, while NeuAc-treated mice slowed the disease progression. Treatment with 6’-sialyllactose led to marked restoration of hyposialylation in muscle and consequently to robust improvement in the muscle size, contractile parameters, and pathology as compared to NeuAc. This is due to the fact that 6’-sialyllactose is longer working as it is further metabolized to free sialic acid after initial absorption. 6’-sialyllactose ameliorated muscle atrophy and degeneration in symptomatic GNE myopathy mice. Our results provide evidence that GNE myopathy can be treated even at a progressive stage and 6’-sialyllactose has more remarkable advantage than free sialic acid, providing a conceptual proof for clinical use in patients.
Introduction
GNE myopathy, also called distal myopathy with rimmed vacuoles or hereditary inclusion body myopathy, is an adult-onset, moderately progressive autosomal recessive myopathy. The disease is characterized clinically by initial atrophy of the tibialis anterior and hamstring muscles and relative sparing of the quadriceps femoris muscle, and pathologically by rimmed vacuoles and intracellular accumulation of amyloid and other proteins (Nishino et al., 2002; Nonaka et al., 2005). GNE myopathy is due to mutations in the GNE gene that encodes a bifunctional enzyme, uridine diphospho-N-acetylglucosamine 2-epimerase and N-acetylmannosamine (ManNAc) kinase (Eisenberg et al., 2001; Nishino et al., 2002), which catalyses the critical two steps in sialic acid biosynthesis. Mutations in the GNE gene lead to significant reduction in either or both of the two enzymatic activities of the gene product (Hinderlich et al., 2004; Noguchi et al., 2004), and lead to reduced sialic acid levels in the serum, skeletal muscle, and cultured cells from patients with GNE myopathy (Noguchi et al., 2004). These findings were further supported by observations in the model mouse (Gne−/− with human GNE Asp176Val-transgenic mouse) that recapitulates the myopathic features in human patients and shows hyposialylation in the serum and other tissues from birth and manifested progressive muscle weakness and atrophy from 21 weeks of age, followed by intracytoplasmic inclusions (∼31 weeks) and rimmed vacuole formation (∼41 weeks) (Malicdan et al., 2007).
To date, patients with GNE myopathy rely on supportive care of symptoms and eventually become wheelchair-bound within ∼12 years after onset (Nishino et al., 2002; Nonaka et al., 2005). We have shown the prophylactic effect of sialic acid-related natural compounds, i.e. N-acetylneuraminic acid (NeuAc, free sialic acid), NeuAc’s glycosyl conjugate, 6’-sialyllactose, and NeuAc’s biosynthetic precursor, ManNAc, in GNE myopathy mice (Malicdan et al., 2009). This study provided basis for clinical trials for patients with GNE myopathy that are currently ongoing, using ManNAc and NeuAc (ClinicalTrials.gov, 2014) but had limitations. As this study was designed to prove prophylactic effect of sialic acid supplementation on the development of myopathy, it is necessary to examine whether myopathic symptoms can actually be recovered by sialic acid treatment once they appear. Given that physicians usually see affected individuals who have already developed muscle atrophy and degeneration, the proof that muscle size and strength are restored in patients at clinical stage is of practical importance. Taking on this endeavor is not without caveat, as preclinical trials using aged, symptomatic mice for a prolonged period of time raise several issues. First, we needed outcome measures to assess the motor performance even in affected mice manifesting severe muscle weakness. Overloaded motor exercise may not be ideal because it may worsen the morbidity or influence the mortality of affected mice. GNE myopathy mice show <50% survival rate and severely worsened motor activity at 50 weeks of age (Malicdan et al., 2007), thus a method that is not physically stressful and one that can be repeated should be considered. Second, there are difficulties with interpreting the results of in vivo experiments using a heterogeneous group of myopathic mice due to some variability in the onset of muscle degeneration, requiring baseline chronological measurement during intended age and period of treatment. In this study, we attempted to evaluate the effect of sialic acid supplementation on myopathic phenotype in the symptomatic GNE myopathy mouse model using an assessment system that might be widely adaptable to studies using other symptomatic mouse models.
Furthermore, the pharmacological properties and cellular incorporation efficiency of sialic acid compounds should be considered. We have recently reported that peracetylated ManNAc was markedly incorporated into human GNE myopathy cells (Malicdan et al., 2012). Peracetylated ManNAc, a synthetic compound, prevented the development of myopathic phenotype in the GNE myopathy mice in a dose-dependent manner but also raised cellular sialic acid levels in various tissues beyond normal levels, raising concerns about hypersialylation, even though it was rapidly excreted into urine like other sialic acid compounds. On the other hand, less metabolized compounds are supposed to circulate through the body for a longer time to give an opportunity to peripheral tissues to incorporate these compounds. In our previous preclinical studies, even with one-third of sialic acid amounts in 6’-sialyllactose in molar ratio, it provided a similar prophylactic effect as free NeuAc and ManNAc in preventing the development of myopathic phenotype in the GNE myopathy mice (Malicdan et al., 2009). When given orally, 6’-sialyllactose has been reported to have a longer retention in blood than NeuAc (Nöule and Schauer, 1981). In this study, we evaluated the effects of a sustained compound, 6’-sialyllactose, in the recovery of cellular sialylation and myopathic phenotype in symptomatic old GNE myopathy mice.
Materials and methods
The GNE myopathy mice were generated as reported previously (Malicdan et al., 2007). Mice were maintained in a barrier-free, specific pathogen-free grade facility on a 12-h light, 12-h dark cycle and had free access to normal chow and water. All animal experiments conducted in this study were approved by and carried out within the rules and regulations of the Ethical Review Committee on the Care and Use of Rodents in the National Institute of Neuroscience, National Centre of Neurology and Psychiatry (NCNP). These policies are based on the ‘Guideline for Animal Experimentation’ as sanctioned by the Council of the Japanese Association of Laboratory Animal Science.
