

ABSTRACT
Viruses require the host translational apparatus to synthesize viral proteins. Host stress response mechanisms that suppress translation, therefore, represent a significant obstacle that viruses must overcome. Here, we report a strategy whereby the mammalian orthoreoviruses compartmentalize the translational machinery within virus-induced inclusions known as viral factories (VF). VF are the sites of reovirus replication and assembly but were thought not to contain ribosomes. It was assumed viral mRNAs exited the VF to undergo translation by the cellular machinery, and proteins reentered the factory to participate in assembly. Here, we used ribopuromycylation to visualize active translation in infected cells. These studies revealed that active translation occurs within VF and that ribosomal subunits and proteins required for translation initiation, elongation, termination, and recycling localize to the factory. Interestingly, we observed components of the 43S preinitiation complex (PIC) concentrating primarily at factory margins, suggesting a spatial and/or dynamic organization of translation within the VF. Similarly, the viral single-stranded RNA binding protein σNS localized to the factory margins and had a tubulovesicular staining pattern that extended a short distance from the margins of the factories and colocalized with endoplasmic reticulum (ER) markers. Consistent with these colocalization studies, σNS was found to associate with both eukaryotic translation initiation factor 3 subunit A (eIF3A) and the ribosomal subunit pS6R. Together, these findings indicate that σNS functions to recruit 43S PIC machinery to the primary site of viral translation within the viral factory. Pathogen-mediated compartmentalization of the translational apparatus provides a novel mechanism by which viruses might avoid host translational suppression.
IMPORTANCE
Viruses lack biosynthetic capabilities and depend upon the host for protein synthesis. This dependence requires viruses to evolve mechanisms to coerce the host translational machinery into synthesizing viral proteins in the face of ongoing cellular stress responses that suppress global protein synthesis. Reoviruses replicate and assemble within cytoplasmic inclusions called viral factories. However, synthesis of viral proteins was thought to occur in the cytosol. To identify the site(s) of viral translation, we undertook a microscopy-based approach using ribopuromycylation to detect active translation. Here, we report that active translation occurs within viral factories and that translational factors are compartmentalized within factories. Furthermore, we find that the reovirus nonstructural protein σNS associates with 43S preinitiation complexes at the factory margins, suggesting a role for σNS in translation. Together, virus-induced compartmentalization of the host translational machinery represents a strategy for viruses to spatiotemporally couple viral protein synthesis with viral replication and assembly.
INTRODUCTION
Translation of mRNAs by eukaryotic cells is a complex energy-dependent process that can be rapidly suppressed in response to cellular stressors such as negative energy balance, starvation, growth factor withdrawal, hypoxia, protein misfolding, and viral infection (reviewed in reference 1). As a consequence, all viruses must subvert cell-mediated suppression of translation to effectively maintain viral protein synthesis (3). Viruses accomplish this by a variety of mechanisms. For example, positive-sense RNA viruses, such as hepatitis C virus (HCV), have specialized RNA structures in the 5′ untranslated region of their genome which serve as internal ribosomal entry sites, allowing translation to initiate without a 5′-methylguanosine cap (4). By then targeting host translation initiation factors for degradation, these viruses can promote their own translation to the detriment of the host (5, 6). In contrast, many DNA viruses produce mRNAs that resemble host mRNAs. These viruses stimulate canonical cap-dependent initiation of translation by promoting eukaryotic translation initiation factor 4 subunit F (eIF4F) assembly while simultaneously stimulating the dephosphorylation of eIF2α to avoid suppression of protein translation (7, 8). Alternatively, two DNA viruses that replicate in the cytosol, poxviruses and asfarviruses, have been suggested to promote viral translation by recruiting host translational factors to the sites of viral replication (9–11).
Compartmentalizing translation within replication sites would likely benefit a virus in several ways. It could concentrate the factors needed for translation of viral mRNAs close to the sites of viral transcription, potentially linking the two processes and increasing the efficiency of gene expression, as occurs in prokaryotes (12). Viral protein synthesis could then also occur in close proximity to the sites of virus assembly, providing an efficient mechanism to recruit newly synthesized viral proteins to the sites of assembly. Importantly, compartmentalization may prevent the cellular mRNA decay machinery from accessing viral mRNAs. RNA viruses, in particular, often produce viral mRNAs with features that trigger mRNA decay pathways (reviewed in reference 13), including transcripts that are uncapped or lack a polyadenylated tail (14).
Reoviruses are nonenveloped viruses that contain a segmented double-stranded RNA (dsRNA) genome. The genome is enclosed within a core particle that is further surrounded by outer capsid proteins that mediate virus entry and penetration (15). Following reovirus entry, transcriptionally active core particles are released into the cytoplasm of the cell and become embedded within a virally encoded matrix protein that forms cytoplasmic inclusion structures called viral factories (VF) (16–18). Within the VF, viral core particles transcribe and release viral mRNAs that possess a dimethylated cap 1 structure at the 5′ terminus but lack a poly(A) tail (19). VF are the sites of viral replication and assembly; however, the sites of viral mRNA translation, as well as the mechanisms surrounding translation, are incompletely understood (20, 21).
Here, we address where translation occurs within reovirus-infected cells using ribosome-bound nascent chain puromycylation, or ribopuromycylation (RPM), to visualize actively translating ribosomes by immunofluorescence microscopy (22, 23). We find that translation occurs within VF and that the translational machinery, including ribosomal subunits and cellular translation factors, localize to VF. We further show that the nonstructural protein σNS strongly colocalizes and immunoprecipitates with two proteins in the 43S preinitiation complex (PIC), eIF3A and pS6R, suggesting a role for σNS in the recruitment or maintenance of ribosomes within VF. Our findings indicate that the cellular ribonucleoprotein complexes required for cap-dependent translation are compartmentalized within VF, highlighting a novel strategy by which RNA viruses gain control of the translational machinery.
RESULTS
Actively translating ribosomes localize to viral factories.
Reovirus core particles are embedded within VF and contain the viral λ3 RNA-dependent RNA polymerase which synthesizes viral mRNA transcripts using the minus strands of the dsRNA genome segments as a template (24). The viral mRNA transcripts are capped, as they are extruded through turret-like structures at the 5-fold axes of symmetry of the core, but lack a polyadenylated tail (25, 26). Viral replication, transcription, and assembly all occur within the VF (18, 21).
Early studies using thin-section electron microscopy failed to detect ribosomes within VF, and it was assumed that viral mRNAs synthesized within factories were exported or diffused out to gain access to the cellular translational machinery (18, 21). This model implies that newly synthesized viral proteins must, by some mechanism(s), be trafficked back into the factory to participate in replication and assembly. Data supporting this model are limited. Therefore, we asked where translation occurs within reovirus-infected cells using the ribopuromycylation method (RPM) to detect the subcellular localization of actively translating ribosomes (23). In this method, cells are briefly treated with emetine, an elongation inhibitor, to immobilize actively translating 80S ribosomes on mRNAs, followed by a brief exposure to puromycin (PMY), a Tyr-tRNA mimetic. PMY is incorporated into the ribosome-associated nascent polypeptide chains, and the subcellular location of ribosome-associated nascent chains can then be detected by indirect immunofluorescence using a monoclonal antibody against PMY (Fig. 1A). Similar to the findings of David et al. (22, 23), our results showed the distribution of actively translating ribosomes in mock-infected CV-1 cells to be predominantly tubulovesicular, with increased perinuclear intensity that likely corresponds to endoplasmic reticulum (ER)-associated polysomes (Fig. 1B, top row). In contrast, PMY labeling strongly colocalized within VF in cells infected with the serotype 3 Dearing (T3D) strain of reovirus for 18 h (Fig. 1B). A series of 0.1-µm confocal sections of infected cells indicated that PMY labeling was distributed throughout the VF and was not simply surface associated (data not shown). Similar results were obtained in infected HeLa cells, suggesting that the localization of PMY in VF is not cell type specific (Fig. 1B). Most reovirus strains form filamentous VF through an association with stabilized microtubules. However, the T3D strain used in these experiments contains a temperature-sensitive mutation in the viral protein μ2 that prevents this association, resulting in the production of globular VF at 37°C (27, 28). Therefore, to evaluate if PMY labeling occurs within filamentous viral factories, we infected cells with the serotype 1 Lang (T1L) strain. As we found for T3D-infected cells, the PMY labeling localized to T1L VF at 18 h postinfection (p.i.) (Fig. 1B).
FIG 1 .

