

ABSTRACT
Group A Streptococcus (GAS, Streptococcus pyogenes) is an ongoing threat to human health as the agent of streptococcal pharyngitis, skin and soft tissue infections, and life-threatening conditions such as necrotizing fasciitis and streptococcal toxic shock syndrome. In animal models of infection, macrophages have been shown to contribute to host defense against GAS infection. However, as GAS can resist killing by macrophages in vitro and induce macrophage cell death, it has been suggested that GAS intracellular survival in macrophages may enable persistent infection. Using isogenic mutants, we now show that the GAS pore-forming toxin streptolysin O (SLO) and its cotoxin NAD-glycohydrolase (NADase) mediate GAS intracellular survival and cytotoxicity for macrophages. Unexpectedly, the two toxins did not inhibit fusion of GAS-containing phagosomes with lysosomes but rather prevented phagolysosome acidification. SLO served two essential functions, poration of the phagolysosomal membrane and translocation of NADase into the macrophage cytosol, both of which were necessary for maximal GAS intracellular survival. Whereas NADase delivery to epithelial cells is mediated by SLO secreted from GAS bound to the cell surface, in macrophages, the source of SLO and NADase is GAS contained within phagolysosomes. We found that transfer of NADase from the phagolysosome to the macrophage cytosol occurs not by simple diffusion through SLO pores but rather by a specific translocation mechanism that requires the N-terminal translocation domain of NADase. These results illuminate the mechanisms through which SLO and NADase enable GAS to defeat macrophage-mediated killing and provide new insight into the virulence of a major human pathogen.
IMPORTANCE
Macrophages constitute an important element of the innate immune response to mucosal pathogens. They ingest and kill microbes by phagocytosis and secrete inflammatory cytokines to recruit and activate other effector cells. Group A Streptococcus (GAS, Streptococcus pyogenes), an important cause of pharyngitis and invasive infections, has been shown to resist killing by macrophages. We find that GAS resistance to macrophage killing depends on the GAS pore-forming toxin streptolysin O (SLO) and its cotoxin NAD-glycohydrolase (NADase). GAS bacteria are internalized by macrophage phagocytosis but resist killing by secreting SLO, which damages the phagolysosome membrane, prevents phagolysosome acidification, and translocates NADase from the phagolysosome into the macrophage cytosol. NADase augments SLO-mediated cytotoxicity by depleting cellular energy stores. These findings may explain the nearly universal production of SLO by GAS clinical isolates and the association of NADase with the global spread of a GAS clone implicated in invasive infections.
INTRODUCTION
Group A Streptococcus (GAS, Streptococcus pyogenes) is a human-restricted pathogen that is responsible for a wide variety of diseases, ranging from superficial infections of the throat (pharyngitis) and skin (impetigo) to invasive and life-threatening conditions such as necrotizing fasciitis and streptococcal toxic shock syndrome, as well as the postinfectious sequelae of acute rheumatic fever and glomerulonephritis (1). Although GAS is generally considered an extracellular pathogen, intracellular reservoirs of viable GAS have been reported in epithelial cells (2–4), neutrophils (5), and macrophages (6). It has been suggested that survival of GAS within host cells may promote the persistence of the organisms during colonization or infection by shielding them from immune effectors and antibiotics.
Macrophages represent one of the earliest immune cells encountered by bacteria during infection. As a front line of the innate immune response, macrophages phagocytose bacteria and produce inflammatory mediators to recruit and activate other effector cells, including neutrophils. Depletion of macrophages in mice by carrageenan treatment resulted in an increased mortality rate after an intravenous challenge with GAS, a result that implies that macrophages contribute to clearance of GAS from the bloodstream (7). In a mouse model of invasive soft tissue infection, clodronate treatment to deplete macrophages was associated with no change in the viable bacterial count at the site of subcutaneous inoculation but increased dissemination, as evidenced by quantitative cultures of blood and spleen (8). These studies support a critical role for macrophages in the control of GAS infection.
However, other studies suggest that GAS virulence determinants may target macrophages and reduce their effectiveness in bacterial clearance. A study of clinical specimens from patients with invasive soft tissue infections suggested that GAS can overcome macrophage-mediated bactericidal mechanisms and survive within macrophages for prolonged periods at sites of severe invasive infection (6). Another study found that M type 1 GAS but not an M protein-negative mutant, survived for at least several hours in macrophages and that viable GAS bacteria were released from infected cells (9). These results imply not only that virulent strains of GAS may resist killing by macrophages but also that macrophages could provide a sanctuary site for intracellular survival and a vehicle for dissemination of infection to the blood, to deep tissues, or to visceral organs.
GAS bacteria secrete several extracellular factors that are thought to contribute to pathogenesis (1, 10). Among these is streptolysin O (SLO), a member of the large family of pore-forming toxins known as cholesterol-dependent cytolysins (CDCs). CDCs share the property of binding to cholesterol-containing membranes, where they oligomerize and insert themselves to form large pores (11–13). CDCs are produced by many Gram-positive bacteria, and some of them have been characterized as important virulence factors (14, 15). Studies from our laboratory have shown that SLO interferes with phagocytosis of GAS by macrophages and contributes to both cytotoxic injury and GAS intracellular survival (16). Others have shown that SLO induces oncosis or apoptosis of macrophages (17, 18).
SLO is intimately associated with another toxin, NAD-glycohydrolase (NADase), a secreted enzyme that catalyzes the hydrolysis of NAD+ to nicotinamide and adenosine diphosphoribose (19). The slo gene forms part of an operon together with nga and ifs, which encode NADase and its intracellular inhibitor IFS, respectively (20, 21). SLO is also functionally linked to NADase in that NADase translocation into epithelial cells after its secretion by extracellular GAS is dependent on SLO (22, 23). After internalization of GAS by keratinocytes, SLO damages the bacterium-containing vacuole and triggers targeted autophagy, a process through which the bacteria are surrounded by an autophagosome-like compartment (24, 25). SLO and NADase inhibit the fusion of this compartment with lysosomes, thereby enhancing GAS survival in keratinocytes (22, 23, 25, 26).
In view of the interdependence of SLO and NADase in the cell biology of GAS infection of epithelial cells, it seems likely that NADase also plays a key role in modulating cytotoxicity and intracellular survival in macrophages. Whether macrophages successfully control GAS infection or rather are exploited by GAS for intracellular survival, persistence, and dissemination likely depends on the effects of virulence factors such as SLO and NADase that regulate bacterial uptake, intracellular survival, cytolytic effects on the macrophage, and the eventual emergence of viable GAS in host tissues.
We investigated the roles of SLO and NADase in the interaction of GAS with human macrophages with the goal of understanding how GAS resists phagocytic killing during infection.