NeuAc was purchased from Japan Food and Liquor Alliance, and ManNAc was purchased from Sigma-Aldrich. 6’-sialyllactose was produced with genetically engineered Escherichia coli expressing α2,6-sialyltransferase gene from Photobacterium sp. JT-ISH-224 and CMP-NeuAc synthase gene from Neisseria meningitides and purified according to the methods reported by Drouillard et al. (2010). The purity and structure was confirmed by HPLC (Endo et al., 2009) and 1H-NMR spectrum, respectively.
Cultured myoblasts from a patient with GNE myopathy carrying homozygous Asp176Val mutations in the GNE gene were obtained with informed consent approved by the Ethical Review Board at the NCNP. Myoblasts were cultured in 10% foetal bovine serum, Dulbecco’s modified Eagle medium (DMEM)/F-12 (Sigma-Aldrich) in a humidified chamber with 5% CO2 at 37°C. Myogenic differentiation was induced at confluence stage by switching the medium to 5% horse serum in DMEM/F-12. Seventy-two hours before sialic acid determination, the medium was replaced with serum-free DMEM/F-12 with or without NeuAc, ManNAc and 6’-sialyllactose and maintained in the humidified chamber for 72 h. Sialic acids were hydrolyzed in 25 mM sulphuric acid for 1 h at 80°C. Released sialic acids were then derivatized with 1,2-diamino-4,5-methylenedioxybenzene and analysed with reversed-phase HPLC as described previously (Hara et al., 1989; Malicdan et al., 2007). Total proteins in tissues and cultured cells were measured using Pierce BCA kit according to the manufacturer’s protocol.
For the experiment of 6’-sialyllactose pharmacokinetics, three wild-type mice were used. After collection of blood from the tail vein and urine for baseline data before administration, 30 mg of 6’-sialyllactose was given via an intragastric route. The urine and blood were then serially collected after 5, 10, 30, 60, 120, 240 and 480 min. At the end of the experiment, the mice were sacrificed. The urine and prepared plasma were frozen and kept at −20°C until processing. To quantify 6’-sialyllactose, samples were labelled with 4-aminobenzoic acid ethyl ester according to the manufacturer’s protocol (ABEE Labelling Kit, J-Oilmills). Fluorescence labelled mono- and disaccharides, and 6’-sialyllactose were chromatographed on an amide-80 in 80% acetonitrile in 0.5 M acetic acid/triethylamine (pH 7.3) at a flow rate of 1.0 ml/min for analysis. Total sialic acids from those prepared plasmas were also measured.
For oral 6’-sialyllactose or NeuAc treatment, GNE myopathy mice, including corresponding littermates, whose ages were ∼50 weeks, were included in the cohort. The GNE myopathy mice were divided into four groups: non-treated (n = 18) given acid water, low dose (n = 10) given 6’-sialyllactose at 100 mg/kg/d, high dose (n = 8) given 6’-sialyllactose at 1000 mg/kg/d, and NeuAc (n = 11) given at 1000 mg/kg/d. 6’-sialyllactose and NeuAc, computed according to the desired dose per day, were mixed with the drinking water and given continuously until the mice reached 80 weeks of age. A few control littermates (Gne+/− or Gne+/− hGNE Asp176Val-Tg) per group were also treated for toxicological examination.
Voluntary exercise within an individual mouse cage was repeatedly measured. A running wheel (SW-15, MELQUEST) was fixed up into a rearing cage to measure spontaneous locomotor activity. The output signal of 1 pulse per revolution by an on/off signal of microswitch was saved to a PC with software for data collection (CIF3Win, Neuroscience). Voluntary exercise was quantified as the total number of wheel revolutions in 72 h. The data were serially collected for mice at 50, 65, 72 and 80 weeks of age. Because of a marked variation among GNE myopathy mice at 50 weeks of age, we used the relative values at 65, 72 and 80 weeks to the initial measurement at 50 weeks for each mouse.
In a preliminary NeuAc treatment study, the motor performance was evaluated using treadmill exercise as previously reported (Malicdan et al., 2008). After 7 days of acclimation on the treadmill, the performance tests were carried out three times, on separate days. The test began with a speed of 20 m/min, which was increased by 10 m/min every minute until the tested mouse was exhausted and could no longer run. The time of exhaustion was used to calculate the distance that the mouse covered during the exercise.
Measurement of the contractile properties of gastrocnemius and tibialis anterior muscles was performed according to previous report (Malicdan et al., 2008). All materials used for in vitro measurement of force were acquired from Nihon Kohden.
Muscle tissues processing and haematoxylin and eosin and modified Gomori trichrome staining for pathological analysis were performed as reported previously (Malicdan et al., 2007). The stained sections were observed on a microscope (Olympus BX51, Olympus) and the images were acquired by DP70 digital CCD (Olympus). The number of rimmed vacuoles was counted on six 10-μm thick serial transverse cryosections with at least 100-μm interval of whole gastrocnemius muscle for each group of mice. For morphometric analyses, we stained the sarcolemma of muscle cryosections with anti-caveolin-3 antibody (Santacruz, 1:200 dilution) for 1 h followed by Alexa Fluor®-conjugated donkey IgG against goat IgG (Invitrogen, 1:800 dilution) for 30 min and imaged six randomly selected fields with a laser-scanning microscope (Olympus). Diameters of myofibres were measured from 600–800 fibres with ImageJ software (NIH). For quantifying the amount of amyloid deposits in myofibres, 10-μm thick cryosections of the gastrocnemius muscle were fixed in 4% paraformaldehyde for 10 min followed by immersion in ice-cold methanol for 10 min at −20°C and stained using a mouse antibody to mouse amyloid-β1–16 (6E10, Covance; 1:400 dilution) with an anti-caveolin-3 antibody. Digitized images at ×200 magnification were captured and used for analysis. Seven-μm thick cryosections from the gastrocnemius muscle were also stained using a rat antibody to mouse laminin-α2 (Alexis, 1:100) and either a mouse antibody to amyloid-β1–40 (Millipore, 1:300 dilution), a mouse antibody to amyloid-β1–42 (Millipore, 1:300 dilution) or a rabbit antibody to LC3 (Cell Signaling, 1:100 dilution). We applied appropriate secondary antibodies labelled with Alexa Fluor® dye according to the previous protocol (Malicdan et al., 2007).