Ribopuromycylated products are synthesized in viral factories. (A) Schematic representation of the ribopuromycylation method (RPM). Addition of the inhibitor emetine “freezes” elongating ribosomes on mRNA (step 1). Puromycin (PMY) is then added (step 2) and becomes incorporated into the nascent chain (step 3). Ribosome-associated puromycylated polypeptide chains are detected with a monoclonal antibody (MAb) against PMY (step 4) and visualized by indirect immunofluorescence or immunoblotting. (B) CV-1 and HeLa cells were mock infected or infected with T3D or T1L at an MOI of 1 for 18 h before RPM processing and immunofluorescence. Representative images are shown (n ≥ 5 independent experiments). Scale bars, 10 µm. (C) CV-1 cells were infected (T1L or T3D, MOI =10) for 18 h p.i. before treatment with 208 µM emetine for 15 m at 37°C followed by RPM labeling. Puromycylated protein levels were assessed in cell lysates by immunoblotting with anti-PMY MAb.
The synthesis of viral proteins exceeds that of cellular protein synthesis at 10.5 h p.i. with T3D (29). As shown in Fig. 1B, the majority of PMY staining is present within VF. Although we have not ruled out the possibility that cellular mRNAs are translated within VF, it seemed likely that the majority of translated mRNAs within factories were viral in origin. To address this, we examined the ratio of puromycylated proteins in lysates from mock-infected and T1L- or T3D-infected CV-1 cells. Consistent with the observation that active translation is more apparent within VF than in other regions of the cell (Fig. 1B), immunoblotting revealed that viral proteins were puromycylated to a greater extent than cellular proteins (Fig. 1C). Together, these data indicate that active translation of reovirus mRNA occurs within VF.
Inhibitors of translation block PMY labeling in viral factories.
PMY can bind to ribosomal subunits that lack an mRNA template, albeit with low affinity (23, 30). However, data presented by David et al. strongly suggest that RPM specifically recognizes translating ribosomes (23). To confirm that PMY labeling of translating ribosomes was specific, we pretreated CV-1 cells with harringtonine (1 µg ml−1), a small-molecule inhibitor of translation that blocks initiation by binding to the E site of the 80S ribosome, which is unoccupied only when the 80S ribosome is bound at the start codon. Once the ribosome begins elongating, the E site is occupied by a deacylated tRNA, and thus harringtonine has no effect on elongating ribosomes (31). In infected cells pretreated with harringtonine prior to RPM, PMY labeling was not evident, likely due to the diminished density of translating ribosomes (see Fig. S1 in the supplemental material). To further confirm specificity, we also assessed PMY labeling in the presence of anisomycin, a structural analog of PMY that binds the large ribosomal subunit and prevents peptide bond formation and subsequent incorporation of PMY into polypeptide chains (32). Pretreatment of cells with anisomycin substantially reduced PMY labeling in infected cells (see Fig. S1). Together, these data suggest that PMY labeling is specific to actively translating ribosomes.
40S and 60S ribosomal subunits are found within viral factories.
Our findings thus far indicated that active translation occurs within VF. However, other authors have argued that ribosomes are not present within VF (33). To address this discrepancy, we coimmunostained with antibodies against μNS to detect VF and ribosomal P (RiboP), which recognizes three phosphoproteins, P0, P1, and P2, present on the 60S ribosome (34). In mock-infected cells, RiboP localized throughout the cytoplasm in a tubulovesicular distribution (Fig. 2). In infected cells, however, we noted a prominent ring of colocalization between RiboP and μNS at the edge of VF that was accompanied by fainter RiboP staining within VF (Fig. 2). A similar localization was observed for the 40S ribosome subunit, phosphorylated ribosomal protein S6 (pS6R) (Fig. 2). In contrast, the L11 subunit of the 60S ribosome strongly colocalized with μNS throughout VF, and the ribosomal protein S3 (rpS3) of the 40S ribosome demonstrated an intermediate phenotype (Fig. 2). This colocalization was independent of factory size, as both small and large factories exhibited the same pattern of staining. We further examined the distribution of ribosomes in infected cells by thin-section electron microscopy and found that free ribosomes were clustered at the margins of VF and associated with membranes within factories (Fig. 2B; see also Fig. S2 in the supplemental material). From this, we conclude that 40S and 60S ribosomal subunits (pS6R, rpS3, RiboP, and L11) localize to VF in reovirus-infected cells. Our observation that ribosomal subunits localized to distinct regions of the VF suggests kinetic and/or spatial distribution of ribosomal subunits within the factory. Similar to how a cell partitions cellular processes, the apparent structural organization within the factory may represent a means to separate viral replication, translation, and assembly. Alternatively, it is possible that some of these ribosomal subunits have extraribosomal functions that contribute to their subcellular localization in infected cells (35).
FIG 2 .

40S and 60S ribosomal subunits are recruited to viral factories. (A) CV-1 cells were infected with T3D at an MOI of 1 for 18 h and processed for RPM and immunofluorescence. Immunostaining was performed for μNS (viral factory), pS6R and rpS3 (40S ribosomal subunits), and RiboP and L11 (60S ribosomal subunits). An enlarged area of the boxed region in the merged images, second row, is shown in the third row with individual channels of μNS and the ribosomal proteins in the rows below. Arrows indicate factories of varied size. Scale bars, 10 µm. (B) CV-1 cells infected with T1L, T3DN, or T3DC were processed at 24 h for electron microscopy. Red arrows indicate ribosomes lining the margins of the factory. Black arrows indicate clusters of ribosomes within the factory. White arrows indicate membrane-associated ribosomes. Scale bars, 0.5 µm.
Translation initiation, elongation, termination, and recycling factors are recruited to viral factories.
Our observations that active translation occurs within and ribosomal subunits localize to VF prompted us to examine the distribution of other cellular factors involved in translation. Eukaryotic translation has four defined stages, initiation, elongation, termination, and ribosome recycling, whereby the 40S and 60S ribosomes are separated and factors are recharged for further rounds of initiation (36, 37). All translation initiation (Fig. 3; see also Table S1 in the supplemental material), elongation, termination, and recycling (Fig. 4; see Table S1) factors tested were detected within VF at 18 h p.i. Despite differences in the capacity of some reovirus strains to suppress host translation, we did not observe differences in the degree of PMY or eIF4E localization to the factory at 18 h following infection with T3D and T1L, compared with localization after infection with the type 3 Abney strain (T3A), which is known to efficiently inhibit host protein synthesis (Fig. 1 and 3 and data not shown). Interestingly, unlike eIF4E and other initiation factors, eIF3A, a member of the 43S preinitiation complex (PIC), localized primarily to the margins of the VF, as was observed for RiboP, pS6R, and rpS3 (Fig. 2 and 3B). Through its interaction with eIF4F, eIF3 recruits activated mRNPs to the 43S PIC for translation (38). The distribution of eIF3A suggests that 43S ribosomal loading onto activated viral mRNPs may occur primarily at the outer margins of the factory (see Fig. 7). It is plausible that the difference in localization observed between eIF3A and eIF4E may reflect differences in the functions of these proteins or how they are recruited to the viral factory. In many instances, the level of staining for translational proteins within the VF was striking. It was unclear if this was a result of increased expression levels or as a consequence of redistribution of the proteins to the factories. To address this, we monitored protein expression levels of eIF4E, eIF4A1, and eIF4G over the course of an infection. As others have found (39), we were unable to detect any difference in the levels of total protein in mock versus infected cells from 0 to 20 h p.i. (Fig. 3C and data not shown). Together, these data suggest that cellular translation proteins are redistributed to the VF.
FIG 3 .
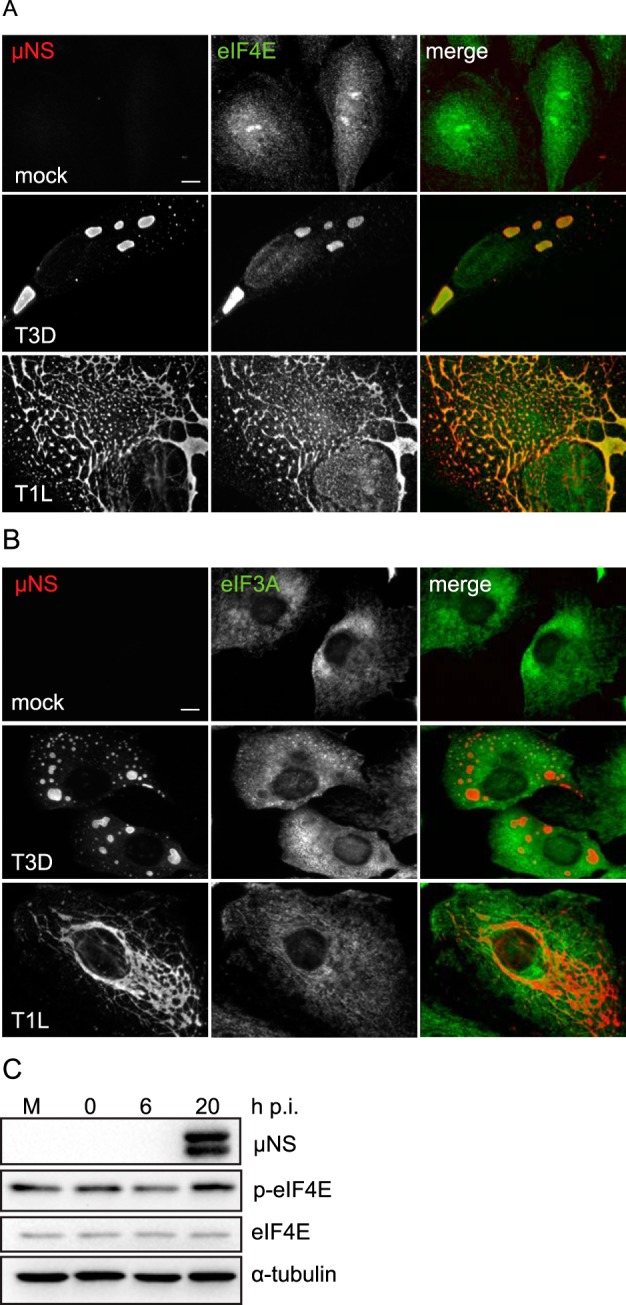
Cellular translation initiation factors colocalize to viral factories. (A, B) CV-1 cells were infected with T3D or T1L at an MOI of 1. At 18 h p.i., RPM-labeled cells were coimmunostained for μNS and eIF4E (A) or eIF3A (B). Scale bars, 10 µm. (C) CV-1 cells were infected with T3D, MOI of 3, for the times indicated. Protein levels were assessed by immunoblotting. M = mock.