Using a virulent strain representative of the M1T1 clonal group widely implicated in invasive GAS disease, we found that both toxins play a critical role in GAS intracellular survival in infected macrophages by two mechanisms. SLO pore formation prevents acidification of the GAS-containing phagolysosome, thereby impairing effective bacterial killing. In addition, SLO-mediated translocation delivers NADase from the phagosome into the macrophage cytosol, where NADase augments the cytotoxic effects of SLO, presumably by inhibiting cellular repair mechanisms. Thus, synergistic effects of the two toxins mediate cytotoxic injury to infected macrophages and enhance GAS intracellular survival.
RESULTS
SLO and NADase promote GAS resistance to killing by macrophages.
To further characterize the roles of SLO and NADase in GAS interactions with macrophages, we compared the intracellular survival of wild-type strain 854 with that of several isogenic mutants. Strain 854 is a clinical isolate from a patient with invasive GAS infection and is representative of the globally disseminated M1T1 clone implicated in many such infections since the 1980s (27–29). We found that mutants deficient in SLO (the Δslo mutant) or NADase (the Δnga mutant) were impaired in intracellular survival in macrophages compared to the parent strain (Fig. 1A and B). To determine whether NADase enzymatic activity is required for optimal survival, we constructed an nga(G330D) mutant in which substitution of aspartic acid for glycine at position 330 reduces enzymatic activity by 5,000-fold, effectively abrogating NADase activity (25, 30). The mutant had survival rates lower than those of the wild-type strain and slightly higher than but not significantly different from those of the Δnga mutant in macrophages (Fig. 1C). This result confirms the requirement of NADase activity for optimal GAS survival in macrophages. Taken together, these data indicate that both SLO and NADase are required for optimal GAS resistance to killing by macrophages.
FIG 1 .

SLO and NADase are required for maximal survival of GAS in macrophages. Values are the mean numbers of CFU recovered immediately after 90 min of exposure to GAS (time zero) and 2 and 4 h later, expressed as a percentage of the value at time zero. Data for wild-type GAS strain 854 and the SLO-deficient Δslo mutant strain are shown in panel A and for clarity in panels B through F in comparison to those of individual mutant strains. Data represent the mean values ± the standard errors from at least four experiments performed in triplicate. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
In separate experiments, to further assess the effect of SLO and NADase expression on GAS interactions with macrophages, we quantified the lactate dehydrogenase (LDH) released by macrophages infected with 854 or its mutants as a marker of cytotoxicity. As shown in Fig. 2, LDH release by macrophages exposed to the wild-type strain increased over the course of infection, whereas cells exposed to the Δslo or Δnga mutant showed minimal cytotoxicity. The nga(G330D) mutant, which expresses enzymatically inactive NADase, also induced minimal cytotoxicity at time zero and at 2 h, with a slight increase at 4 h that may be explained by the low level of residual NADase activity of the mutant enzyme (30). These data indicate that both SLO and NADase contribute to GAS cytotoxicity for macrophages.
FIG 2 .
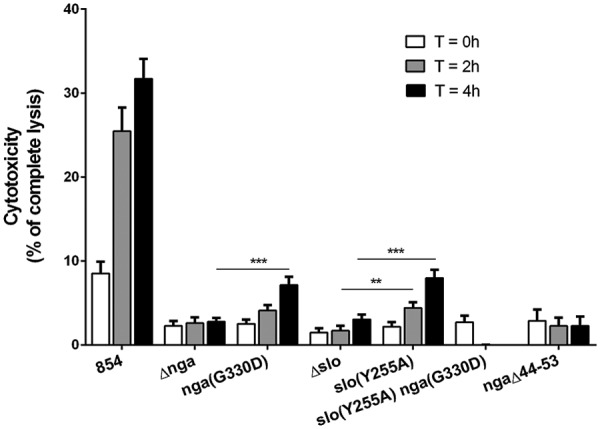
SLO and NADase mediate GAS cytotoxicity for macrophages. Cell culture supernatants were collected to measure the LDH released by macrophages at 0, 2, and 4 h after infection. Cytotoxicity is expressed as a percentage of the amount of LDH released by uninfected macrophages lysed with 10% Triton X-100. Shown are the mean values ± the standard errors of four independent experiments performed in triplicate. Values for all of the mutant strains were significantly different from those for strain 854 at the corresponding time points. Asterisks indicate other comparisons with statistically significant differences. **, P < 0.01; ***, P < 0.001.
SLO pore formation promotes cytotoxicity and GAS survival in macrophages.
The experiments described above implicate NADase as playing a central role in both the cytotoxic effects of GAS on macrophages and in GAS intracellular survival. Because NADase delivery into the host cell cytosol depends on SLO, we considered whether the role of SLO in cytotoxicity and intracellular survival might be simply to translocate NADase, rather than direct cellular injury due to SLO pore formation. To assess the contribution of SLO pore formation to cytotoxicity and intracellular survival, we constructed a SLO mutant that harbors a single amino acid substitution, slo(Y255A), that locks SLO in a prepore state (31). SLO(Y255A) retains the abilities to insert itself into cholesterol-containing membranes and to translocate NADase as efficiently as wild-type SLO, but it lacks pore-forming or hemolytic activity (25, 31). The slo(Y255A) mutant was less cytotoxic for macrophages than parent strain 854 but more cytotoxic than the Δslo mutant (Fig. 2). Similarly, results in Fig. 1D show an intermediate level of survival of the slo(Y255A) mutant compared to that of wild-type strain 854. These results indicate that both SLO pore formation and its role in delivery of NADase contribute to GAS intracellular survival and cytotoxicity in macrophages.
We inferred that the higher levels of cytotoxicity and intracellular survival observed in macrophages exposed to the slo(Y255A) mutant than in those exposed to the Δslo mutant were attributable to the intracellular enzymatic activity of NADase, which is delivered to cells infected with the slo(Y255A) mutant but not to those infected with the Δslo mutant. To test this hypothesis directly, we constructed a slo(Y255A) nga(G330D) double mutant that does not form SLO pores and expresses an inactive NADase. As expected, macrophages exposed to the double mutant were indistinguishable from those exposed to the Δslo mutant with respect to defective intracellular survival and the complete absence of cytotoxicity (Fig. 1E and 2, respectively). Taken together, the data indicate that both SLO pore formation and NADase activity are required for GAS to evade killing by macrophages.
SLO and NADase do not inhibit lysosomal fusion with GAS-containing phagosomes.