Samples for ELISA to measure amyloid-β1–40 and amyloid-β1–42 amounts in the gastrocnemius muscles were prepared according to the manufacturer’s protocol with slight modifications. Muscle homogenates were extracted using equal volumes of 0.4% diethylamine and 100 mM NaCl. After centrifugation at 20 000g for 1 h at 4°C, the supernatant was neutralized with 0.5 M Tris base, pH 6.8. Amyloid-β1–40 and amyloid-β1–42 amounts were measured with ELISA kits for amyloid-β1–40 (IBL) and amyloid-β1–42 (IBL). The data were normalized with protein concentration.
After measurement of force, the mice were euthanized and the organs were harvested and immediately frozen on dry ice and kept at −80°C until use. The tissues were homogenized using a Potter homogenizer in a buffer containing 75 mM KCl, 10 mM Tris, 2 mM MgCl2, 2 mM EGTA, and protease inhibitor mixture (Complete Mini Protease Inhibitor Tablet, Roche Applied Science), pH 7.4. The homogenized tissues were centrifuged for 1 h at 30 000g at 4°C. The pellet, which contained the membrane fractions, was used for measurement of the sialic acid and protein. After washing with the same buffer twice, the pellet was resuspended in 50 mM H2SO4, hydrolyzed and subjected to sialic acid measurement. We used the other pelleted fraction for analysis of the total protein amount by extracting protein with SDS buffer (2% SDS, 10% glycerol, 10 mM EDTA, 5% 2-mercaptoethanol, 0.0625 M Tris-HCl, pH 6.8).
We determined the activity of plasma creatine kinase, aspartate aminotransferase and alkaline phosphatase according to the manufacturer’s protocol (Cicaliquid CK, AST and ALP, Kanto Chemical). We measured the blood urea nitrogen (BUN) level using a rate assay with urease and leucine dehydrogenase (Urutrateliquid BUN, Toyobo).
All values were expressed as means ± standard error (SE), or means ± standard deviation (SD) for plasma creatine kinase, aspartate aminotransferase, alkaline phosphatase and BUN values, as appropriate. For survival analysis, we used the Kaplan-Meier methods to draw the survival curve. For analysis of spontaneous locomotion activity, we used Wilcoxon’s signed-rank test to compare the data recorded at 65, 72 and 80 weeks of age, respectively with that at 50 weeks in each mouse. For other analyses, we used one-way ANOVA with Dunnett’s post-test or Kruskal-Wallis analysis with Dunn’s post-test to compare the treatment group with non-treated group. A P-value of 0.05 was considered as the threshold for significance.
Results
We analysed the incorporation of sialic acid compounds into differentiated myotubes from a patient with GNE myopathy with increasing concentrations of NeuAc, ManNAc or 6’-sialyllactose. We found that the cellular sialic acid levels were similarly recovered by addition of any of those compounds to medium (Fig. 1A). To analyse the pharmacokinetics of 6’-sialyllactose, we collected the blood and urine samples serially before and 5, 10, 30, 60, 120, 240 and 480 min after giving a single dose to mice via an intragastric route. After intragastric administration, 6’-sialyllactose appeared in blood within 5 min, peaked at 10 min, and returned to the baseline within 240 min (Fig. 1B). Large amounts of 6’-sialyllactose were excreted into urine at 60–120 min after administration, but it took 480 min for the 6’-sialyllactose to return to the baseline levelin urine, showing slower excretion into urine (Fig. 1C) than either NeuAc or ManNAc (Malicdan et al., 2009). We also measured free NeuAc, which is the metabolized form of 6’-sialyllactose, and detected bipolar peaks in the blood at 5 and 60 min (Fig. 1D), while the peak was maximal in the urine at 120–240 min (Fig. 1E).
Figure 1.
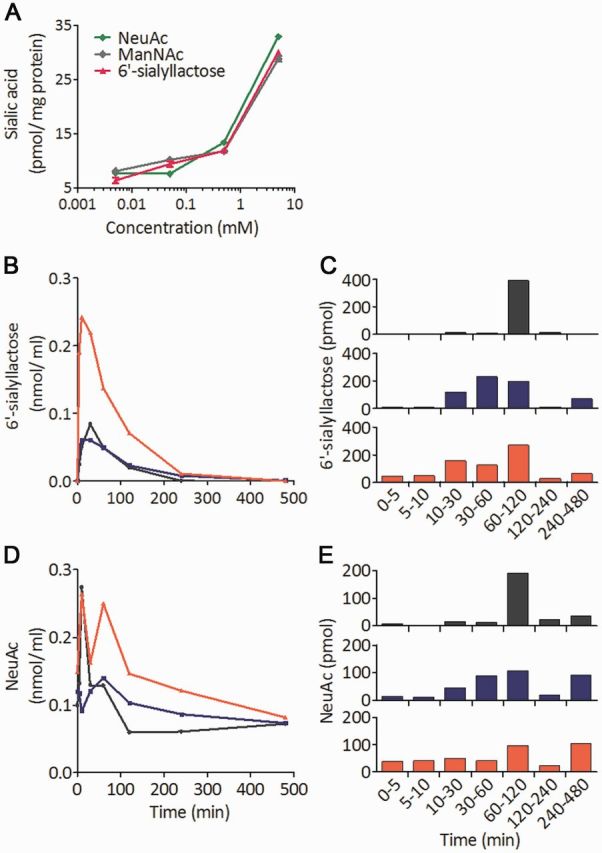
Comparison among compounds to increase cell sialylation and pharmacokinetics of 6’-sialyllactose. (A) Total sialic acid levels in myotubes from a patient with GNE myopathy treated with ManNAc, NeuAc and 6’-sialyllactose. Three compounds were added to condition medium at the concentration shown on the x-axis. (B and C) Phamacokinetics of orally administrated 6’-sialyllactose concentration in blood (B) and its excreted amounts in urine (C) before and at 5, 10, 30, 60, 120, 240 and 480 min after its oral administration. (D and E) Profiles of NeuAc concentration in blood (D) and its excreted amounts in urine (E). For the experiment of 6’-sialyllactose pharmacokinetics, three wild-type mice were used. Each colour (grey, blue, and red) denotes one mouse in B–F.