FIG 4 .
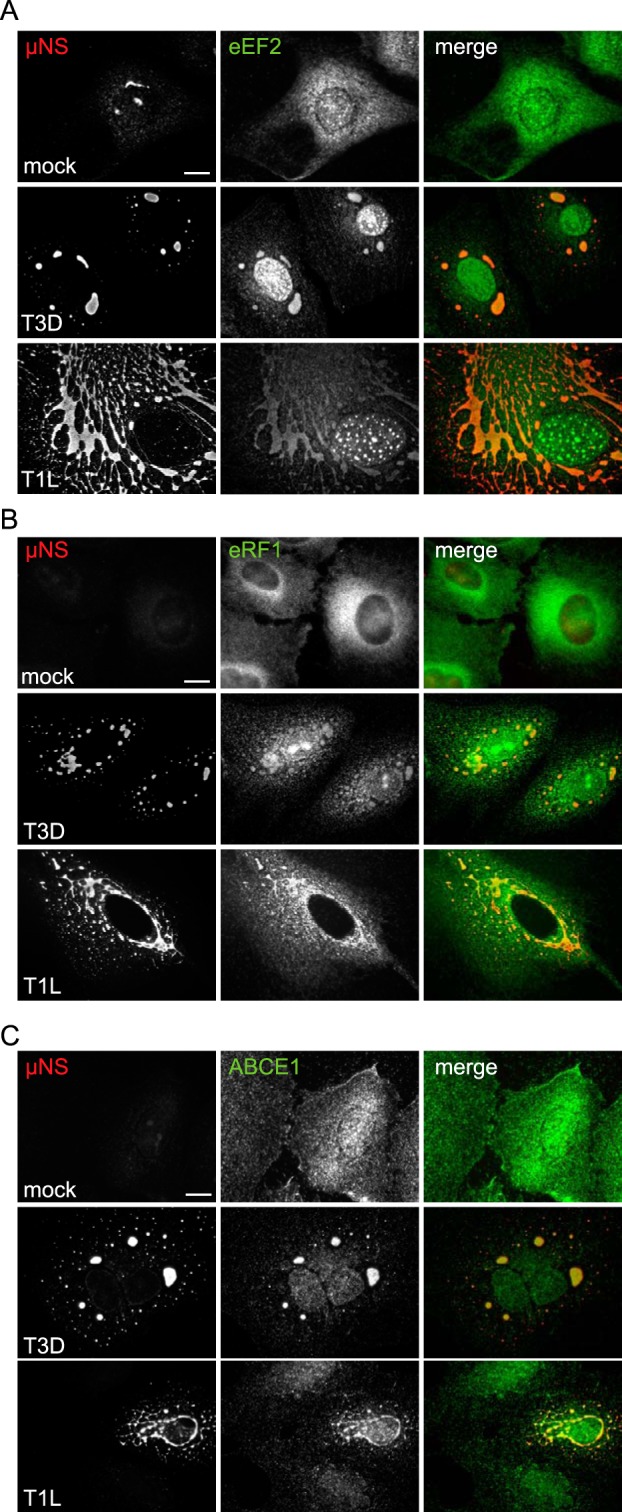
Translation elongation, termination, and recycling factors are recruited to viral factories. CV-1 cells were infected with T3D or T1L at an MOI of 1. At 18 h p.i., RPM-labeled cells were coimmunostained for μNS and eEF2 (A), eRF1 (B), or ABCE1 (C). Scale bars, 10 µm.
FIG 7 .

Reovirus retargets the cellular translational apparatus to the viral factory where viral translation, replication, and assembly occur. Following infection, cellular proteins involved in translation retarget to the viral factory (VF), possibly through interactions with viral proteins. Newly synthesized viral mRNAs are released from transcriptionally active core particles into the surrounding VF (1), where they become bound by eIF4F to form activated viral mRNPs (2). Viral mRNPs are loaded onto 43S ribosomes at the margins of the VF, possibly via σNS, and in association with the ER (3). This is followed by joining of the 60S ribosome to form the 80S ribosome (4), which permeates through the VF as translation ensues (5), resulting in accumulation of newly synthesized viral proteins within the VF (6).
Viral factories are dynamic and allow vesicular traffic to pass through them.
The patterns of localization of PMY, initiation factors, and ribosomal subunits suggested that viral mRNPs might be loaded onto ribosomes at the margins of the factory and then trafficked through the factory as active translation proceeded (see Fig. 7). If this model were correct, the matrix of the VF would have to be dynamic to allow movement of ribosomes. Fixed images of VF do not provide information about the dynamics of the factory, and although vesicular structures were seen within factories, the dynamics of the VF matrix in infected cells are not known. Therefore, we examined the dynamics of the µNS matrix by live-cell microscopy. As we were unable to recover a recombinant virus expressing GFP-µNS, we transiently transfected CV-1 cells with a µNS construct with enhanced green fluorescent protein (eGFP) fused at its N terminus and then infected those cells with T1L intermediate subviral particles (ISVPs). ISVPs are similar to the partially uncoated virions isolated from infected cells and are infectious (40–42). Living infected cells expressing GFP-µNS were mounted within a temperature- and CO2-controlled chamber, and at 12 h p.i., images were collected at approximately 3-s intervals over the course of 8 to 9 min (see Fig. S3 in the supplemental material). The GFP-labeled µNS provided sufficient contrast to clearly detect trafficking of vesicular structures through the VF (see Fig. S3, left, and Movie S1 in the supplemental material). Vesicular trafficking depends on an intact microtubule network. To confirm that movement was due to vesicular trafficking, we treated cells with a microtubule-depolymerizing agent, nocodazole. In the presence of nocodazole, vesicular trafficking through factories was substantially reduced (see Fig. S3, right; note that the same factory is imaged in both panels and Movie S2 in the supplemental material). We conclude from these findings that at 12 h p.i., the VF matrix is deformable such that vesicles can traffic through the matrix.
RPM staining within viral factories is not dependent on the microtubule network.
Assembly of filamentous VF is dependent on an intact microtubule network, and disruption of this network alters the size and distribution of factories in T1L-infected cells (43). In cells infected with the T3D strain, which forms globular VF, depolymerization of microtubules leads to an increase in the number of factories, with a corresponding decrease in their size. Despite changes in factory morphology, treatment with nocodazole has little to no effect on viral yield (44). The impact of microtubule depolymerization on viral translation, however, has not been examined. We therefore tested whether depolymerization of microtubules would affect the recruitment of the translational machinery to VF. HeLa cells were infected with T3D for 6 h before being treated with 10 µM nocodazole as previously described (27). As expected, nocodazole treatment of infected cells led to depolymerization of microtubules with small pinpoint VF instead of the larger factories typical of T3D infection (see Fig. S4A in the supplemental material). Although nocodazole treatment altered the size and distribution of the factories, PMY staining remained and appeared more pronounced in cells where the microtubule network was disrupted compared to that in untreated cells (see Fig. S4B). These data suggest that active translation within VF is not dependent on the microtubule network.
Ribosomal subunits colocalize with viral protein σNS.