To better understand how GAS survives in macrophages, we investigated the mechanism by which SLO and NADase promote GAS resistance to phagocytic killing. After phagocytic uptake of a microbe, effective killing depends on the maturation of the phagosome through a series of membrane fusions with other vacuolar structures to convert the phagosome into a microbicidal organelle. Pathogens have evolved various strategies to counteract the host defense and to resist phagocytic killing. Among these is the arrest or reprogramming of phagosomal maturation and modification of or escape from the phagosome (32, 33). These mechanisms have been well studied for intracellular pathogens; however, less is known about the fate and the intracellular localization in macrophages of primarily extracellular pathogens such as GAS. We used confocal microscopy to investigate the intracellular localization of GAS in macrophages after phagocytosis. To determine whether the GAS-containing vacuole progresses through the classical stages of phagosome maturation, we examined GAS-infected macrophages for colocalization of the bacteria with lysosome-associated membrane protein 1 (LAMP-1), an abundant marker of late endosomes and lysosomes. As a positive control to assess the specificity of LAMP-1 staining and the functionality of the phagosome maturation pathway, we used macrophages exposed to heat-killed cells of GAS strain 854. As anticipated, more than 85% of the heat-killed bacteria colocalized with LAMP-1 at 1 h after infection (Fig. 3A and B). Unexpectedly, internalized wild-type and Δslo mutant bacteria were also associated with the late endosomal/lysosomal marker LAMP-1 at the same and later time points. This colocalization suggests that GAS bacteria are quickly trafficked to phagolysosomes and that SLO and NADase do not prevent fusion of the phagosome and LAMP-1-positive lysosomes.
FIG 3 .
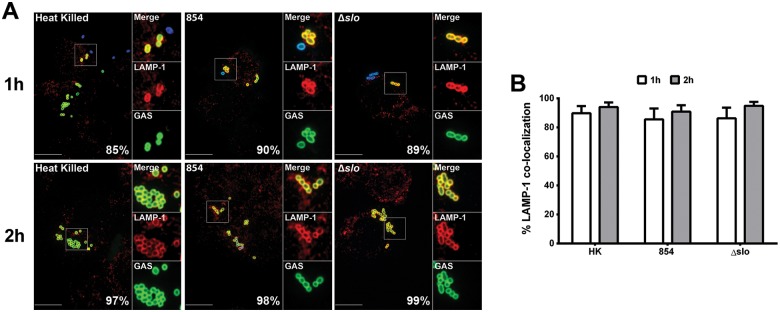
SLO and NADase do not prevent lysosome fusion to the GAS-containing phagosome. (A) Confocal microscopy images showing colocalization of LAMP-1 (Alexa 568, red) and heat-killed GAS strain 854 (HK), live 854, or the Δslo mutant (Alexa 488, green). Extracellular bacteria were stained both green and blue (Alexa 660). Representative images from one experiment are shown. Scale bars = 10 µm. The percentage of intracellular bacteria of each strain colocalized with LAMP-1 is indicated. (B) Percentage of intracellular GAS associated with LAMP-1 after 1 h (white bars) or 2 h (gray bars) of infection. Mean values ± the standard errors of at least three independent experiments are shown.
SLO prevents phagolysosomal acidification.
The greater intracellular survival of wild-type 854 than its isogenic mutants (Fig. 1) suggested that SLO and/or NADase interfere with phagolysosomal killing. However, as discussed above, wild-type GAS bacteria were rapidly trafficked to compartments marked by LAMP-1, i.e., compartments consistent with mature phagolysosomes. This finding was unanticipated because, in human keratinocytes, GAS production of SLO is associated with impaired fusion of the GAS-containing vacuole with lysosomes and failure to acidify this compartment for effective bacterial killing (25, 34, 35). As trafficking to a mature phagolysosome appeared to be intact in macrophages infected with wild-type GAS, we investigated the possibility that these GAS-containing compartments were not fully functional. The phagolysosome is biochemically defined by a markedly acidic pH (4.5 to 5) that is required for lysosomal hydrolase activation and for cargo degradation (36). We assessed the ability of bacterium-containing phagolysosomes to acidify by using the acidotropic probe LysoTracker red DND-99. We found that more than 40% of the intracellular Δslo mutant bacteria colocalized with the acidotropic probe after 1 h of infection, whereas only 12% of the wild-type GAS bacteria were associated with LysoTracker (Fig. 4). A similar pattern was observed after 2 h: 80% colocalization with LysoTracker for the Δslo mutant compared to 24% for the wild-type strain. These data indicate that the wild-type strain interferes with phagolysosome acidification and implicate SLO in this process.
FIG 4 .

SLO impairs acidification of GAS-containing phagolysosomes. (A) Confocal microscopy images demonstrating the association between the acidotropic probe LysoTracker Red DND-99 (red) and GFP-expressing GAS (green) at 1 and 2 h of infection. Extracellular GAS bacteria are also stained blue (Alexa 660). Representative images from one experiment are shown. Scale bars = 10 µm. The percentage of intracellular bacteria of each strain that colocalized with LysoTracker is shown. (B) Percentage of heat-killed (HK) or live GAS colocalized with LysoTracker at 1 h (white bars) or 2 h (gray bars) of infection. Mean values ± the standard errors of at least three independent experiments are shown. Black asterisks indicate values that differ significantly from that for strain 854; red asterisks indicates values that differ significantly from that for the Δslo mutant. *, P < 0.05; **, P < 0.01.
To further investigate the role of SLO and/or NADase in inhibiting phagolysosomal acidification, we examined the association of the nga(G330D) mutant with LysoTracker in infected macrophages. This mutant behaved similarly to the wild-type parent strain with respect to its failure to colocalize with LysoTracker, a result that suggests that SLO production, even in the absence of enzymatically active NADase, is sufficient to impair phagolysosomal acidification (Fig. 4). To test whether SLO pore formation is required to inhibit acidification, we examined cells infected with the non-pore-forming slo(Y255A) mutant. The slo(Y255A) mutant strain also was located predominantly in nonacidic compartments, similar to wild-type GAS (Fig. 4). Two hypotheses could explain this result. (i) Because SLO(Y255A) does not form pores but does insert itself into cell membranes as a prepore complex (31), it is possible that the prepore complexes might perturb membrane integrity sufficiently to disrupt the proton gradient responsible for acidification of the phagolysosome. (ii) Alternatively, NADase produced by the slo(Y255A) mutant could be responsible for inhibiting phagolysosome acidification by depleting NAD+ and, secondarily, ATP, which is required to power the vacuolar proton-ATPase pumps that establish the proton gradient. To distinguish between these hypotheses, we examined cells infected with the nga(G330D) slo(Y255A) double mutant, which expresses non-pore-forming SLO and enzymatically inactive NADase (Fig. 4). We found that this mutant was also located in nonacidic vacuoles, suggesting that NADase is not required for inhibition of acidification and that insertion of SLO(Y255A) is sufficient to disrupt the phagolysosomal proton gradient.
SLO pore formation damages the phagolysosomal membrane.