We conducted a preliminary NeuAc treatment study for symptomatic GNE myopathy mice and performed repetitive treadmill tests to assess their motor performance (Supplementary Fig. 1A). NeuAc treatment prevented the progression of symptoms on the loss of body weight and running performance of symptomatic GNE myopathy mice as compared to non-treated mice (Supplementary Fig. 1B and C), but this repetitive treadmill test led to physical exhaustion even in control littermates and increased mortality in the non-treated group (Supplementary Fig. 1A and C). Thus, in the next study, we measured voluntary running as a spontaneous locomotion activity to assess the motor performance to avoid causing further injury in symptomatic mice.
We analysed the progression of disease in GNE myopathy mice from 50 to 80 weeks of age. GNE myopathy mice were smaller than their littermates at 50 weeks of age, ranging from 28.6 to 32.5 g (Fig. 2A), and did not show body weight gain up to 80 weeks of age (Fig. 2B and Supplementary Fig. 2). Three (16.7%) of 18 GNE myopathy mice died by 80 weeks of age (Fig. 2C). Although marked variation in spontaneous locomotion activity was observed even in affected mice having the same genotype, control littermates maintained locomotion activity from 50–80 weeks of age (Fig. 2D). In contrast, GNE myopathy mice showed a decrease in activity with ageing, especially ∼70–80 weeks (Fig. 2D and Supplementary Fig. 3B). At 80 weeks of age, the weight and cross-sectional area (CSA) of gastrocnemius and tibialis anterior muscles were smaller, and absolute isometric (Pt), tetanic (Po) forces, and the size-normalized isometric force (Pt/CSA), tetanic force (Po/CSA) of those muscles were markedly lower in GNE myopathy mice as compared to those of control littermates (Fig. 3A–F and Supplementary Fig. 5A–F), suggesting that the muscles were highly degenerated. Gastrocnemius muscles of 80-week-old GNE myopathy mice showed an increased number of atrophic fibres (Fig. 3G), accompanied by increased amyloid-β1–40 and amyloid-β1–42 deposits and LC3-positive rimmed vacuoles (Fig. 4A, D and E, and Supplementary Fig. 4) compared to previously characterized GNE myopathy mice at 55 weeks of age (Malicdan et al., 2007, 2009). It should be noted that some of the GNE myopathy mice showed extremely numerous rimmed vacuoles (>300) in six muscle transverse sections at this age (Fig. 4B) (Malicdan et al., 2009). The creatine kinase level was only mildly elevated; 80.5 ± 65.5 (mean ± SD, n = 15) versus 50.5 ± 11.7 IU/l (n = 5) (Supplementary Table 1).
Figure 2.

Phenotypes of treated and non-treated GNE myopathy mice. (A) Mean body weights of all GNE myopathy (GM) and littermate (LM) mice at 50 weeks of age. (B) Growth curves. The body weights at 55, 60, 65, 70, and 75 weeks of age are plotted relative to each at 50 weeks of age: GNE myopathy mice with high dose 6’-sialyllactose (HD, red upward closed triangles, n = 8); GNE myopathy mice with low dose 6’-sialyllactose (LD, blue downward closed triangles, n = 10); GNE myopathy mice with NeuAc (green closed diamonds, n = 10); non-treated GNE myopathy mice, non-treated (yellow open circles, n = 18); and control littermates (black closed circles, n = 5). (C) Survival curves: high dose (red), low dose (blue), NeuAc (green), non-treated (yellow) and littermates (black). (D) Spontaneous locomotion activities. The locomotion activities at 65, 72 and 80 weeks of age are plotted relative to those at 50 weeks of age: high dose (dark grey bars, n = 5), low dose (grey bars, n = 7), NeuAc (light grey bars, n = 8), non-treated (white bars, n = 14) and littermates (black bars, n = 14). *P < 0.05, **P < 0.01.
Figure 3.
