

Abstract
Oxidative stress is a major and recurring cause of damage during inflammation, especially following organ transplantation. Initial ischaemia–reperfusion injury causes the production of many reactive oxygen and nitrogen species, and subsequent recruitment and activation of inflammatory cells can lead to further oxidative stress. This stress is well known to cause damage at the cellular level, for example by induction of senescence leading to the production of a characteristic senescence-associated secretory phenotype. Chemokines are an important component of the senescence-associated secretory phenotype, recruiting further leucocytes and reinforcing the stress and senescence responses. As well as inducing the production of proteins, including chemokines, oxidative stress can alter proteins themselves, both directly and by induction of enzymes capable of modification. These alterations can lead to important modifications to their biological activity and also alter detection by some antibodies, potentially limiting the biological relevance of some immunochemical and proteomic biomarkers. Peroxynitrite, a reactive nitrogen species generated during inflammation and ischaemia, can cause such modifications by nitrating chemokines. Matrix metalloproteinases, released by many stressed cells, can cleave chemokines, altering function, while peptidylarginine deiminases can inactivate certain chemokines by citrullination. This review discusses the relationship between inflammation and post-translational modification, focusing on the functional modulation of transplant-relevant pro-inflammatory chemokines.
Keywords: chemokines, inflammation, post-translational modification, stress
Introduction
It is well known that inflammation is a complex response to both infection and tissue injury. It is necessary for the resolution of damage, but if uncontrolled, chronic inflammation can occur. The sustained recruitment of leucocytes and consequent oxidative stress can lead to worsening injury; tight regulation of inflammation is therefore crucial.
The migration of leucocytes to sites of inflammation is mediated by a specific class of cytokines termed chemokines. These chemokines are produced in response to the various stresses that occur during inflammation. What is increasingly clear is that these stresses can have profound effects on the capacity of chemokines to elicit targeted immune cell recruitment. The present review will summarize the stresses occurring during inflammation, particularly those which transplanted organs experience, and what affects these and other stress-related molecules have on chemokine biology.
Stress
There are many causes of stress and damage to tissues linked to inflammation, and even more linked to transplantation. During all stages of organ transplantation there are opportunities for cellular stress and this has consequences for graft outcome. Oxidative stress is the major cause of cellular stress and can occur at multiple points throughout the life of the graft. For example, when the blood supply returns to tissues after a period of surgical ischaemia, ischaemia–reperfusion injury can occur.1 This is a major cause of organ dysfunction following surgery. Intracellular oxidative stress under these conditions is generated by mitochondria and oxidases in epithelial and endothelial cells.
Pathological oxidative stress arises due to an imbalance of the producers and scavengers (antioxidants) of reactive oxygen species and reactive nitrogen species (ROS/RNS), altering the normal redox state of cells. These reactive species accumulate in cells or tissues, affecting numerous cellular pathways and macromolecules. A major source is electron leakage from the mitochondria during aerobic respiration.2–4 Peroxisomes, the site of lipid metabolism, and NADPH oxidase in the plasma membrane of activated macrophages also produce ROS.5 Vascular adhesion protein-1, an amine oxidase present at increased levels on inflamed endothelium, for example in the liver, produces hydrogen peroxide and is linked to leucocyte recruitment during inflammation.6 During chronic inflammation nitric oxide synthase 2 (NOS2) is up-regulated in almost all liver cells,7 further increasing the potential for peroxynitrite formation.
The infiltration of immune cells able to perform oxidative burst, for example macrophages and neutrophils, further increases stress by the release of multiple reactive species. Macrophages produce nitric oxide and superoxide by inducible NOS and NADPH oxidase, which react to form peroxynitrite. This aids macrophage antimicrobial function8 and is triggered by l-arginine depletion occurring in inflammation. This is an innate immune effector mechanism to aid the killing of pathogens but contributes to inflammation and stress with excess release of reactive species.