In light of our findings that SLO is required to inhibit phagolysosome acidification, it is likely that SLO secreted by phagocytosed GAS forms pores in the phagolysosomal membrane and disrupts the proton gradient by permitting diffusion of solutes across the damaged membrane. To further assess membrane damage in GAS-containing phagolysosomes, we immunostained infected macrophages for galectin 8. Galectin 8 is a cytosolic lectin that binds galactosides normally located on the interior surface of vacuolar membranes. Damage to the vacuolar membrane can expose these galactosides to the cytosol, thereby promoting galectin 8 binding to the damaged vacuole (37). As a negative control, we used macrophages containing heat-killed wild-type bacteria that are expected not to damage the phagolysosome. As expected, compartments containing heat-killed GAS did not associate with galectin 8. Similar negative results were obtained with cells infected with live organisms of the Δslo mutant strain, consistent with the location of these bacteria in intact, fully acidified phagolysosomes (Fig. 5). In contrast, more than 50% of the intracellular wild-type GAS bacteria were associated with galectin 8 by 1 h of infection. These data support a model in which SLO pores damage the phagolysosomal membrane, thereby disabling phagolysosome acidification (Fig. 6). We observed an intermediate level of galectin 8 binding to phagolysosomes containing the nga(G330D) mutant, which produces SLO and enzymatically inactive NADase (28% at 1 h, 18% at 2 h). This result implies that NADase contributes to SLO-mediated injury to the phagosomal membrane by inhibiting cellular repair mechanisms. Phagolysosomes containing the slo(Y255A) mutant accumulated slightly, though not significantly, greater amounts of galectin 8 than those containing the Δslo mutant. The fact that infection with the slo(Y255A) mutant impairs phagolysosomal acidification but has minimal effects on galectin 8 binding implies that damage to the vacuolar membrane caused by insertion of the SLO(Y255A) prepore complex is sufficient to at least partially disrupt the proton gradient but insufficient to expose inner membrane galactosides to the cytosol. Together, these results implicate NADase as contributing to SLO-mediated cytotoxicity by interfering with the cellular repair of SLO-mediated membrane damage and possibly by inhibiting the vacuolar proton-ATPase.
FIG 5 .

SLO mediates membrane damage to GAS-containing phagolysosomes. (A) Confocal microscopy images of the association between GAS (Alexa 568, red) and galectin 8 (Alexa 488, green) during the infection of macrophages. Extracellular GAS bacteria are both red and blue (Alexa 568 and 660, respectively). Images representative of one experiment are shown. Scale bars = 10 µm. The percentage of intracellular bacteria of each strain associated with galectin 8 is indicated. (B) Percentage of colocalization of GAS and galectin 8 after 1 h (white bars) or 2 h (gray bars) of infection. Mean values ± the standard errors of at least three independent experiments are shown. Black asterisks indicate values that differ significantly from that of strain 854, and red asterisks indicate values that differ significantly from that of the Δslo mutant strain. *, P < 0.05; **, P < 0.01; ***, P < 0.001. HK, heat killed.
FIG 6 .
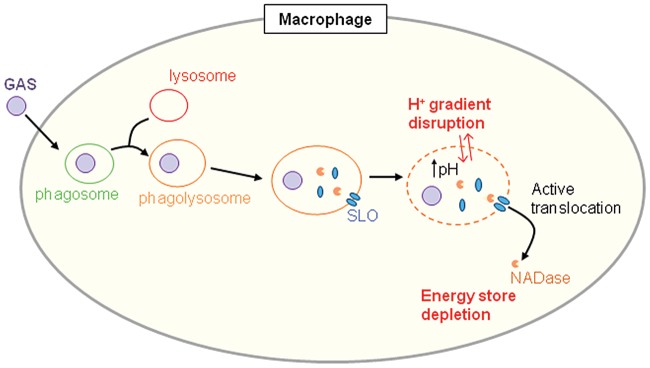
Model of GAS survival in macrophages. GAS is internalized by phagocytosis. The GAS-containing phagosome fuses with lysosomes to form a phagolysosome. SLO and NADase are secreted by GAS into this compartment. SLO damages the phagolysosome membrane, preventing phagolysosome acidification. NADase is translocated by SLO from the phagolysosome into the macrophage cytosol, where it depletes NAD+ and ATP, thereby inhibiting cellular repair of SLO-mediated membrane damage. The combined action of SLO and NADase interferes with phagolysosomal killing and promotes GAS intracellular survival.
Delivery of NADase to the macrophage cytosol depends on SLO-mediated translocation rather than passive diffusion through SLO pores.
NADase intoxication of epithelial cells is thought to occur by SLO-mediated translocation of NADase from the extracellular space across the cell membrane (22, 23). In contrast, in macrophages, SLO-mediated cytotoxicity depends on internalization of the bacteria, an observation that indicates that the source of NADase intoxication of macrophages is intracellular bacteria contained within phagosomes (16). Accordingly, we were interested in determining whether delivery of NADase from GAS contained within phagosomes occurs by specific SLO-mediated translocation or by simple diffusion through SLO pores in the phagosomal membrane. To distinguish between these possibilities, we constructed a mutant of strain 854 that expresses NADase harboring a 10-amino-acid (aa) deletion in the N-terminal domain that is required for SLO-mediated translocation (38). The ngaΔ44-53 mutant strain secreted the same amounts of NADase activity as wild-type 854 (see Fig. S1 in the supplemental material). However, the mutant was impaired both in induction of macrophage cytotoxicity and in intracellular survival to a degree similar to that of mutants lacking enzymatically active NADase (Fig. 1F and 2). These results imply that delivery of NADase from the phagosome depends on SLO-mediated translocation rather than simple diffusion through SLO pores in the vacuolar membrane.
DISCUSSION
Macrophages constitute an important element of innate immunity to bacterial infections, including those due to GAS. By phagocytosing bacteria, these cells limit bacterial proliferation, and upon activation, they secrete inflammatory cytokines that recruit and activate other effector cells, including neutrophils. The success of GAS as a pathogen depends in part on its ability to evade capture and killing by macrophages. Production of the secreted CDC SLO is strongly associated with the capacity of GAS to incapacitate macrophage function: SLO-producing GAS bacteria induce apoptosis of murine and human macrophages in vitro and resist macrophage-mediated bacterial killing, in contrast to isogenic SLO-deficient mutants (16, 18). We now find that SLO secreted by phagocytosed bacteria damages GAS-containing phagolysosomes. In addition, SLO mediates the translocation of the cotoxin NADase into the macrophage cytosol. Both toxins contribute to GAS-induced macrophage cytotoxicity and enhance GAS survival.
In human epithelial keratinocytes exposed to GAS in vitro, SLO and NADase production is associated with increased GAS intracellular survival (25, 34). Both toxins prevent maturation of the GAS-containing vacuole to a functional bactericidal compartment by inhibiting fusion with lysosomes (25, 34, 35). We anticipated that enhanced survival of SLO- and NADase-producing GAS in macrophages would reflect a similar mechanism. Unexpectedly, we found that phagosomes containing wild-type GAS followed the same maturation pathway as phagosomes containing SLO- or NADase-deficient mutants, readily fusing with LAMP-1-positive lysosomes. Hertzén et al. also found that GAS could survive in human macrophages (9). In contrast to our results, their study found that GAS expression of the M1 protein was associated with failure of the GAS-containing phagosome to fuse with lysosomes. Possible reasons for the discrepancy in GAS intracellular trafficking between that study and ours could be differences in the GAS strains used, the macrophages, or their preparation or different experimental methodologies. In our studies, phagosomes containing wild-type GAS fused with lysosomes; however, SLO appeared to be highly effective in preventing phagolysosomal acidification, as assessed by localization of the acidotropic probe LysoTracker. Whereas heat-killed GAS or live SLO-deficient bacteria were efficiently trafficked to compartments that had the acidic pH associated with mature phagolysosomes, phagolysosomes containing wild-type GAS failed to acidify. Furthermore, this SLO-associated inhibition of phagolysosomal acidification did not require complete pore formation, as we observed failure of acidification in macrophages infected with a strain expressing SLO(Y255A), a mutant protein that oligomerizes and inserts itself into cholesterol-containing membranes but does not form pores, lyse erythrocytes, or produce overt cytotoxicity. The sublytic membrane disruption caused by SLO(Y255A) was sufficient to defeat phagolysosomal acidification.