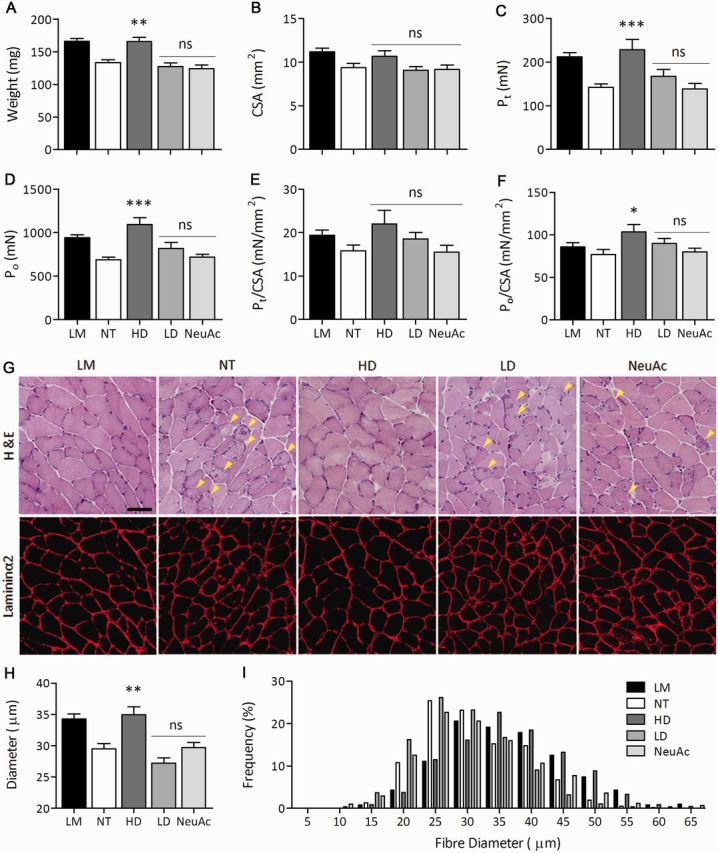
Muscle size and contractile properties of gastrocnemius muscle in GNE myopathy mice after 6’-sialyllactose treatment. (A) Muscle weight. (B) Whole-muscle CSA. (C–F) Contractile properties of the gastrocnemius: (C) isometric force (Pt), (D) tetanic force (Po), (E) specific isometric force (Pt/CSA), and (F) specific tetanic force (Po/CSA). (G) Haematoxylin and eosin staining and immunohistochemistry for laminin-α2 in transverse sections. Arrowheads denote rimmed vacuoles. Scale bar = 50 μm. (H) Mean diameters of myofibres after treatment. Control littermates (LM, black bars, n = 14); non-treated GNE myopathy mice (NT, white bars, n = 15); GNE myopathy mice with high dose 6’-sialyllactose (HD, dark grey bars, n = 5); GNE myopathy mice with low dose 6’-sialyllactose (LD, grey bars, n = 7); GNE myopathy mice with NeuAc (light grey bars, n = 9). (I) Histogram of myofibre diameters. High dose (HD, dark grey bars), low dose (grey bars), NeuAc (light grey bars), non-treated (white bars) and littermates (black bars). The histogram in high dose is shifted to the right, indicating the fibre size is increased after treatment. *P < 0.05, **P < 0.01, ***P < 0.001, ns = not significant.
Figure 4.
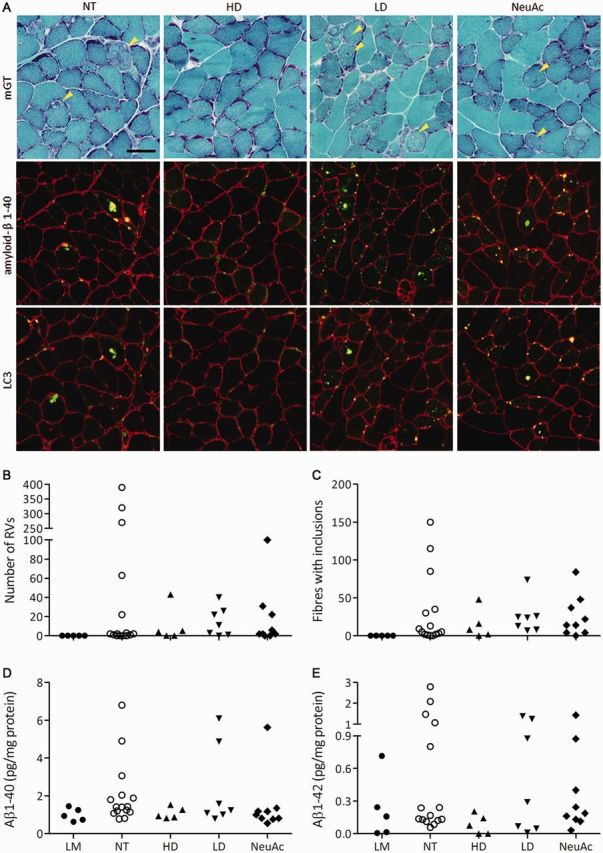
Number of rimmed vacuoles (RVs) and amyloid deposits after 6’-sialyllactose treatment. (A) Modified Gomori trichrome staining of gastrocnemius transverse sections. Arrowheads denote rimmed vacuoles. Immunosignals of amyloid-β1-40 and LC3 are localized within rimmed vacuoles. Scale bar = 50 μm. (B) Number of rimmed vacuoles. (C) Amyloid inclusions. (D and E) Measurement of amyloid-β1-40 (D) and amyloid-β1-42 (E) in muscle by ELISA. Control littermates (LM, closed circles, n = 5); non-treated GNE myopathy mice, (NT, open circles, n = 15); GNE myopathy mice with high dose 6’-sialyllactose (HD, upward closed triangles, n = 5); GNE myopathy mice with low dose 6’-sialyllactose (LD, downward closed triangles, n = 7); GNE myopathy mice with NeuAc (closed diamonds, n = 9).
For sialic acid supplementation in symptomatic mice, GNE myopathy mice at 50 weeks of age were divided into four groups: non-treated (n = 18), given water; low dose of 6’-sialyllactose at 100 mg/kg/d (n = 10); high dose of 6’-sialyllactose at 1000 mg/kg/d (n = 8); and NeuAc at 1000 mg/kg/d (NeuAc, n = 11). The treatment effect was measured by comparing the data from treated and non-treated mice with control littermates (n = 15).
In the high dose group, body weight was increased up to a level almost similar to that of the littermates during treatment (Fig. 2B). Such a body weight gain was not observed either in the low dose or NeuAc group (Fig. 2B). Mortality rate was almost equal among all the treated groups, as in this study, two to three mice in each of three treated groups died before the end of the study (Fig. 2C). Chronological examination of spontaneous locomotion activity revealed that the activity was well maintained in the high dose group (Fig. 2D), of which one mouse showed a marked increase (Supplementary Fig. 3C). A slight increase in activity was seen in the NeuAc group at 65 weeks of age (Fig. 2D), but by 80 weeks both low dose and NeuAc groups showed a significant decrease in locomotion.