It has been suggested that σNS and σ3 viral proteins, encoded by the S3 and S4 genome segments, respectively, may be involved in promoting viral translation (45, 46). σNS is observed in VF as early as 6 h p.i., a time when factories are first detected (47). σNS is an RNA binding protein with a strong affinity for single-stranded RNA (ssRNA) and is essential for viral replication (48, 49). Immunostaining of infected cells for σNS often shows a strong concentration of σNS at the outer margins of VF (50). From this, we hypothesized that σNS might act as a functional link between viral ribonucleoprotein (mRNP) complexes and components of the 43S PIC. To test this, we first assessed the localization of σNS in cells infected with T3D for 18 h. As expected, σNS staining was distributed throughout viral factories but was most intense at the outer margins of the factories (see Fig. S5 in the supplemental material). We also noted that the cytosolic staining pattern for σNS was distributed in a tubulovesicular staining pattern, similar to that observed for eIF3A and ribosomal subunits. We therefore assessed whether σNS colocalized with any of the components of the translational machinery that we had previously localized to the margins of the VF. At 18 h p.i., we observed a striking colocalization between σNS and eIF3A, RiboP, pS6R, and rpS3, suggesting that σNS may be specifically involved in ribosomal recruitment to, or retention within, the factory (see Fig. S5). Our initial findings with wide-field fluorescence microscopy suggested that the viral nonstructural protein σNS colocalized with markers of the 43S PIC on the margins of the factories. However, at the resolution limit of light microscopy (~0.2 µm in xy dimension) it was not possible to discern individual ribosomes, which are approximately 35 nm in diameter. Therefore, to get higher-resolution images, we repeated the experiments using superresolution structured illumination microscopy (SR-SIM). SR-SIM allows xy resolutions approaching 100 nm and increases both the lateral and axial resolutions by a factor of two (51). Consistent with our conventional wide-field immunofluorescence data, we observed σNS localization primarily at the margins of VF in close association with eIF3A and pS6R (Fig. 5A).
FIG 5 .

Viral protein σNS colocalizes with the ribosomal subunit pS6R and 43S-PIC member eIF3A. (A) At 18 h p.i., T3D-infected CV-1 cells were processed for SR-SIM using a Zeiss Elyra superresolution 3D-SIM microscope. Standard wide-field and Z-stack images were acquired with a 110-nm interval at 32 nm/pixel. Scale bars, 5 µm. (B) Immunoblot analysis reveals that σNS in T3D- and T1L-infected CV-1 cell lysates (bottom) coimmunoprecipitates (top) with endogenous eIF3A and pS6R. M, mock; D, T3D; L, T1L.
σNS interacts in a complex with pS6R and eIF3A.
Given its role as an RNA binding protein, the observation that σNS strongly colocalized to the outer margins of the VF in association with eIF3A and ribosomal subunits (Fig. 5; see also Fig. S5 in the supplemental material) led us to hypothesize that σNS may act to facilitate an interaction between the activated viral mRNP and the 43S PIC (see Fig. 7). To assess potential interactions between σNS and eIF3A or the ribosomal subunit pS6R, we performed coimmunoprecipitation (CoIP) studies in T3D- and T1L-infected CV-1 cells. Cell lysates were subjected to immunoprecipitation with antibodies directed against eIF3A and pS6R, and antibody-protein complexes were resolved by SDS-PAGE before immunoblotting for σNS. We found that σNS precipitated with eIF3A and pS6R, revealing that an interaction exists between σNS and members of the 43S PIC, potentially as part of a complex (Fig. 5B). Probing for μNS yielded similar results (data not shown). This was expected given the previous finding that σNS and μNS interact and further supports that these interactions likely occur within the factory (47). The association between σNS and eIF3A and pS6R is not dependent on the expression of other viral proteins, as σNS precipitated with both of these proteins in 293T cells ectopically expressing S3 (see Fig. S5c).
Ribosomal subunits and σNS are closely associated with the endoplasmic reticulum.
Colocalization between σNS, eIF3A, and ribosomal subunits was strongest at the outer margins of and in the area immediately surrounding the factory, organizing along tubulovesicular structures analogous to the endoplasmic reticulum. To confirm that the structures surrounding viral inclusions represented the endoplasmic reticulum, CV-1 cells were transfected for 24 h with DsRed2-ER, a marker for the ER, followed by infection with either T3D or T1L for 24 h. Despite weak colocalization between DsRed2-ER and µNS, definitive colocalization was observed between DsRed2-ER and σNS, primarily at the margins of factories (Fig. 6). Consistent with this, calnexin, an integral ER protein, also localized to VF (Fig. 6C). These findings are in agreement with a recent report that markers for the ER codistribute with VF in reovirus-infected cells (52). Together, these data suggest that σNS colocalizes with ribosomal subunits and eIF3A in association with ER membranes.
FIG 6 .
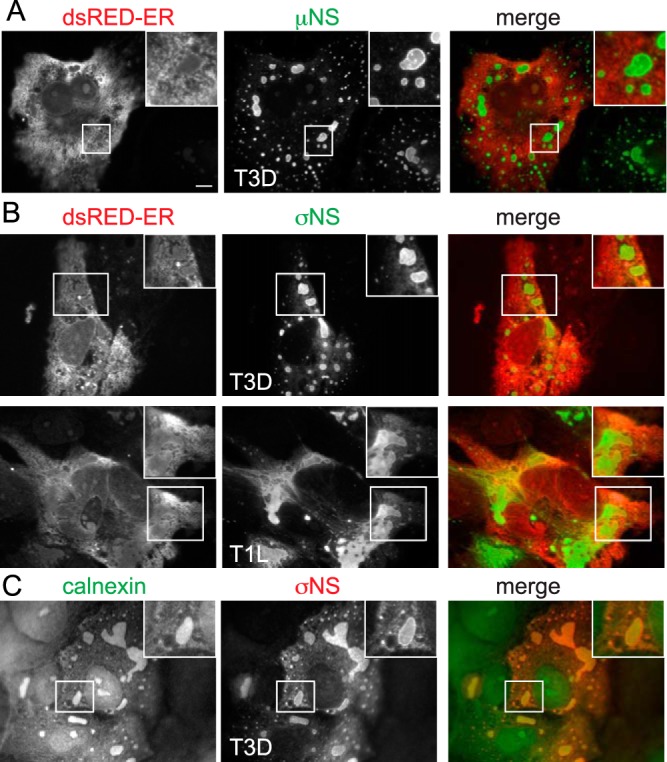
The viral factory is closely associated with the endoplasmic reticulum. (A) CV-1 cells were transfected with dsRedER-2 for 24 h before being infected with T3D at an MOI of 10 for 24 h. Following infection, cells were fixed and immunostained for μNS. The boxed area in each panel is enlarged to show detail (inset). Scale bars, 10 µm. (B) Cells were transfected and infected as described above with T1L or T3D. Following infection, cells were fixed and immunostained for σNS. The boxed area in each panel is enlarged to show detail (inset). (C) Cells were infected with T3D for 18 h before being fixed and immunostained for calnexin and anti-σNS. The boxed area in each panel is enlarged to show detail (inset).
DISCUSSION
Viruses depend upon the host translational apparatus for synthesis of viral proteins. Perhaps as a consequence, a major component of the innate antiviral defense mechanism acts to limit protein synthesis through phosphorylation of eIF2α by the double-stranded RNA-activated protein kinase R (PKR) (53). Despite robust activation of PKR and phosphorylation of eIF2α, reovirus proteins are translated efficiently, while host cell protein synthesis declines (15). The mechanism(s) by which reovirus mRNAs are preferentially translated has heretofore remained unclear. The data presented herein indicate that reovirus mRNPs are in part preferentially synthesized by being transcribed within compartments (VF), which concentrate the host protein synthesis machinery.
We found that the majority of proteins involved in translation (initiation factors, elongation factors, termination factors, and recycling factors) localized diffusely throughout the VF matrix. However, some ribosomal subunits and eIF3A distributed primarily to the limiting margins of the VF. Based on these observations, we propose that reovirus mRNPs are spatially segregated and translated within VF. Our findings indicate that cellular translation initiation factors redistribute to VF. We propose that newly transcribed viral mRNAs become bound by the eIF4F cap-binding complex within VF to form activated viral mRNP complexes, perhaps in association with the viral σNS protein that binds single-stranded RNA (ssRNA). The viral mRNPs would then engage the 43S PIC at the margins of the factory aided by an interaction with σNS. Upon 60S joining, the viral mRNA polysomes would move through the factory as translation ensues. In this way, viral proteins would be synthesized in situ, allowing their efficient incorporation into replication and particle assembly complexes (Fig. 7). This model provides a rational platform for future study of the mechanisms involved but leaves several questions unanswered. One important question is how translation is first initiated at the very earliest stages of infection after transcribing core particles are deposited in the cytosol. An important clue may be the localization of these core particles to stress granules (SG) at early times postinfection (54). SG form in response to phosphorylation of eIF2α, which occurs early after reovirus infection (54). SG contain stalled 48S preinitiation complexes, 40S ribosome subunits, eIF2, eIF3, eIF4A, eIF4B, eIF4E, eIF4G, and eIF5, together with various RNA binding proteins, including the SG markers, T-cell restricted intracellular antigen 1 (TIA1), TIA-related protein (TIAR), and rasGAP SH3 domain binding protein 1 (G3BP1) (55). The mRNPs within SG are stable, but active translation does not occur. The recent finding that ectopically expressed µNS protein localizes to SG that are induced by sodium arsenite suggests that viral factories may form in association with SG (56). Because SG act to compartmentalize components of the translational machinery, it is possible that reoviruses have usurped this host cell stress response to initially compartmentalize the translational machinery before viral factories form. The mechanism by which 48S complexes are suppressed is thought to be through sequestration of RNA by the SG-associated RNA-binding proteins, G3BP, TIA1, and TIAR (57). It is possible that suppression of viral mRNP translation may be overcome through competition of σNS with these proteins.