Production of SLO prevented phagolysosomal acidification but was insufficient to support maximal GAS survival in the absence of enzymatically active NADase. Experiments with galectin 8 binding to evaluate damage to GAS-containing vacuoles revealed that maximal damage occurred in phagolysosomes that contained wild-type GAS. Damage was reduced in cells infected with GAS that produced wild-type SLO but enzymatically inactive NADase and was minimal in cells infected with SLO-deficient GAS or with GAS producing SLO(Y255A). The fact that NADase production was associated with increased phagolysosomal membrane damage is compatible with previous observations that NADase depletes intracellular NAD+ and ATP in epithelial cells exposed to GAS (26). Such depletion of cellular energy stores is predicted to inhibit cellular repair of SLO pores in the phagolysosomal membrane. The contribution of NADase to phagolysosomal membrane damage correlates with GAS survival: optimal survival was observed only in GAS producing both wild-type SLO and enzymatically active NADase.
The finding that NADase contributes to GAS evasion of killing by macrophages raised the question of how NADase is delivered to the macrophage cytosol during the bacterium-macrophage encounter. NADase intoxication of epithelial cells is thought to reflect binding of GAS to the cell surface, secretion of NADase and SLO, association of the two toxins on the cell membrane, and SLO-mediated translocation of NADase into the host cell. The situation is quite different for macrophages: GAS bacteria are efficiently phagocytosed, and SLO and NADase are secreted into the GAS-containing phagosome. Because SLO damages the phagolysosomal membrane, it seemed possible that NADase could diffuse passively through SLO pores into the macrophage cytosol. However, GAS bacteria that produced enzymatically active NADase with an N-terminal deletion that prevents SLO-mediated translocation were defective in intracellular survival, a result that implies that active SLO-mediated translocation is required to deliver NADase from the phagosome into the cytosol.
These results support a model of GAS evasion of macrophage killing that depends on both SLO and its cotoxin NADase. While SLO and NADase have been shown to enhance GAS intracellular survival in epithelial cells, the present data identify important differences in the mechanisms through which the toxins modulate GAS survival in macrophages. In particular, NADase intoxication of macrophages appears to occur predominantly or exclusively from intracellular GAS contained within phagosomes or phagolysosomes. SLO secreted by GAS within these compartments does not prevent the fusion of GAS-containing vacuoles with lysosomes, as occurs in keratinocytes. However, SLO damages the phagolysosomal membrane and interferes with the development or maintenance of an acidic intraluminal pH. Despite the presence of SLO-mediated damage to the phagolysosomal membrane, translocation of NADase into the macrophage cytosol does not occur by passive diffusion but requires SLO-mediated translocation. We conclude that the coordinated action of SLO and NADase disables macrophage bactericidal mechanisms and mediates GAS evasion of this key effector of host innate immunity. The ability of GAS to escape macrophage killing favors persistence of the organism in colonization of the human pharynx and may be particularly important in invasive infection where clinical isolates often harbor spontaneous inactivating mutations in the CsrRS or RopB (Rgg) regulatory system, resulting in marked upregulation of both SLO and NADase (39–41).
MATERIALS AND METHODS
Bacterial strains, macrophage cell line, and growth conditions.
The GAS strains used in this study are listed in Table 1. GAS 854 is an M type 1 strain isolated from a patient with a retroperitoneal abscess (42). GAS strains were grown in Todd-Hewitt broth (Difco) supplemented with 0.5% yeast extract (THY) at 37°C without shaking or on THY agar supplemented with 5% defibrinated sheep blood (BD Biosciences). For cloning experiments, Escherichia coli strain DH5α was grown in Luria-Bertani broth (Difco) at 37°C. When required, antibiotics were added to the medium at the following concentrations: for E. coli, erythromycin at 100 µg/ml, spectinomycin at 100 µg/ml, and carbenicillin at 100 µg/ml; for GAS, penicillin at 20 µg/ml, gentamicin at 200 µg/ml, spectinomycin at 100 µg/ml, and erythromycin at 1 µg/ml. THP-1 human monocyte-macrophage cells were purchased from the American Type Culture Collection. Monocytes were cultured in RPMI medium (Life Technologies) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Life Technologies) at 37°C in 5% CO2.
TABLE 1 .
Bacterial strains used in this study
| Strain | Genotype/relevant properties | Reference |
|---|---|---|
| 854 | Wild-type, M type 1, invasive clinical isolate | 45 |
| 854 Δnga | nga deletion mutant | This study |
| 854 nga(G330D) | G330D point mutant of NADase, lacks NADase activity | This study |
| 854 Δslo | slo deletion mutant | This study |
| 854 slo(Y255A) | SLO Y255A point mutant that lacks SLO pore formation but retains NADase translocation | This study |
| 854 slo(Y255A) nga(G330D) | Double point mutant that lacks SLO pore formation and NADase activity | This study |
| 854 ngaΔ44-53 | Deletion of 10-aa region of NADase translocation domain that preserves enzymatic activity but prevents NADase translocation | This study |
| 854/phasp-gfp | 854 expressing GFP from plasmid phasp-gfp, Spcra | This study |
| 854 Δnga/phasp-gfp | 854 Δnga mutant expressing GFP from plasmid phasp-gfp, Spcr | This study |
| 854 nga(G330D)/phasp-gfp | 854 nga(G330D) mutant expressing GFP from plasmid phasp-gfp, Spcr | This study |
| 854 Δslo/phasp-gfp | 854 Δslo mutant expressing GFP from plasmid phasp-gfp, Spcr | This study |
| 854 slo(Y255A)/phasp-gfp | 854 slo(Y255A) mutant expressing GFP from plasmid phasp-gfp, Spcr | This study |
| 854 slo(Y255A) nga(G330D)/phasp-gfp | 854 slo(Y255A) nga(G330D) double mutant expressing GFP from plasmid phasp-gfp, Spcr | This study |
Spcr, spectinomycin resistance.
Reagents.