Gastrocnemius muscle size in the high dose group at 80 weeks of age was increased up to the level of control littermates, while there was no change in the low dose and NeuAc groups (Fig. 3A and B). Likewise, tibialis anterior muscle size was increased to the level of littermates in the high dose group but not both in the low dose and NeuAc groups (Supplementary Fig. 5A and B). In terms of contractile properties, 6’-sialyllactose showed increases in twitch Pt and tetanic Po forces of gastrocnemius muscle dose-dependently, and especially the high dose group presented increases in both muscle power beyond the levels of control littermates (Fig. 3C and D). Twitch force (Pt/CSA) and tetanic force (Po/CSA) were also improved with 6’-sialyllactose in a dose-dependent manner, with a notable complete normalization in the high dose group (Fig. 3E and F). The NeuAc group showed mild improvement of Po force and Po/CSA (Fig. 3D and 4F), whereas Pt/CSA remained similar to the level of non-treated group (Fig. 3E). In the analyses of tibialis anterior muscles, we found a near-complete recovery of all contractile parameters in the high dose group (Supplementary Fig. 5). In the low dose group, twitch Pt and tetanic Po forces were increased similar to the levels of control littermates in the absence of increase in muscle size, indicating a marked improvement of Pt/CSA and Po/CSA. We did not observe any favourable results in the NeuAc group (Supplementary Fig. 5).
On muscle pathology, the number of rimmed vacuole-containing fibres was decreased in all treated groups, especially in the high dose group (Fig. 3G). The high dose group also showed a smaller number of atrophic fibres than those in other treated groups (Fig. 3G). Figure 3I shows the representative data of histogram of myofibre diameters in gastrocnemius muscle of one mouse from each group. The average and distribution of myofibre size in gastrocnemius muscle in the high dose group were completely restored similarly to control littermates, but not in the low dose group (Fig. 3H and Supplementary Fig. 6). The NeuAc group showed no increase in fibre size (Fig. 3H) and the histogram was similar to that of the non-treated group (Supplementary Fig. 6).
By all treatment modalities, almost all of the GNE myopathy mice had few or only a small number of rimmed vacuoles, except for one mouse in the NeuAc group (Fig. 4B). Furthermore, amyloid-β1–40 and amyloid-β1–42 amounts, in addition to the number of amyloid-β inclusions, were also almost totally diminished by all three treatments (Fig. 4C–E). Overall, 6’-sialyllactose dose-dependently restored muscle degeneration in symptomatic GNE myopathy mice, while free NeuAc ameliorated muscle degeneration to some degree.
The sialic acid levels in blood were elevated by all treatments, high and low doses of 6’-sialyllactose, and NeuAc (Fig. 5A). Of note, sialic acid levels in blood did not reach the level of littermates either in the high dose or NeuAc group, but we found one mouse in the low dose group that had a similar level of blood sialic acid to littermates for unknown reason. Similarly, all three treatments increased sialic acid amounts in various tissues (Fig. 5B, C and D). In the high dose group, sialic acid levels in skeletal muscle and kidney were significantly and most efficiently increased among all three treatment groups (Fig. 5B and C). An increased level of sialic acid in skeletal muscle was also found in the NeuAc group (Fig. 5B).
Figure 5.

Total sialic acid levels in blood and tissues. (A) Total sialic acid concentration in blood. (B–D) Sialic acid levels in membranous fraction prepared from skeletal muscle (B), kidney (C), and liver (D). Control littermates (LM, black bars, n = 14); non-treated GNE myopathy mice (NT, white bars, n = 15); GNE myopathy mice with high dose 6’-sialyllactose (HD, dark grey bars, n = 5); GNE myopathy mice with low dose 6’-sialyllactose (LD, grey bars, n = 7); GNE myopathy mice with NeuAc (light grey bars, n = 9). *P < 0.05, **P < 0.01, ns = not significant.
In this study, we did not detect any improvement of the creatine kinase level in all treated groups: high dose group, 79.0 ± 55.8 (n = 5); low dose group, 80.9 ± 53.5 (n = 7); NeuAc group, 85.1 ± 48.8 IU/l (n = 9). We also confirmed no hepatic and renal toxicity by measurements of plasma aspartate aminotransferase, alkaline phosphatase, and blood urea nitrogen (Supplementary Table 1).
Discussion
We have reported the prophylactic effect of NeuAc, ManNAc, and 6’-sialyllactose against the development of GNE myopathy, but there were some limitations in the study (Malicdan et al., 2009). First, the study design was prophylactic, limiting interpretation for clinical application. Second, we did not detect apparent dose-effect as the expected GNE myopathy phenotype was completely precluded, even at low doses. Third, to completely evaluate the choice of the compounds, there was a need to use severely affected animals. We address these issues in the present study. We explored a therapeutic strategy in symptomatic GNE myopathy mice, keeping in consideration the pharmacological properties of sialic acid in aiming to efficiently raise sialic acid levels in the blood and various tissues. We should take into account two major properties of potential therapeutic compounds, including effective incorporation into cells and pharmacokinetic properties. In this study, we tested 6’-sialyllactose, a compound that is slowly metabolized, allowing longer retention time in the circulation, and thereby effectively increasing sialic acid in the muscle.
In cultured human GNE muscle cells, a dose-dependent increase in sialic acid levels was seen by the addition of 6’-sialyllactose, similar to previous reports with NeuAc and ManNAc (Malicdan et al., 2012). These results indicate that the incorporation rate or dosage of 6’-sialyllactose were not different from those of free NeuAc. It has been suggested that there is a difference in metabolism between 6’-sialyllactose and NeuAc or ManNAc. In the present pharmacokinetic results of oral 6’-sialyllactose, 6’-sialyllactose in blood was returned to the baseline at 240 min after its administration as compared to 120 min when NeuAc was administered (Fig. 1B) (Malicdan et al., 2009). Similarly, 48–91% of 6’-sialyllactose was excreted into urine after 60 to 480 min (Fig. 1C), whereas 75% of NeuAc was excreted within 60 min (Malicdan et al., 2009), showing that 6’-sialyllactose is less metabolized as compared to NeuAc. Previous reports also support these results that it took 24 h to excrete almost all amounts of sialyllactose and 6 h for 60–90% of NeuAc into urine when they were orally administered (Nöule and Schauer, 1981). Furthermore, the peak of free NeuAc in blood at 60 min after administration of 6’-sialyllactose (Fig. 1D) suggests that while circulating through the body, 6’-sialyllactose might be slowly metabolized into NeuAc and providing NeuAc source in the circulation within 50 min of delay from the time 6’-sialyllactose is given. In other words, giving 6’-sialyllactose can extend the metabolizing time of sialic acid (compounds). These results support our pharmacological hypothesis that slowly metabolized compounds can circulate and stay in the body longer, allowing the tissues more time to incorporate the circulating sialic acid.