The effects of oxidative stress are varied, depending on the reactive species involved as well as the concentration, cell type and environment. Changes at the cellular and organ level can lead to inflammation, fibrosis and cancer but many of these alterations are the result of changes at a molecular level, for example to DNA and proteins.5
Damage to DNA due to oxidative stress includes both single-base damage and strand breaks.9,10 These can introduce genetic mutations, cause downstream signalling effects and activate DNA damage repair mechanisms, potentially resulting in apoptosis or senescence.10 Peroxidation is the main form of lipid damage, causing cell death both by peroxidation of membrane lipoproteins, disrupting membrane integrity, and by generation of reactive aldehydes including 4-hydroxynonenal, which interact with Fas pathways, including Jun N-terminal kinase, leading to apoptosis.2 This lipid modification is crucial in the pathogenesis of multiple diseases including atherosclerosis.5
Superoxide (•O2−) is often the first free radical produced; it is short-lived, damaging only macromolecules in the immediate vicinity. In contrast, hydrogen peroxide has a longer half-life and can damage molecules directly as well as acting as a signalling molecule, activating protein kinase C, for example.9 The effects of the RNS peroxynitrite have been extensively reviewed by Szabo et al.11 This reactive species is involved in multiple aspects of inflammation and ischaemia–reperfusion injury, both modifying proteins directly and altering transcription factors such as nuclear factor-κB, resulting in, for example, altered cell adhesion molecule expression.12 Oxidative stress can alter other transcription factors such as hypoxia inducible factor-1α and p53, further developing the stress response, and can also increase mitochondrial permeability allowing signalling molecules to transition to the nucleus and vice versa.13
All of these mechanisms enable stress to greatly alter protein production, leading to induction of apoptosis, necrosis and senescence, and in these altered cellular states there is further amplification of stress and inflammation. For example, senescent cells cause the production of the senescence-associated secretory phenotype including multiple chemokines, proteases and other pro-inflammatory factors,14,15 creating a vicious circle of worsening damage, potentially spreading senescence. Cellular senescence is known to play a key role in liver allograft rejection.16 Increased expression of chemokine is particularly important in such senescent and inflammatory environment situations because chemokines can recruit inflammatory cells, further adding to the damaging feedback loop.
This review, however, will focus on the modifications that happen to chemokines during such stress, in terms of both post-translational residue modification and protein expression.
Chemokines
Chemokines, chemoattractant cytokines, are small (8000–14 000 molecular weight) proteins secreted by many cell types primarily to direct leucocyte navigation and so have an important role in stressful and inflammatory conditions such as transplantation.17 Chemokines are a large protein family consisting of at least 45 ligands and 20 receptors. These ligands are classified both functionally and structurally. Inflammatory chemokines direct cells to the site of injury and are expressed in response to stress and infection, homeostatic chemokines are involved in development and tissue maintenance and their expression is more constitutive. Structural classification is based on the position of the N-terminal cysteines, creating the four classes: CC, CXC, CX3C and C, with receptor classes named appropriately.18,19
Chemokine regulation is highly complex and, because of their role in leucocyte recruitment, this tight regulation is crucial. This ensures, for example, that inflammation does not become uncontrolled and pathogenic. The regulation occurs at multiple levels as shown in Fig. 1. Briefly, chemokines can be regulated at the transcriptional and translational level, with inflammatory chemokines especially being up-regulated in response to stress. Some pre-formed chemokines are stored in endothelial cells, in secretory granules including Weibel–Palade bodies, and released for rapid response to insult.20 Once produced, the functions of chemokines are also closely regulated by post-translational modification, receptor expression and glycosaminoglycan (GAG) binding. All of these can be modified during stress. Chemokines typically bind and signal through G-protein-coupled receptors but decoy, or atypical, receptors can also bind chemokines, generally with anti-inflammatory effects.21 To form the chemokine gradients needed for in vivo function,22 chemokines bind to GAGs such as heparan sulphate. This chemokine immobilization increases the concentration at the site of production, aiding the infiltration of cells. Endothelial expression of these sugars increases during stresses produced by transplantation, altering the potential to bind chemokines and so alter the chemokine function.23
Figure 1.