Restriction enzymes were purchased from Thermo Scientific, and cell culture reagents were from Life Technologies. Unless otherwise specified, reagents were purchased from Sigma-Aldrich. Phorbol 12-myristate 13-acetate (PMA) was diluted in dimethyl sulfoxide and stored at −20°C (16). The primary antibodies (rabbit anti-GAS group A carbohydrate antibody conjugated to Alexa Fluor 660, 488, or 568; mouse anti-LAMP-1 antibody; anti-galectin 8 affinity-purified goat polyclonal IgG) and secondary antibodies (conjugated to Alexa Fluor 488 or 568) used were previously described (25, 35). LysoTracker Red DND-99 was purchased from Molecular Probes/Life Technologies and freshly diluted in RPMI medium containing 10% FBS prior to use.
DNA manipulations.
To generate the G330D substitution in NADase and the Y255A substitution in SLO, GAS strain 854 was transformed by electroporation with plasmids pJRS233-ngaG330D and pJRS233-sloY255A, respectively (25). Transformants were then subjected to allelic exchange to replace the wild-type gene with the mutated allele as previously described (43). Plasmids pngaΔ (22) and pJRS233-ngaG330D, respectively, were used to delete nga from 854 and to introduce the nga(G330D) mutation into the slo(Y255A) mutant to create the slo(Y255A) nga(G330D) double mutant. The Δslo mutant was generated in a previous study (16).
To construct the NADase translocation ngaΔ44-53 mutant, a plasmid expressing ifs and nga carrying a deletion of 30 bp, from position 129 to position 159, was synthesized by Genewiz, Inc. This mutation introduces a deletion of amino acids 44 to 53 into the N-terminal translocation domain of NADase. Oligonucleotide primers BBS97 and BBS4 (GGATCCGTGTTTACAAACCAATGGATG and GTCGACCCCTGATGGACCTCTGTTACC, respectively) were used to amplify the nga DNA fragment carrying the deletion. The resulting 1-kb fragment was subsequently digested with BamHI and SalI and ligated into pJRS233. The newly formed pJRS233-ngaΔ44-53 plasmid was then transformed into strain 854. Transformants were then subjected to allelic gene replacement (43).
Cytotoxicity and survival assays.
Macrophage cytotoxicity was assessed by LDH release measurements. THP-1 cells were seeded into 24-well plates and differentiated into macrophages with PMA as previously described (16). Infections were performed as previously described, with the following modifications (16). GAS bacteria were grown in THY to an A600 of 0.3 to 0.4, collected by centrifugation, washed with phosphate-buffered saline (PBS), and resuspended and diluted in RPMI medium with 10% FBS. Macrophages were washed once and infected with GAS at a multiplicity of infection (MOI) of 4. After 90 min of infection, penicillin and gentamicin were added to the medium and kept throughout the experiment to kill extracellular bacteria. Thirty minutes (T0), 2 h (T2), or 4 h (T4) later, supernatants were collected for LDH measurements with a cytotoxicity kit from Roche. Uninfected cells lysed with 10% Triton X-100 were used as a standard of complete cell lysis.
Bacterial survival assays were performed as previously described (16). Briefly, macrophages were infected with GAS for 90 min. After infection, extracellular bacteria were killed by the addition of antibiotics to the medium. Macrophages were then washed and returned to antibiotic-free medium for 2 or 4 h. To determine the percentage of survival, bacteria that survived intracellularly and bacteria that escaped from macrophages were quantified by quantitative culture of macrophage lysates and cell culture medium. Survival was calculated as follows: (CFU recovered at 2 or 4 h/CFU recovered at time zero) × 100%.
Immunofluorescence.
THP-1 monocytes were seeded onto coverslips in 24-well plates at 1 × 106/ml in RPMI medium with 10% FBS in the presence of 25 nM PMA to induce their differentiation into macrophages. After a 24-h incubation at 37°C in 5% CO2, cells were washed three times with PBS and then incubated in fresh RPMI medium containing 10% FBS for an additional 24 h prior to infection. GAS strains were grown in THY until the early exponential phase (A600, ~0.25), collected by centrifugation, washed once in PBS, and resuspended in 1 ml of RPMI medium supplemented with 10% FBS prior to infection. Macrophages were infected at a MOI of 10 for 1 h. After infection, samples were washed in PBS and extracellular bacteria were stained with Alexa Fluor 660-conjugated anti-group A carbohydrate antibody for 15 min. Samples were then either immediately subjected to the immunostaining protocol described below or treated first with an antibiotic mixture (penicillin at 20 µg/ml and gentamicin at 200 µg/ml) to kill extracellular bacteria, washed three times with PBS, and incubated for an additional hour postinfection at 37°C in 5% CO2 before immunostaining. Unless otherwise specified, all of the following steps were performed in the dark at room temperature. Infected cells were fixed with 2% paraformaldehyde (PFA) in PBS (pH 7.4) for 30 min. Cells were then washed three times with PBS, permeabilized for 15 min with 0.1% Triton X-100 in PBS, washed again three times in PBS, and blocked for 30 min in PBS containing 0.5% bovine serum albumin (BSA). Cells were then incubated for 1 h with the primary (mouse anti-LAMP-1 or goat anti-galectin 8) antibody diluted in 0.5% BSA in PBS. After three washes in PBS, cells were incubated for 1 h with Alexa Fluor 488- or 568-conjugated rabbit anti-GAS group A carbohydrate and either goat anti-mouse Alexa Fluor 568-conjugated IgG or donkey anti-goat Alexa Fluor 488-conjugated IgG. Slides were mounted in ProLong Gold medium (Molecular Probes) and incubated for 16 to 24 h at room temperature in the dark before imaging.
LysoTracker Red labeling.
Cells were infected as described above with GAS transformed with plasmid phasp-gfp, which encodes green fluorescent protein (GFP), as previously described (35, 44). Cells were incubated with 50 nM LysoTracker Red DND-99 freshly diluted in RPMI medium plus 10% FBS for 5 to 10 min after either 1 or 2 h of infection. Cells were then washed and fixed with 2% PFA. Coverslips were then immediately mounted in Prolong gold medium and incubated for 16 to 24 h at room temperature in the dark before confocal imaging.
Confocal microscopy.
Confocal microscopy was performed at the Harvard Digestive Diseases Center core facility as previously described (25, 35). Image acquisition and analysis were performed with Slidebook 5 (Intelligent Imaging Innovations, Denver, CO). To quantify the percentage of GAS colocalization with cell markers, three to five independent experiments were performed and at least 300 intracellular bacteria were counted for each experiment.
Statistical analysis.
Data are expressed as mean values ± the standard errors of the means. The statistical significance of differences between groups was evaluated by two-tailed Student t test with GraphPad Prism. A P value of ≤0.05 was considered statistically significant.
SUPPLEMENTAL MATERIAL
The NADase translocation ngaΔ44-53 mutant strain secretes the same amount of NADase activity as wild-type strain 854. Shown are the mean NADase activities ± the standard errors of culture supernatants from the strains indicated. No activity was detected in supernatants from the NADase deletion Δnga mutant or the enzymatically inactive nga(G330D) mutant. Download
ACKNOWLEDGMENTS
This work was supported in part by Public Health Service grant AI070926 from the National Institute of Allergy and Infectious Diseases.