In this study, using a less intensive method to assess the motor performance in symptomatic mice, we showed that motor performance continues to deteriorate after 50 weeks of age, presumably because of severe muscle degeneration. By applying this method to all treatment groups, we found that a high dose of 6’-sialyllactose remarkably prevented further deterioration of motor performance. Our findings support the use of this less invasive and reliable tool in the evaluation of other models of myopathy.
As previously reported, GNE myopathy mice manifest muscle weakness and atrophy from 21 weeks of age, and show decreased specific twitch force after intracellular inclusions appear and parallel reduction in specific twitch and tetanic forces after rimmed vacuoles appear (Malicdan et al., 2007, 2008). Indeed, 80-week-old GNE myopathy mice showed physiological findings congruent with progressive muscle degeneration. In terms of muscle contractile parameters, gastrocnemius muscles in the high dose group showed almost complete normalization in size and in both tetanic and twitch forces, suggesting that the properties of contractile machinery as well as contraction regulating systems have been repaired. Thus, these results imply that atrophy and weakness in older GNE myopathy mice are reversible by maintaining sufficient sialic acid levels in the muscle. We propose that muscle atrophy and weakness in patients with GNE myopathy can be rescued by sialic acid supplementation.
The low dose treatment did not restore muscle atrophy but ameliorated the contractile properties of gastrocnemius and tibialis anterior muscles and in the NeuAc group, sialic acid levels in skeletal muscle were increased to the level in the low dose group, but only minimal beneficial effect were seen on contractile properties and there was no effect on muscle size (Fig. 3E, F and H, and Supplementary Fig. 4). These findings suggest that there is a positive correlation between recovery of hyposialylation and improvement of muscle atrophy and weakness, and that the restoration of muscle size in GNE myopathy requires a high and steady amount of sialic acid incorporated into muscle cells.
The presence of intracellular protein deposits and rimmed vacuoles in the central portion of myofibres presumably interferes with the muscle force generation in rimmed vacuolar and autophagic vacuolar myopathies (Malicdan and Nishino, 2012; Raben et al., 2012). Rimmed vacuoles are likely a secondary event to protein misfolding or aggregation in GNE myopathy (Malicdan et al., 2008). However, intracellular inclusions and rimmed vacuoles almost disappeared in the affected mice after all three treatments (Fig. 4B and C), suggesting again that hyposialylation in muscles is associated with formation of such intracellular inclusions.
From the biochemical point of view, we highlight our finding that the high dose group had higher sialic acid levels in skeletal muscle than the NeuAc group (Fig. 5B), leading to near-complete recovery of muscle function. Given that large amounts of sialic acid may be needed to rescue muscle atrophy, in addition to contractile property, there are several points that we must consider to achieve an efficient increase in sialic acid level in peripheral tissues, including the pharmacological advantages of the less metabolized compounds such as 6’-sialyllactose; high cellular uptake of compounds such as peracetylated ManNAc (Malicdan et al., 2012); and the use of sustained-released preparations of free sialic acid. In addition, the finding that 6’-sialyllactose and peracetylated ManNAc remarkably increased sialic acid level in kidney (Malicdan et al., 2012) also suggest that the other diseases associated with GNE gene mutation, such as severe renal disorders in Gne mutants (Galeano et al., 2007; Ito et al., 2012) and other transgenic models (Clement et al., 2011), would be treatable strategies.
In this study, survival rate did not differ among all GNE myopathy groups during treatment (Fig. 4C). We also attempted to characterize respiratory and cardiac function in older mice by using whole body plethysmography and ECG, however, we did not detect physiological deterioration in the non-treated group compared to the littermates (data not shown). As previously demonstrated, GNE myopathy mice started to die from 3 weeks of age and showed a gradual increment of mortality from 25–55 weeks, but thereafter GNE myopathy mice showed a reduced mortality (Malicdan et al., 2007). The surviving GNE myopathy mice older than 55 weeks of age might be resistant to the death related to vital organ functions, such as lung and heart, and that the survival rate may not be informative in evaluating efficacy of treatment in the experiments using symptomatic myopathic older mice; specific methods should be chosen on their phenotypes. In this study, spontaneous locomotion activity was applicable to older GNE myopathy mice when repeatedly performed, and indeed we demonstrated that the high dose group showed reversed motor function in the same GNE myopathy mice.
Our results emphasize that GNE myopathy is a treatable disease, requiring enhanced sialylation for recovery of skeletal muscle function. This study provided a proof of concept in the use of slowly metabolized sialic acid in the clinical trial of patients with GNE myopathy.
Supplementary Material
Acknowledgements
The authors acknowledge Fumiko Funato and Nozomi Matsuyama for their technical assistance.
Glossary
Abbreviations
- CSA
cross-sectional area
- ManNAc
N-acetylmannosamine
- NeuAc
N-acetylneuraminic acid
Funding
This study was partially supported by Intramural Research Grant (25-5) for Neurological and Psychiatric Disorders of NCNP, by Comprehensive Research on Disability Health and Welfare from the Ministry of Health, Labour and Welfare, and by JSPS KAKENHI (23390236).
Supplementary material
Supplementary material is available at Brain online.