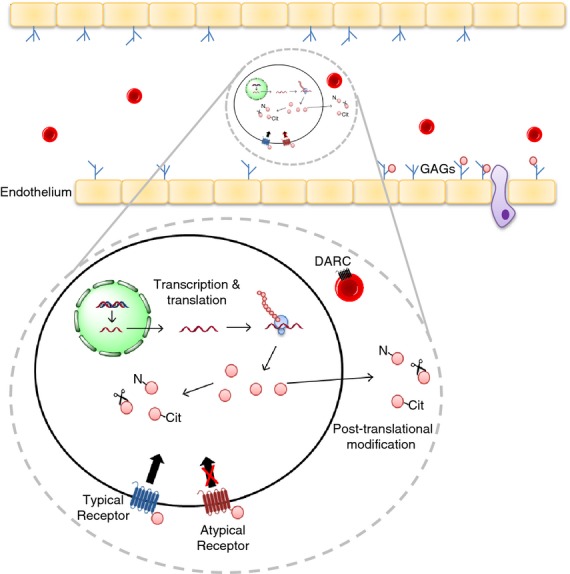
Mechanisms of chemokine regulation. Chemokine function is regulated at many levels. Protein production is regulated at both the transcriptional and translational level, with some microRNAs regulating mRNA levels. Chemokine post-translational modification occurs both intra- and extra-cellularly for example nitration, citrullination and protein cleavage, all of which can alter chemokine function. Chemokines bind and signal through ‘typical’ G-protein-coupled receptors, this triggers downstream signalling and causes cell migration. Atypical receptors, including duffy antigen/chemokine receptor (DARC), however, bind chemokine, reducing bioavaliability, but do not signal in the normal manner. Chemokines need to bind glycosaminoglycans (GAGs), for example heparan sulphate, for in vivo function. GAGs are presented on the endothelium and bind chemokine, forming a chemokine gradient, enabling cell migration. N, chemokine nitration; Cit, chemokine citrullination.
Events throughout transplantation effect the production of chemokines and therefore inflammation and graft outcome as reviewed by El-Sawy et al.17 Tissue stress as early as brain death of the donor can lead to endothelial dysfunction and chemokine release, and ischaemia–reperfusion injury also causes the production of pro-inflammatory factors. Upon reperfusion, pro-inflammatory cytokines are produced, including tumour necrosis factor-α and interleukin-1β. These stimulate the release of preformed chemokines from vascular endothelium, including CCL2 and CXCL8, recruiting macrophages and neutrophils, respectively, to the graft.17 The interferon-γ-inducible chemokines CXCL9, CXCL10 and CXCL11 are rapidly increased following reperfusion, and are abundant in rejecting allografts. However, controversy surrounds the role of their receptor, CXCR3, in transplantation.24
Epithelial cells including those of the liver and kidney also have potent chemokine-producing ability. Such compartments are therefore particularly capable of eliciting an increase in lymphocyte recruitment, causing the bile-duct and peritubular inflammation seen in rejection.17 Later stages of transplant rejection, including arteriosclerosis and fibrosis, are increased by chemokines25,26 and a growing body of research shows that chemokine levels could be used as biomarkers, such as CXCL10 for monitoring graft inflammation and predicting liver fibrosis.27
Chemokines are involved in senescence, with particular focus on components of senescence-associated secretory phenotype including the CXCR2 ligands,14 CXCL1 and CXCL8. Production of these, and other pro-inflammatory molecules, further increases inflammation, senescence and, in the case of transplantation, potentially organ loss.
Post-translational modification of chemokines
The function of chemokines, as with many proteins, can be profoundly altered by post-translational modification, as reviewed in detail by Mortier et al.28 These modifications can abrogate or enhance, and in a few cases are mandatory for, chemokine function and so act as a powerful regulatory mechanism to fine tune chemokine function. Modification can be both enzymatic and chemical, altering peptide length or modifying amino acids directly. These modifications can profoundly alter chemokine behaviour, changing which receptors they target, their presentation by GAGs and our ability to detect them with current antibodies. Multiple factors involved in tissue stress influence these modifications.
Cleavage
Multiple enzyme families can cleave chemokines, particularly at the disordered N-terminus, although some examples of C-terminal cleavage have been found.
Matrix metalloproteinases
Matrix metalloproteinases (MMPs), zinc-dependent endopeptidases, are involved in extracellular matrix degradation and remodelling and multiple members of the family are up-regulated following transplantation and during inflammation. MMP-2 and MMP-9, for example, are up-regulated during ischaemia–reperfusion.29,30 Other MMPs are strongly associated with immune cells including the neutrophil-associated MMP-8 and the macrophage-specific MMP-12.31 Reactive nitrogen species can also up-regulate MMPs.32 As well as involvement in tissue remodelling these enzymes also have important roles in modifying bioactive molecules such as chemokines.