We thank Maghnus O’Seaghdha for technical advice and helpful discussions and Ramiro Massol and the Harvard Digestive Diseases Imaging Core for assistance with confocal microscopy.
Footnotes
Citation Bastiat-Sempe B, Love JF, Lomayesva N, Wessels MR. 2014. Streptolysin O and NAD-glycohydrolase prevent phagolysosome acidification and promote group A Streptococcus survival in macrophages. mBio 5(5):e01690-14. doi:10.1128/mBio.01690-14.
REFERENCES
- 1. Cunningham MW. 2000. Pathogenesis of group A streptococcal infections. Clin. Microbiol. Rev. 13:470–511. 10.1128/CMR.13.3.470-511.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Osterlund A, Popa R, Nikkilä T, Scheynius A, Engstrand L. 1997. Intracellular reservoir of Streptococcus pyogenes in vivo: a possible explanation for recurrent pharyngotonsillitis. Laryngoscope 107:640–647. 10.1097/00005537-199705000-00016 [DOI] [PubMed] [Google Scholar]
- 3. Spinaci C, Magi G, Varaldo PE, Facinelli B. 2006. Persistence of erythromycin-resistant group A streptococci in cultured respiratory cells. Pediatr. Infect. Dis. J. 25:880–883. 10.1097/01.inf.0000238136.63851.4a [DOI] [PubMed] [Google Scholar]
- 4. Rohde M, Chhatwal GS. 2013. Adherence and invasion of streptococci to eukaryotic cells and their role in disease pathogenesis. Curr. Top. Microbiol. Immunol. 368:83–110. 10.1007/82_2012_281 [DOI] [PubMed] [Google Scholar]
- 5. Medina E, Rohde M, Chhatwal GS. 2003. Intracellular survival of Streptococcus pyogenes in polymorphonuclear cells results in increased bacterial virulence. Infect. Immun. 71:5376–5380. 10.1128/IAI.71.9.5376-5380.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Thulin P, Johansson L, Low DE, Gan BS, Kotb M, McGeer A, Norrby-Teglund A. 2006. Viable group A streptococci in macrophages during acute soft tissue infection. PLoS Med. 3(3):e53. 10.1371/journal.pmed.0030053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Goldmann O, Rohde M, Chhatwal GS, Medina E. 2004. Role of macrophages in host resistance to group A streptococci. Infect. Immun. 72:2956–2963. 10.1128/IAI.72.5.2956-2963.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mishalian I, Ordan M, Peled A, Maly A, Eichenbaum MB, Ravins M, Aychek T, Jung S, Hanski E. 2011. Recruited macrophages control dissemination of group A streptococcus from infected soft tissues. J. Immunol. 187:6022–6031. 10.4049/jimmunol.1101385 [DOI] [PubMed] [Google Scholar]
- 9. Hertzén E, Johansson L, Wallin R, Schmidt H, Kroll M, Rehn AP, Kotb M, Mörgelin M, Norrby-Teglund A. 2010. M1 protein-dependent intracellular trafficking promotes persistence and replication of Streptococcus pyogenes in macrophages. J. Innate Immun. 2:534–545. 10.1159/000317635 [DOI] [PubMed] [Google Scholar]
- 10. Kwinn LA, Nizet V. 2007. How group A streptococcus circumvents host phagocyte defenses. Future Microbiol. 2:75–84. 10.2217/17460913.2.1.75 [DOI] [PubMed] [Google Scholar]
- 11. Alouf JE. 2000. Bacterial protein toxins. An overview. Methods Mol. Biol 145:1–26. 10.1385/1-59259-052-7:1 [DOI] [PubMed] [Google Scholar]
- 12. Bhakdi S, Tranum-Jensen J, Sziegoleit A. 1985. Mechanism of membrane damage by streptolysin-O. Infect. Immun. 47:52–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Palmer M. 2001. The family of thiol-activated, cholesterol-binding cytolysins. Toxicon 39:1681–1689. 10.1016/S0041-0101(01)00155-6 [DOI] [PubMed] [Google Scholar]
- 14. Hamon MA, Ribet D, Stavru F, Cossart P. 2012. Listeriolysin O: the Swiss army knife of Listeria. Trends Microbiol. 20:360–368. 10.1016/j.tim.2012.04.006 [DOI] [PubMed] [Google Scholar]
- 15. Marriott HM, Mitchell TJ, Dockrell DH. 2008. Pneumolysin: a double-edged sword during the host-pathogen interaction. Curr. Mol. Med. 8:497–509. 10.2174/156652408785747924 [DOI] [PubMed] [Google Scholar]
- 16. Love JF, Tran-Winkler HJ, Wessels MR. 2012. Vitamin D and the human antimicrobial peptide LL-37 enhance group A streptococcus resistance to killing by human cells. mBio 3(5):e00394-12. 10.1128/mBio.00394-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Goldmann O, Sastalla I, Wos-Oxley M, Rohde M, Medina E. 2009. Streptococcus pyogenes induces oncosis in macrophages through the activation of an inflammatory programmed cell death pathway. Cell. Microbiol. 11:138–155. 10.1111/j.1462-5822.2008.01245.x [DOI] [PubMed] [Google Scholar]
- 18. Timmer AM, Timmer JC, Pence MA, Hsu LC, Ghochani M, Frey TG, Karin M, Salvesen GS, Nizet V. 2009. Streptolysin O promotes group A streptococcus immune evasion by accelerated macrophage apoptosis. J. Biol. Chem. 284:862–871. 10.1074/jbc.M804632200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Karasawa T, Yamakawa K, Tanaka D, Gyobu Y, Nakamura S. 1995. NAD(+)-glycohydrolase productivity of haemolytic streptococci assayed by a simple fluorescent method and its relation to T serotype. FEMS Microbiol. Lett. 128:289–292. 10.1111/j.1574-6968.1995.tb07538.x [DOI] [PubMed] [Google Scholar]
- 20. Kimoto H, Fujii Y, Hirano S, Yokota Y, Taketo A. 2006. Genetic and biochemical properties of streptococcal NAD-glycohydrolase inhibitor. J. Biol. Chem. 281:9181–9189. 10.1074/jbc.M506879200 [DOI] [PubMed] [Google Scholar]
- 21. Meehl MA, Pinkner JS, Anderson PJ, Hultgren SJ, Caparon MG. 2005. A novel endogenous inhibitor of the secreted streptococcal NAD-glycohydrolase. PLoS Pathog. 1:e35. 10.1371/journal.ppat.0010035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bricker AL, Cywes C, Ashbaugh CD, Wessels MR. 2002. NAD+-glycohydrolase acts as an intracellular toxin to enhance the extracellular survival of group A streptococci. Mol. Microbiol. 44:257–269. 10.1046/j.1365-2958.2002.02876.x [DOI] [PubMed] [Google Scholar]
- 23. Madden JC, Ruiz N, Caparon M. 2001. Cytolysin-mediated translocation (CMT): a functional equivalent of type III secretion in Gram-positive bacteria. Cell 104:143–152. 10.1016/S0092-8674(01)00198-2 [DOI] [PubMed] [Google Scholar]