References
- Clement LC, Avila-Casado C, Macé C, Soria E, Bakker WW, Karsten S, et al. Podocyte-secreted angiopoietin-like-4 mediates proteinuria in glucocorticoid-sensitive nephrotic syndrome. Nat Med. 2011;17:117–23. doi: 10.1038/nm.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ClinicalTrials.gov [Internet] A service of the U.S. National Institute of Health. Available from: http://www.clinicaltrials.gov./ (18 April 2014, date last accessed) [Google Scholar]
- Drouillard S, Mine T, Kajiwara H, Yamamoto T, Samain E. Efficient synthesis of 6’-sialyllactose, 6,6’-disialyllactose, and 6’-KDO-lactose by metabolically engineered E. coli expressing a multifunctional sialyltransferase from the Photobacterium sp. JT-ISH-224. Carbohydr Res. 2010;345:1394–9. doi: 10.1016/j.carres.2010.02.018. [DOI] [PubMed] [Google Scholar]
- Eisenberg I, Avidan N, Potikha T, Hochner H, Chen M, Olender T, et al. The UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion body myopathy. Nat Genet. 2001;29:83–7. doi: 10.1038/ng718. [DOI] [PubMed] [Google Scholar]
- Endo S, Morita M, Ueno M, Maeda T, Terabayashi T. Fluorescent labeling of a carboxyl group of sialic acid for MALDI-MS analysis of sialyloligosaccharides and ganglioside. Biochem Biophys Res Commun. 2009;378:890–4. doi: 10.1016/j.bbrc.2008.12.011. [DOI] [PubMed] [Google Scholar]
- Galeano B, Klootwijk R, Manoli I, Sun M, Ciccone C, Darvish D, et al. Mutation in the key enzyme of sialic acid biosynthesis causes severe glomerular proteinuria and is rescued by N-acetylmannosamine. J Clin Invest. 2007;117:1585–94. doi: 10.1172/JCI30954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara S, Yamaguchi M, Takemori Y, Furuhata K, Ogura H, Nakamura M. Determination of mono-O-acetylated N-acetylneuraminic acids in human and rat sera by fluorometric high-performance liquid chromatography. Anal Biochem. 1989;179:162–6. doi: 10.1016/0003-2697(89)90218-2. [DOI] [PubMed] [Google Scholar]
- Hinderlich S, Salama I, Eisenberg I, Potikha T, Mantey LR, Yarema KJ, et al. The homozygous M712T mutation of UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase results in reduced enzyme activities but not in altered overall cellular sialylation in hereditary inclusion body myopathy. FEBS Lett. 2004;566:105–9. doi: 10.1016/j.febslet.2004.04.013. [DOI] [PubMed] [Google Scholar]
- Ito M, Sugihara K, Asaka T, Toyama T, Yoshihara T, Furuichi K, et al. Glycoprotein hyposialylation gives rise to a nephrotic-like syndrome that is prevented by sialic acid administration in GNE V572L point-mutant mice. PLoS One. 2012;7:e29873. doi: 10.1371/journal.pone.0029873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malicdan MC, Nishino I. Autophagy in lysosomal myopathies. Brain Pathol. 2012;22:82–8. doi: 10.1111/j.1750-3639.2011.00543.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malicdan MC, Noguchi S, Hayashi KY, Nishino I. Muscle weakness correlates with muscle atrophy and precedes the development of inclusion body or rimmed vacuoles in the mouse model of DMRV/hIBM. Physiol Gnenomics. 2008;35:106–15. doi: 10.1152/physiolgenomics.90219.2008. [DOI] [PubMed] [Google Scholar]
- Malicdan MC, Noguchi S, Nonaka I, Hayashi KY, Nishino I. A Gne knockout mouse expressing human GNE D176V mutation develops features similar to distal myopathy with rimmed vacuoles or hereditary inclusion body myopathy. Hum Mol Genet. 2007;16:2669–82. doi: 10.1093/hmg/ddm220. [DOI] [PubMed] [Google Scholar]
- Malicdan MC, Noguchi S, Hayashi KY, Nonaka I, Nishino I. Prophylactic treatment with sialic acid metabolites precludes the development of the myopathic phenotype in the DMRV-hIBM mouse model. Nat Med. 2009;15:690–5. doi: 10.1038/nm.1956. [DOI] [PubMed] [Google Scholar]
- Malicdan MC, Noguchi S, Tokutomi T, Goto Y, Nonaka I, Hayashi KY, et al. Peracetylated N-acetylmannosamine, a synthetic sugar molecule, efficiently rescue muscle phenotype and biochemical defects in mouse model of sialic acid-deficient myopathy. J Biol Chem. 2012;287:2689–705. doi: 10.1074/jbc.M111.297051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino I, Noguchi S, Murayama K, Driss A, Sugie K, Oya Y, et al. Distal myopathy with rimmed vacuoles is allelic to hereditary inclusion body myopathy. Neurology. 2002;59:1689–93. doi: 10.1212/01.wnl.0000041631.28557.c6. [DOI] [PubMed] [Google Scholar]
- Noguchi S, Keira Y, Murayama K, Ogawa M, Fujita M, Kawahara G, et al. Reduction of UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase activity and sialylation in distal myopathy with rimmed vacuoles. J Biol Chem. 2004;279:11402–7. doi: 10.1074/jbc.M313171200. [DOI] [PubMed] [Google Scholar]
- Nonaka I, Noguchi S, Nishino I. Distal myopathy with rimmed vacuoles and hereditary inclusion body myopathy. Curr Neurol Neurosci Rep. 2005;5:61–5. doi: 10.1007/s11910-005-0025-0. [DOI] [PubMed] [Google Scholar]
- Nöule U, Schauer R. Uptake, metabolism and excretion of orally and intravenously administered, 14C-and 3H-labeled N-acetylneuraminic acid mixture in the mouse and rat. Hoppe Seylers Z Physiol Chem. 1981;362:1495–506. doi: 10.1515/bchm2.1981.362.2.1495. [DOI] [PubMed] [Google Scholar]
- Raben N, Wong A, Ralston E, Myerowitz R. Autophagy and mitochondria in Pompe disease: nothing is so new as what has long been forgotten. Am J Med Genet C Semin Med Genet. 2012;160C:13–21. doi: 10.1002/ajmg.c.31317. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.