Infection with several viruses, including poliovirus, Sindbis virus, and turnip mosaic virus, results in compartmentalization of specific cellular proteins involved in translation, but thus far active translation has not been shown to occur at these sites (58, 59). In addition, spatial organization of the translational machinery within the VF has also been observed within replication complexes of the cytosolic DNA viruses, such as African swine fever virus (ASFV) and vaccinia virus (VV) (9, 10). The distribution of the translational machinery at late time points during ASFV infection is similar to our finding that ribosomal subunits and 43S PIC members localize to the margins of the factory at 18 h p.i. (9). The specific organization of ribosomal subunits and 43S PIC members at the margins of the factory could reflect a means to segregate viral plus-stranded RNAs destined for incorporation into nascent viral core particles from those destined to be used for protein synthesis. This would ensure efficiency within the viral life cycle. Importantly, because reovirus mRNAs are noncanonical and lack a polyadenylated tail, compartmentalization within the factory could protect viral mRNAs from recognition by the cellular mRNA decay machinery.
Initiation of cap-dependent translation, in particular recruitment of the eIF4E cap-binding protein to the mRNA, is considered to be a rate-limiting step for protein synthesis (60). Translation initiation is therefore a major target for RNA and DNA viruses alike to modulate in order to maintain efficient viral translation (3). It has been noted that DNA viruses promote translation by increasing total cell concentration of available eIF4F complexes; however, another strategy, suggested for VV, involves retargeting translation proteins to the factory, increasing their local concentration, and promoting eIF4F complex formation within the VF without altering total cell concentrations (8, 10, 11). Our observation that the distribution of, and not the expression level of, eIF4E was altered during infection suggests that reovirus may adopt a similar strategy to ensure an increased concentration of initiation factors at sites of active viral translation.
Despite our observation that VF are the sites of active translation, it remains unclear how the translational machinery is recruited to viral factories. As suggested above, it may relate to interactions between µNS and components of SG, consistent with our observation that TIAR localizes to VF throughout infection (E. A. Desmet and J. S. L. Parker, unpublished data). Rotavirus NSP3 protein has previously been shown to interact with eIF4G; however, until now, no interactions between reovirus proteins and cellular translational proteins involved in initiation have been detected (61). Our finding that σNS interacts with eIF3A and pS6R suggests that translational machinery is recruited to the factory by viral proteins. This is consistent with the finding of others that σNS cosediments with 40S and 60S ribosomes (62) and suggests that σNS is directly involved in viral translation. Similarly, the transactivator/viroplasmin (TAV) of cauliflower mosaic virus forms a complex with the 43S PIC through an interaction with eIF3 and the 60S ribosome (63).
Recently, it was found that reovirus factories are permeated with a meshwork of cellular membranes that bear markers for the ER and ER-Golgi intermediate compartments (ERGIC) (52). Consistent with our findings, the authors noted that the rough endoplasmic reticulum (RER) made numerous contacts with VF, which they suggested may indicate a role for RER in the transport of newly synthesized viral proteins to the VF, as is the case for rubella virus (64). While this is consistent with the previous model for reovirus translation, our current data suggest that the RER at the margins of the factory serves instead as a source of ribosomes for the translation of viral mRNAs within the VF. This is consistent with our observation that membranes are distributed throughout the factories and are associated with ribosomes.
The work presented herein that translation occurs within VF and that cellular factors required for translation are compartmentalized within viral factories suggests that reoviruses may couple transcription of viral mRNA with translation. Furthermore, our data provide an explanation for the association of ER membranes with VF as a source of ribosomes and translational machinery. Finally, our finding that σNS interacts with eIF3A and pS6R reveals a novel role for σNS in viral translation. Importantly, the strategies used by reovirus to reorganize the cellular translational machinery may help to uncover novel approaches or targets used by viruses to subvert the host antiviral response.
MATERIALS AND METHODS
Viruses.
Reoviruses T1L and T3D laboratory stocks originated from the T1/human/Ohio/Lang/1953 and T3/human/Ohio/Dearing/1955 isolates, respectively (65). The superscript N in T3DN denotes a clone obtained from M. L. Nibert (Harvard Medical School) and distinguishes it from that from L. W. Cashdollar (Medical College of Wisconsin), designated T3DC. The two clones differ in the nucleotide sequence of M1 and viral inclusion morphology (27). All infections were performed with T3DN (abbreviated T3D in the text) unless otherwise noted. Viruses were plaque purified, and third-passage cell lysate stocks were used for subsequent experiments. T1L ISVPs were prepared as described (66).
Infections and transfections.
CV-1 or HeLa cells were seeded the day before an experiment such that cells obtained 50 to 70% confluence at the time of the infection. Virus was absorbed to cells at a multiplicity of infection (MOI) of 1 to 10 for 1 h at room temperature (RT) in phosphate-buffered saline (PBS; pH 7.4), supplemented with 2 mM MgCl2. Following absorption, virus was removed and cells were incubated in growth medium at 37°C for the indicated time. Transfections in CV-1 cells were carried out with 2.5 µg of DNA and 6 µl of FuGene 6 according to the manufacturer’s instructions. At 24 h posttransfection, cells were infected as described above at an MOI of 10 for 24 h. Transfections in 293T cells were carried out with 1.8 µg of DNA and 4 µl of Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions.
Ribopuromycylation method.
The ribopuromycylation method (RPM) was performed as previously described with slight modification (23). Briefly, cells were seeded and infected as described above. At the indicated times, cells were either left untreated or treated with emetine (208 µM) for 15 min at 37°C. Cells were rinsed with warm Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 7.5% fetal bovine serum (FBS) (Atlanta Biologicals), followed by a 5-min incubation at 37°C in DMEM-FBS supplemented with 182 µM puromycin (PMY) and 208 µM emetine. Following incubation, cells were immediately placed on ice and washed twice with ice-cold PBS and then extracted and fixed for 20 min on ice in polysome buffer (50 mM Tris-HCl [pH 7.5], 5 mM MgCl2, 25 mM KCl, 355 µM cycloheximide, EDTA-free protease inhibitors, and 10 U/ml RNaseOut) supplemented with 200 mM NaCl, 0.1% Triton X-100, and 3% paraformaldehyde (PFA). In experiments examining the microtubule network, RPM was performed as described above with the exception that following RPM, cells were incubated for 3 min in 100% methanol at −20°C followed by 3 washes for 5 min each in PBS containing 0.1% Triton X-100.
Immunofluorescence and SR-SIM microscopy.
Fixed and permeabilized cells on glass coverslips were incubated in staining buffer (SB; 0.05% saponin, 10 mM glycine, 5% FBS, and PBS) for 15 min at RT before incubation with primary antibodies diluted in SB for 1 h at RT. Coverslips were then washed before being incubated with the appropriate secondary antibodies diluted in SB for 1 h at RT. Coverslips were mounted onto glass slides with ProLong Gold Anti-Fade reagent with DAPI (4[prime],6-diamidino-2-phenylindole; Invitrogen). Images were obtained using a Nikon TE2000 inverted microscope equipped with phase and fluorescence optics through a PlanApo 60× 1.40-numerical-aperture oil objective with type A immersion liquid (Nikon) ± a ×1.5 optical zoom. Images were collected digitally with a Coolsnap HQ charge-coupled-device (CCD) camera (Roper) and NIS-elements software (version 4.12; Nikon) and were processed and prepared for presentation using Photoshop (CS3; Adobe) and Illustrator (CS6; Adobe) software. For SR-SIM, CV-1 cells were grown on high-performance no. 1.5 coverslips (Carl Zeiss Microscopy). Cells were infected, fixed, and immunostained as described above with the exception that coverslips were mounted onto glass slides with Vectashield mounting medium. Images were collected with a Zeiss Elyra superresolution three-dimensional SIM (3D-SIM) microscope. Z-stack images were acquired with a 110-nm interval at 32 nm/pixel. Each SR image results from 15 images (5 phases × 3 rotations of a diffraction grating). Images were acquired at 50 ms and were analyzed using Zen2012 software (Zeiss).
Immunoprecipitation.