- 24. Nakagawa I, Amano A, Mizushima N, Yamamoto A, Yamaguchi H, Kamimoto T, Nara A, Funao J, Nakata M, Tsuda K, Hamada S, Yoshimori T. 2004. Autophagy defends cells against invading group A streptococcus. Science 306:1037–1040. 10.1126/science.1103966 [DOI] [PubMed] [Google Scholar]
- 25. O’Seaghdha M, Wessels MR. 2013. Streptolysin O and its co-toxin NAD-glycohydrolase protect group A streptococcus from xenophagic killing. PLoS Pathog. 9(6):e1003394. 10.1371/journal.ppat.1003394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Michos A, Gryllos I, Håkansson A, Srivastava A, Kokkotou E, Wessels MR. 2006. Enhancement of streptolysin O activity and intrinsic cytotoxic effects of the group A streptococcal toxin, NAD-glycohydrolase. J. Biol. Chem. 281:8216–8223. 10.1074/jbc.M511674200 [DOI] [PubMed] [Google Scholar]
- 27. Aziz RK, Kotb M. 2008. Rise and persistence of global M1T1 clone of Streptococcus pyogenes. Emerg. Infect. Dis. 14:1511–1517. 10.3201/eid1410.071660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cleary PP, Kaplan EL, Handley JP, Wlazlo A, Kim MH, Hauser AR, Schlievert PM. 1992. Clonal basis for resurgence of serious Streptococcus pyogenes disease in the 1980s. Lancet 339:518–521. 10.1016/0140-6736(92)90339-5 [DOI] [PubMed] [Google Scholar]
- 29. Musser JM, Kapur V, Szeto J, Pan X, Swanson DS, Martin DR. 1995. Genetic diversity and relationships among Streptococcus pyogenes strains expressing serotype M1 protein: recent intercontinental spread of a subclone causing episodes of invasive disease. Infect. Immun. 63:994–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chandrasekaran S, Ghosh J, Port GC, Koh EI, Caparon MG. 2013. Analysis of polymorphic residues reveals distinct enzymatic and cytotoxic activities of the Streptococcus pyogenes NAD+ glycohydrolase. J. Biol. Chem. 288:20064–20075. 10.1074/jbc.M113.481556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Magassa N, Chandrasekaran S, Caparon MG. 2010. Streptococcus pyogenes cytolysin-mediated translocation does not require pore formation by streptolysin O. EMBO Rep. 11:400–405. 10.1038/embor.2010.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Flannagan RS, Cosío G, Grinstein S. 2009. Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat. Rev. Microbiol. 7:355–366. 10.1038/nrmicro2128 [DOI] [PubMed] [Google Scholar]
- 33. Flannagan RS, Jaumouillé V, Grinstein S. 2012. The cell biology of phagocytosis. Annu. Rev. Pathol. 7:61–98. 10.1146/annurev-pathol-011811-132445 [DOI] [PubMed] [Google Scholar]
- 34. Håkansson A, Bentley CC, Shakhnovic EA, Wessels MR. 2005. Cytolysin-dependent evasion of lysosomal killing. Proc. Natl. Acad. Sci. U. S. A. 102:5192–5197. 10.1073/pnas.0408721102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Logsdon LK, Håkansson AP, Cortés G, Wessels MR. 2011. Streptolysin O inhibits clathrin-dependent internalization of group A streptococcus. mBio 2(1):e00332-00310. 10.1128/mBio.00332-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yates RM, Russell DG. 2005. Phagosome maturation proceeds independently of stimulation of Toll-like receptors 2 and 4. Immunity 23:409–417. 10.1016/j.immuni.2005.09.007 [DOI] [PubMed] [Google Scholar]
- 37. Thurston TL, Wandel MP, von Muhlinen N, Foeglein A, Randow F. 2012. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 482:414–418. 10.1038/nature10744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ghosh J, Caparon MG. 2006. Specificity of Streptococcus pyogenes NAD(+) glycohydrolase in cytolysin-mediated translocation. Mol. Microbiol. 62:1203–1214. 10.1111/j.1365-2958.2006.05430.x [DOI] [PubMed] [Google Scholar]
- 39. Ikebe T, Ato M, Matsumura T, Hasegawa H, Sata T, Kobayashi K, Watanabe H. 2010. Highly frequent mutations in negative regulators of multiple virulence genes in group A streptococcal toxic shock syndrome isolates. PLoS Pathog. 6(4):e1000832. 10.1371/journal.ppat.1000832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tran-Winkler HJ, Love JF, Gryllos I, Wessels MR. 2011. Signal transduction through CsrRS confers an invasive phenotype in group A streptococcus. PLoS Pathog. 7(10):e1002361. 10.1371/journal.ppat.1002361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Treviño J, Perez N, Ramirez-Peña E, Liu Z, Shelburne SA, III, Musser JM, Sumby P. 2009. CovS simultaneously activates and inhibits the CovR-mediated repression of distinct subsets of group A streptococcus virulence factor-encoding genes. Infect. Immun. 77:3141–3149. 10.1128/IAI.01560-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gryllos I, Grifantini R, Colaprico A, Cary ME, Hakansson A, Carey DW, Suarez-Chavez M, Kalish LA, Mitchell PD, White GL, Wessels MR. 2008. PerR confers phagocytic killing resistance and allows pharyngeal colonization by group A streptococcus. PLoS Pathog. 4(9):e1000145. 10.1371/journal.ppat.1000145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ashbaugh CD, Warren HB, Carey VJ, Wessels MR. 1998. Molecular analysis of the role of the group A streptococcal cysteine protease, hyaluronic acid capsule, and M protein in a murine model of human invasive soft-tissue infection. J. Clin. Invest. 102:550–560. 10.1172/JCI3065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gryllos I, Cywes C, Shearer MH, Cary M, Kennedy RC, Wessels MR. 2001. Regulation of capsule gene expression by group A streptococcus during pharyngeal colonization and invasive infection. Mol. Microbiol. 42:61–74. 10.1046/j.1365-2958.2001.02635.x [DOI] [PubMed] [Google Scholar]
- 45. Gryllos I, Tran-Winkler HJ, Cheng MF, Chung H, Bolcome R, III, Lu W, Lehrer RI, Wessels MR. 2008. Induction of group A streptococcus virulence by a human antimicrobial peptide. Proc. Natl. Acad. Sci. U. S. A. 105:16755–16760. 10.1073/pnas.0803815105 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The NADase translocation ngaΔ44-53 mutant strain secretes the same amount of NADase activity as wild-type strain 854. Shown are the mean NADase activities ± the standard errors of culture supernatants from the strains indicated. No activity was detected in supernatants from the NADase deletion Δnga mutant or the enzymatically inactive nga(G330D) mutant. Download