At 18 h, CV-1 cells were treated with 208 µM emetine for 15 min at 37°C before cross-linking with 0.75% paraformaldehyde for 10 min at RT. The reaction was quenched with the addition of 125 mM glycine for 5 min at RT, and cells were lysed for 30 min on ice in lysis buffer containing 150 mM Tris-HCl, 50 mM NaCl, 5 mM MgCl2, 1% NP-40, EDTA-free protease inhibitors (Roche), 1 mM phenylmethylsulfonyl fluoride (PMSF), and 10 U/ml RNase Out (Invitrogen). Cell lysates were incubated overnight at 4°C with protein G beads (Life Technologies) that were preincubated for 2 h at 4°C with eIF3A or pS6R monoclonal antibodies. After being washed, samples were resuspended in SDS sample buffer, boiled, and resolved by SDS-PAGE.
SUPPLEMENTAL MATERIAL
Supplemental methods. Download
Degree of colocalization of cellular factors to the viral factory (μNS).
RPM staining within viral factories is absent in the presence of translation inhibitors. CV-1 cells were either mock infected or infected with T3D for 18 h. Before processing for RPM, cells were either left untreated (lower panels) or were treated with harringtonine (1 µg ml−1) (upper panels) or anisomycin 9.4 µM (middle panels) for 15 min at 37°C. Cells were then washed with PBS and processed for RPM and immunofluorescence. Cells were immunostained with a chicken polyclonal antiserum to μNS and a mouse MAb PMY (2A4) followed by Alexa594-conjugated goat anti-chicken IgG and Alexa488-conjugated goat anti-mouse IgG. Images were collected digitally with exposure times of 700 ms. Representative images are shown (n ≥ 3 independent experiments). Scale bar, 10 µm. Download
Ribosomes localize to the margins of the factory and can be observed studding membranes and clustered within the factory. CV-1 cells were infected with T3DC and at the indicated times after infection were processed for electron microscopy to visualize viral factories. Within viral factories, full and empty viral particles, coated microtubules, and membranes can be observed. Black arrows indicate clustering of free ribosomes, white arrows indicate ribosomes associated with membranes, and red arrows indicate ribosomes lining the margins of the factory. (A, C, D) Scale bar, 0.5 µm; (B) scale bar, 1 µm. Download
Microtubule-dependent vesicular traffic passes through GFP-µNS-labeled viral factories. CV-1 cells transiently expressing GFP-µNS were infected with T1L ISVPs at an effective MOI of 5. At 12 h p.i., the movement of GFP-µNS incorporated into viral factories was followed over the course of 9 min using live-cell microscopy, with images collected approximately every 3 s. Images from a representative cell are shown (top). Close-up images of GFP-µNS within a single “factory” are shown in the bottom panels. Following an initial series of images, 10 µM nocodazole was added to the culture medium to depolymerize microtubules. A second series of images was then collected at 8 min, 60 min, and 120 min after nocodazole treatment. Images collected at 60 min after nocodazole treatment are shown at the bottom right. Each subpanel from left to right and top to bottom was collected at ~32-s intervals. Download
RPM labeling within viral factories is not dependent on an intact microtubule network. HeLa cells were infected with T3D at an MOI of 1. At 6 h p.i., cells were left untreated or were treated with 10 µM nocodazole. At 18 h p.i., cells were processed for RPM and then immunostained for μNS by using a chicken polyclonal antiserum and for α-tubulin (A) or puromycin (PMY) (B) by using MAbs followed by Alexa594-conjugated goat anti-chicken IgG and Alexa488-conjugated goat anti-mouse IgG. Download
Viral protein σNS colocalizes with ribosomal subunits and eIF3A. (A) CV-1 cells were infected with T3D at an MOI of 1 for 18 h. Following infection, cells were subjected to RPM labeling and processed for immunofluorescence. Cells were stained with a chicken polyclonal antiserum to μNS followed by Alexa594-conjugated goat anti-chicken IgG before being stained with a guinea pig MAb to σNS followed by Alexa488-conjugated donkey anti-guinea pig IgG. (B) Similarly, eIF3A, RiboP, pS6R, and rpS3 were detected before staining for σNS as described above. The boxed area in each panel is enlarged to show detail (inset). Scale bar, 10 µm. (C) 293T cells were transfected with empty vector (V) or S3 (σNS) for 24 h before being harvested for coimmunoprecipitation. Immunoblot analysis revealed that σNS precipitated with eIF3A and, to a lesser extent, pS6R. Download
Vesicular traffic passes through GFP-μNS-labeled viral factories. CV-1 cells transiently expressing GFP-µNS were infected with T1L ISVPs at an effective MOI of 5. At 12 h p.i., the movement of GFP-µNS incorporated into viral factories was followed over the course of 9 min using live-cell microscopy with images collected approximately every 3 s. Digital images (200) were collected at approximately 3-s intervals using a Nikon PlanApo 60× objective and contrast adjusted using Volocity (Improvision) software, and QuickTime movies were prepared. Download
Vesicular traffic through viral factories is microtubule dependent. Following the initial series of images taken in Movie S1, 10 µM nocodazole was added to the culture medium to depolymerize microtubules. A second series of images was then collected at 60 min after nocodazole treatment, collected at ~32-s intervals, and images were collected and processed as described above. Download
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (grants R56AI090076 and R21AI105520 to J.S.L.P.).
We thank Shu-bing Qian for the generous gifts of harringtonine, anti-ribosomal P protein, and anti-PMY (clone 2A4) antibodies, Terence Dermody for the anti-σNS specific antibody, Alexander David and Jonathan Yewdell for experimental insights, and Rebecca Williams for support with SR-SIM. We are grateful for the technical support provided by Lucy Hardwick and Alexander J. Polino.
Footnotes
Citation Desmet EA, Anguish LJ, Parker JSL. 2014. Virus-mediated compartmentalization of the host translational machinery. mBio 5(5):e01463-14. doi:10.1128/mBio.01463-14.
REFERENCES
- 1. Corradetti MN, Guan KL. 2006. Upstream of the mammalian target of rapamycin: do all roads pass through mTOR? Oncogene 25:6347–6360. 10.1038/sj.onc.1209885 [DOI] [PubMed] [Google Scholar]
- 2. Reference deleted.
- 3. Walsh D, Mohr I. 2011. Viral subversion of the host protein synthesis machinery. Nat. Rev. Microbiol. 9:860–875. 10.1038/nrmicro2655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Otto GA, Puglisi JD. 2004. The pathway of HCV IRES-mediated translation initiation. Cell 119:369–380. 10.1016/j.cell.2004.09.038 [DOI] [PubMed] [Google Scholar]
- 5. Connor JH, Lyles DS. 2002. Vesicular stomatitis virus infection alters the eIF4F translation initiation complex and causes dephosphorylation of the eIF4E binding protein 4E-BP1. J. Virol. 76:10177–10187. 10.1128/JVI.76.20.10177-10187.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rose JK, Trachsel H, Leong K, Baltimore D. 1978. Inhibition of translation by poliovirus: inactivation of a specific initiation factor. Proc. Natl. Acad. Sci. U. S. A. 75:2732–2736. 10.1073/pnas.75.6.2732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Walsh D, Mohr I. 2004. Phosphorylation of eIF4E by Mnk-1 enhances HSV-1 translation and replication in quiescent cells. Genes Dev. 18:660–672. 10.1101/gad.1185304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Walsh D, Perez C, Notary J, Mohr I. 2005. Regulation of the translation initiation factor eIF4F by multiple mechanisms in human cytomegalovirus-infected cells. J. Virol. 79:8057–8064. 10.1128/JVI.79.13.8057-8064.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Castelló A, Quintas A, Sánchez EG, Sabina P, Nogal M, Carrasco L, Revilla Y. 2009. Regulation of host translational machinery by African swine fever virus. PLoS Pathog. 5:e1000562. 10.1371/journal.ppat.1000562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Katsafanas GC, Moss B. 2007. Colocalization of transcription and translation within cytoplasmic poxvirus factories coordinates viral expression and subjugates host functions. Cell Host Microbe 2:221–228. 10.1016/j.chom.2007.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Walsh D, Arias C, Perez C, Halladin D, Escandon M, Ueda T, Watanabe-Fukunaga R, Fukunaga R, Mohr I. 2008. Eukaryotic translation initiation factor 4F architectural alterations accompany translation initiation factor redistribution in poxvirus-infected cells. Mol. Cell. Biol. 28:2648–2658. 10.1128/MCB.01631-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Proshkin S, Rahmouni AR, Mironov A, Nudler E. 2010. Cooperation between translating ribosomes and RNA polymerase in transcription elongation. Science 328:504–508. 10.1126/science.1184939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Moon SL, Barnhart MD, Wilusz J. 2012. Inhibition and avoidance of mRNA degradation by RNA viruses. Curr. Opin. Microbiol. 15:500–505. 10.1016/j.mib.2012.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cao D, Parker R. 2001. Computational modeling of eukaryotic mRNA turnover. RNA 7:1192–1212. 10.1017/S1355838201010330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dermody T, Parker JSL, Sherry B. 2013. Orthoreoviruses. In Knipe DM, Howley PM. (ed), Fields virology, 6th ed. Lippincott Williams and Wilkins, Philadelphia, PA. [Google Scholar]
- 16. Broering TJ, McCutcheon AM, Centonze VE, Nibert ML. 2000. Reovirus nonstructural protein µNS binds to core particles but does not inhibit their transcription and capping activities. J. Virol. 74:5516–5524. 10.1128/JVI.74.12.5516-5524.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ivanovic T, Agosto MA, Chandran K, Nibert ML. 2007. A role for molecular chaperone Hsc70 in reovirus outer capsid disassembly. J. Biol. Chem. 282:12210–12219. 10.1074/jbc.M610258200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rhim JS, Jordan LE, Mayor HD. 1962. Cytochemical, fluorescent-antibody and electron microscopic studies on the growth of reovirus (echo 10) in tissue culture. Virology 17:342–355. 10.1016/0042-6822(62)90125-3 [DOI] [PubMed] [Google Scholar]
- 19. Furuichi Y, Muthukrishnan S, Shatkin AJ. 1975. 5′-Terminal m-, p 7G(5')ppp(5')G-m-p in vivo: identification in reovirus genome RNA. Proc. Natl. Acad. Sci. U. S. A. 72:742–745. 10.1073/pnas.72.2.742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fields BN, Raine CS, Baum SG. 1971. Temperature-sensitive mutants of reovirus type 3: defects in viral maturation as studied by immunofluorescence and electron microscopy. Virology 43:569–578. 10.1016/0042-6822(71)90282-0 [DOI] [PubMed] [Google Scholar]
- 21. Miller CL, Arnold MM, Broering TJ, Hastings CE, Nibert ML. 2010. Localization of mammalian orthoreovirus proteins to cytoplasmic factory-like structures via nonoverlapping regions of µNS. J. Virol. 84:867–882. 10.1128/JVI.01571-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. David A, Bennink JR, Yewdell JW. 2013. Emetine optimally facilitates nascent chain puromycylation and potentiates the ribopuromycylation method (RPM) applied to inert cells. Histochem. Cell Biol. 139:501–504. 10.1007/s00418-012-1063-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. David A, Dolan BP, Hickman HD, Knowlton JJ, Clavarino G, Pierre P, Bennink JR, Yewdell JW. 2012. Nuclear translation visualized by ribosome-bound nascent chain puromycylation. J. Cell Biol. 197:45–57. 10.1083/jcb.201112145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Skehel JJ, Joklik WK. 1969. Studies on the in vitro transcription of reovirus RNA catalyzed by reovirus cores. Virology 39:822–831. 10.1016/0042-6822(69)90019-1 [DOI] [PubMed] [Google Scholar]
- 25. Bartlett NM, Gillies SC, Bullivant S, Bellamy AR. 1974. Electron microscopy study of reovirus reaction cores. J. Virol. 14:315–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gillies S, Bullivant S, Bellamy AR. 1971. Viral RNA polymerases: electron microscopy of reovirus reaction cores. Science 174:694–696. 10.1126/science.174.4010.694 [DOI] [PubMed] [Google Scholar]
- 27. Parker JS, Broering TJ, Kim J, Higgins DE, Nibert ML. 2002. Reovirus core protein µ2 determines the filamentous morphology of viral inclusion bodies by interacting with and stabilizing microtubules. J. Virol. 76:4483–4496. 10.1128/JVI.76.9.4483-4496.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yin P, Keirstead ND, Broering TJ, Arnold MM, Parker JS, Nibert ML, Coombs KM. 2004. Comparisons of the M1 genome segments and encoded mu2 proteins of different reovirus isolates. Virol. J. 1:6. 10.1186/1743-422X-1-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zweerink HJ, Joklik WK. 1970. Studies on the intracellular synthesis of reovirus-specified proteins. Virology 41:501–518. 10.1016/0042-6822(70)90171-6 [DOI] [PubMed] [Google Scholar]
- 30. Lührmann R, Bald R, Stöffler-Meilicke M, Stöffler G. 1981. Localization of the puromycin binding site on the large ribosomal subunit of Escherichia coli by immunoelectron microscopy. Proc. Natl. Acad. Sci. U. S. A. 78:7276–7280. 10.1073/pnas.78.12.7276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fresno M, Jiménez A, Vázquez D. 1977. Inhibition of translation in eukaryotic systems by harringtonine. Eur. J. Biochem. 72:323–330. 10.1111/j.1432-1033.1977.tb11256.x [DOI] [PubMed] [Google Scholar]
- 32. Hansen JL, Moore PB, Steitz TA. 2003. Structures of five antibiotics bound at the peptidyl transferase center of the large ribosomal subunit. J. Mol. Biol. 330:1061–1075. 10.1016/S0022-2836(03)00668-5 [DOI] [PubMed] [Google Scholar]
- 33. Sharpe AH, Chen LB, Fields BN. 1982. The interaction of mammalian reoviruses with the cytoskeleton of monkey kidney CV-1 cells. Virology 120:399–411. 10.1016/0042-6822(82)90040-X [DOI] [PubMed] [Google Scholar]
- 34. Elkon KB, Parnassa AP, Foster CL. 1985. Lupus autoantibodies target ribosomal P proteins. J. Exp. Med. 162:459–471. 10.1084/jem.162.2.459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Warner JR, McIntosh KB. 2009. How common are extraribosomal functions of ribosomal proteins? Mol. Cell 34:3–11. 10.1016/j.molcel.2009.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dever TE, Green R. 2012. The elongation, termination, and recycling phases of translation in eukaryotes. Cold Spring Harb. Perspect. Biol. 4:a013706. 10.1101/cshperspect.a013706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jackson RJ, Hellen CU, Pestova TV. 2010. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 11:113–127. 10.1038/nrm2838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hinnebusch AG. 2006. eIF3: a versatile scaffold for translation initiation complexes. Trends Biochem. Sci. 31:553–562. 10.1016/j.tibs.2006.08.005 [DOI] [PubMed] [Google Scholar]
- 39. Duncan RF. 1990. Protein synthesis initiation factor modifications during viral infections: implications for translational control. Electrophoresis 11:219–227. 10.1002/elps.1150110305 [DOI] [PubMed] [Google Scholar]
- 40. Chang CT, Zweerink HJ. 1971. Fate of parental reovirus in infected cell. Virology 46:544–555. 10.1016/0042-6822(71)90058-4 [DOI] [PubMed] [Google Scholar]
- 41. Shatkin AJ, LaFiandra AJ. 1972. Transcription by infectious subviral particles of reovirus. J. Virol. 10:698–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Silverstein SC, Astell C, Levin DH, Schonberg M, Acs G. 1972. The mechanisms of reovirus uncoating and gene activation in vivo. Virology 47:797–806. 10.1016/0042-6822(72)90571-5 [DOI] [PubMed] [Google Scholar]
- 43. Broering TJ, Parker JS, Joyce PL, Kim J, Nibert ML. 2002. Mammalian reovirus nonstructural protein µNS forms large inclusions and colocalizes with reovirus microtubule-associated protein µ2 in transfected cells. J. Virol. 76:8285–8297. 10.1128/JVI.76.16.8285-8297.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Carvalho J, Arnold MM, Nibert ML. 2007. Silencing and complementation of reovirus core protein µ2: functional correlations with µ2-microtubule association and differences between virus- and plasmid-derived µ2. Virology 364:301–316. 10.1016/j.virol.2007.03.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Giantini M, Seliger LS, Furuichi Y, Shatkin AJ. 1984. Reovirus type 3 genome segment S4: nucleotide sequence of the gene encoding a major virion surface protein. J. Virol. 52:984–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schmechel S, Chute M, Skinner P, Anderson R, Schiff L. 1997. Preferential translation of reovirus mRNA by a σ3-dependent mechanism. Virology 232:62–73. 10.1006/viro.1997.8531 [DOI] [PubMed] [Google Scholar]
- 47. Miller CL, Broering TJ, Parker JS, Arnold MM, Nibert ML. 2003. Reovirus σNS protein localizes to inclusions through an association requiring the µNS amino terminus. J. Virol. 77:4566–4576. 10.1128/JVI.77.8.4566-4576.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gillian AL, Schmechel SC, Livny J, Schiff LA, Nibert ML. 2000. Reovirus protein σNS binds in multiple copies to single-stranded RNA and shares properties with single-stranded DNA binding proteins. J. Virol. 74:5939–5948. 10.1128/JVI.74.13.5939-5948.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kobayashi T, Chappell JD, Danthi P, Dermody TS. 2006. Gene-specific inhibition of reovirus replication by RNA interference. J. Virol. 80:9053–9063. 10.1128/JVI.00276-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Becker MM, Goral MI, Hazelton PR, Baer GS, Rodgers SE, Brown EG, Coombs KM, Dermody TS. 2001. Reovirus σNS protein is required for nucleation of viral assembly complexes and formation of viral inclusions. J. Virol. 75:1459–1475. 10.1128/JVI.75.3.1459-1475.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gustafsson MG, Shao L, Carlton PM, Wang CJ, Golubovskaya IN, Cande WZ, Agard DA, Sedat JW. 2008. Three-dimensional resolution doubling in wide-field fluorescence microscopy by structured illumination. Biophys. J. 94:4957–4970. 10.1529/biophysj.107.120345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. de Castro IF, Zamora PF, Ooms L, Fernandez JJ, Lai CM, Mainou BA, Dermody TS, Risco C. 2014. Reovirus forms neo-organelles for progeny particle assembly within reorganized cell membranes. mBio 5(1):e00931-13. 10.1128/mBio.00931-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Proud CG. 2005. eIF2 and the control of cell physiology. Semin. Cell Dev. Biol. 16:3–12. 10.1016/j.semcdb.2004.11.004 [DOI] [PubMed] [Google Scholar]
- 54. Qin Q, Hastings C, Miller CL. 2009. Mammalian orthoreovirus particles induce and are recruited into stress granules at early times postinfection. J. Virol. 83:11090–11101. 10.1128/JVI.01239-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Reineke LC, Lloyd RE. 2013. Diversion of stress granules and P-bodies during viral infection. Virology 436:255–267. 10.1016/j.virol.2012.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Carroll K, Hastings C, Miller CL. 2014. Amino acids 78 and 79 of mammalian orthoreovirus protein µNS are necessary for stress granule localization, core protein λ2 interaction, and de novo virus replication. Virology 448:133–145. 10.1016/j.virol.2013.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. White JP, Lloyd RE. 2012. Regulation of stress granules in virus systems. Trends Microbiol. 20:175–183. 10.1016/j.tim.2012.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sanz MA, Castelló A, Ventoso I, Berlanga JJ, Carrasco L. 2009. Dual mechanism for the translation of subgenomic mRNA from sindbis virus in infected and uninfected cells. PLoS One 4:e4772. 10.1371/journal.pone.0004772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Thivierge K, Cotton S, Dufresne PJ, Mathieu I, Beauchemin C, Ide C, Fortin MG, Laliberté JF. 2008. Eukaryotic elongation factor 1A interacts with turnip mosaic virus RNA-dependent RNA polymerase and VPg-Pro in virus-induced vesicles. Virology 377:216–225. 10.1016/j.virol.2008.04.015 [DOI] [PubMed] [Google Scholar]
- 60. Sonenberg N, Hinnebusch AG. 2009. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 136:731–745. 10.1016/j.cell.2009.01.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Groft CM, Burley SK. 2002. Recognition of eIF4G by rotavirus NSP3 reveals a basis for mRNA circularization. Mol. Cell 9:1273–1283. 10.1016/S1097-2765(02)00555-5 [DOI] [PubMed] [Google Scholar]
- 62. Huismans H, Joklik WK. 1976. Reovirus-coded polypeptides in infected cells: isolation of two native monomeric polypeptides with affinity for single-stranded and double-stranded RNA, respectively. Virology 70:411–424. 10.1016/0042-6822(76)90282-8 [DOI] [PubMed] [Google Scholar]
- 63. Thiébeauld O, Schepetilnikov M, Park HS, Geldreich A, Kobayashi K, Keller M, Hohn T, Ryabova LA. 2009. A new plant protein interacts with eIF3 and 60S to enhance virus-activated translation re-initiation. EMBO J. 28:3171–3184. 10.1038/emboj.2009.256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Fontana J, López-Iglesias C, Tzeng WP, Frey TK, Fernández JJ, Risco C. 2010. Three-dimensional structure of rubella virus factories. Virology 405:579–591. 10.1016/j.virol.2010.06.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Goral MI, Mochow-Grundy M, Dermody TS. 1996. Sequence diversity within the reovirus S3 gene: reoviruses evolve independently of host species, geographic locale, and date of isolation. Virology 216:265–271. 10.1006/viro.1996.0059 [DOI] [PubMed] [Google Scholar]
- 66. Chandran K, Parker JS, Ehrlich M, Kirchhausen T, Nibert ML. 2003. The delta region of outer-capsid protein µ1 undergoes conformational change and release from reovirus particles during cell entry. J. Virol. 77:13361–13375. 10.1128/JVI.77.24.13361-13375.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental methods. Download
Degree of colocalization of cellular factors to the viral factory (μNS).
RPM staining within viral factories is absent in the presence of translation inhibitors. CV-1 cells were either mock infected or infected with T3D for 18 h. Before processing for RPM, cells were either left untreated (lower panels) or were treated with harringtonine (1 µg ml−1) (upper panels) or anisomycin 9.4 µM (middle panels) for 15 min at 37°C. Cells were then washed with PBS and processed for RPM and immunofluorescence. Cells were immunostained with a chicken polyclonal antiserum to μNS and a mouse MAb PMY (2A4) followed by Alexa594-conjugated goat anti-chicken IgG and Alexa488-conjugated goat anti-mouse IgG. Images were collected digitally with exposure times of 700 ms. Representative images are shown (n ≥ 3 independent experiments). Scale bar, 10 µm. Download
Ribosomes localize to the margins of the factory and can be observed studding membranes and clustered within the factory. CV-1 cells were infected with T3DC and at the indicated times after infection were processed for electron microscopy to visualize viral factories. Within viral factories, full and empty viral particles, coated microtubules, and membranes can be observed. Black arrows indicate clustering of free ribosomes, white arrows indicate ribosomes associated with membranes, and red arrows indicate ribosomes lining the margins of the factory. (A, C, D) Scale bar, 0.5 µm; (B) scale bar, 1 µm. Download
Microtubule-dependent vesicular traffic passes through GFP-µNS-labeled viral factories. CV-1 cells transiently expressing GFP-µNS were infected with T1L ISVPs at an effective MOI of 5. At 12 h p.i., the movement of GFP-µNS incorporated into viral factories was followed over the course of 9 min using live-cell microscopy, with images collected approximately every 3 s. Images from a representative cell are shown (top). Close-up images of GFP-µNS within a single “factory” are shown in the bottom panels. Following an initial series of images, 10 µM nocodazole was added to the culture medium to depolymerize microtubules. A second series of images was then collected at 8 min, 60 min, and 120 min after nocodazole treatment. Images collected at 60 min after nocodazole treatment are shown at the bottom right. Each subpanel from left to right and top to bottom was collected at ~32-s intervals. Download
RPM labeling within viral factories is not dependent on an intact microtubule network. HeLa cells were infected with T3D at an MOI of 1. At 6 h p.i., cells were left untreated or were treated with 10 µM nocodazole. At 18 h p.i., cells were processed for RPM and then immunostained for μNS by using a chicken polyclonal antiserum and for α-tubulin (A) or puromycin (PMY) (B) by using MAbs followed by Alexa594-conjugated goat anti-chicken IgG and Alexa488-conjugated goat anti-mouse IgG. Download
Viral protein σNS colocalizes with ribosomal subunits and eIF3A. (A) CV-1 cells were infected with T3D at an MOI of 1 for 18 h. Following infection, cells were subjected to RPM labeling and processed for immunofluorescence. Cells were stained with a chicken polyclonal antiserum to μNS followed by Alexa594-conjugated goat anti-chicken IgG before being stained with a guinea pig MAb to σNS followed by Alexa488-conjugated donkey anti-guinea pig IgG. (B) Similarly, eIF3A, RiboP, pS6R, and rpS3 were detected before staining for σNS as described above. The boxed area in each panel is enlarged to show detail (inset). Scale bar, 10 µm. (C) 293T cells were transfected with empty vector (V) or S3 (σNS) for 24 h before being harvested for coimmunoprecipitation. Immunoblot analysis revealed that σNS precipitated with eIF3A and, to a lesser extent, pS6R. Download
Vesicular traffic passes through GFP-μNS-labeled viral factories. CV-1 cells transiently expressing GFP-µNS were infected with T1L ISVPs at an effective MOI of 5. At 12 h p.i., the movement of GFP-µNS incorporated into viral factories was followed over the course of 9 min using live-cell microscopy with images collected approximately every 3 s. Digital images (200) were collected at approximately 3-s intervals using a Nikon PlanApo 60× objective and contrast adjusted using Volocity (Improvision) software, and QuickTime movies were prepared. Download
Vesicular traffic through viral factories is microtubule dependent. Following the initial series of images taken in Movie S1, 10 µM nocodazole was added to the culture medium to depolymerize microtubules. A second series of images was then collected at 60 min after nocodazole treatment, collected at ~32-s intervals, and images were collected and processed as described above. Download