MMP-8 is released from many inflammatory cells including infiltrating neutrophils. N-terminal cleavage of CXCL5 and CXCL8 by this enzyme increases chemotactic activity.33 CXCL8 further increases MMP release from neutrophils, and matrix component degradation by the enzyme further facilitates cell migration into the tissue, creating a feedback loop, amplifying cell recruitment and potentially leading to worsening damage if unresolved. MMP-1 and MMP-9 also cleave CXCL8.34,35
Chemokine inactivation also occurs by MMP cleavage. MMP-12 cleaves and inactivates ELR (glutamic acid-leucine-arginine) containing CXC chemokines, including CXCL8,31 at the important motif E-LR. Later in inflammation, cleavage of CCL2, -7, -8 and -13 results in loss of activity and the formation of potent antagonists31 that may actively suppress macrophage recruitment, promoting resolution and preventing damage. There is specificity between ligand and substrate, indicating tight control of chemokine regulation. An example of this is CCL2 and CCL7, which are co-expressed and share the receptor CCR2. CCL2 can be cleaved by MMP-1 and MMP-3 but not by MMP-2. MMP-2, however, can cleave and inactivate other chemokines including CCL7.36
CD26
Several other enzymes can also process chemokines. For example, the serine protease CD26 (dipeptidyl peptidase-4; DPP4). CD26 is a membrane glycoprotein expressed by numerous cell types. It is highly expressed on T cells and is associated with several aspects of immune regulation including T-cell proliferation, and inhibition of this enzyme has been investigated as a therapy for transplant rejection.37 CXCL11 cleavage by CD26 prevents chemotaxis but the chemokine is still able to desensitize the receptor CXCR3 and so can decrease the local activity of full-length CXCL11.38 CCL5 is also subject to post-translational processing by CD26, which cleaves the first two N-terminal amino acids, preventing binding to CCR139 and CCR3,40 but CCR5 signalling is still intact, skewing the chemokine towards T-cell recruitment. Interestingly, CD26 cleavage increases the anti-viral properties of CCL5 against HIV.39 CCL4 also has altered receptor usage following CD26 cleavage, it still binds CCR5 but unlike the full form also binds CCR1 and CCR2,41 altering the cell types that it can recruit. To further complicate chemokine processing in vivo, GAG binding protects CXCL12 from proteolysis by CD26.42 Cleavage by other enzymes is also impaired by GAG binding, including the plasmin cleavage of CCL11.43
Infection
As well as enabling fine tuning of the chemokine system during inflammation, chemokine cleavage has been used by bacteria to evade the immune response. The pathogen Streptococcus pyogenes can express SpyCEP (S. pyogenes cell envelope protease), which cleaves chemokines containing an ELR motif, including CXCL8.44 This cleavage inactivates the chemokine, unusually at the C-terminus, with functional effects most likely due to disruption of GAG binding and dimerization rather than receptor binding. SpyCEP is a potent virulence factor, helping the bacteria to evade the immune response by decreasing neutrophil recruitment, and is crucial to aggressive bacterial dissemination,45 increasing, for example, lethal necrotizing fasciitis.44
Porphyromonas gingivalis, a bacterium implicated in periodontal disease, also produces enzymes capable of chemokine cleavage called gingipains.46,47 These enzymes can both cleave CXCL8 into more potent forms of the chemokine and degrade it, abrogating function, depending on whether the enzyme is soluble or membrane bound.
Although functional alterations produced by the modifications discussed so far have been researched, the effects on epitope loss and therefore detection by many common methods have not been characterized. This is a fundamental problem as discussed later.
Residue modification
Modification of chemokines by direct alteration of amino acid residues will now be discussed. This can be either non-enzymatic or enzyme-catalysed.
Nitration
As discussed previously, transplantation and associated inflammation involve oxidative stress. The RNS peroxynitrite is formed by the rapid reaction of nitric oxide and superoxide and is present in many inflammatory situations.11 Like other components of oxidative stress, peroxynitrite has both molecular and cellular effects, inducing apoptosis and senescence. Peroxynitrite can selectively oxidize and nitrate several residues, including the oxidation of histidine and the nitration of tyrosine and tryptophan. As shown in Fig. 2(a), nitration of tyrosine results in the formation of 3-nitrotyrosine. This gives the modified residue a 45-Da mass increase but the overall charge remains unaltered. 3-Nitrotyrosine is a marker of nitro-oxidative stress and the presence of peroxynitrite and is seen in many pathological conditions including diabetes.11 Antibodies have been raised against 3-nitrotyrosine and are used to identify nitrated proteins and the presence of peroxynitrite or related species.11 Figure 3 demonstrates the use of such antibodies to show the presence of peroxynitrite in a murine model of kidney ischaemia–reperfusion injury (unpublished data from our group).
Figure 2.
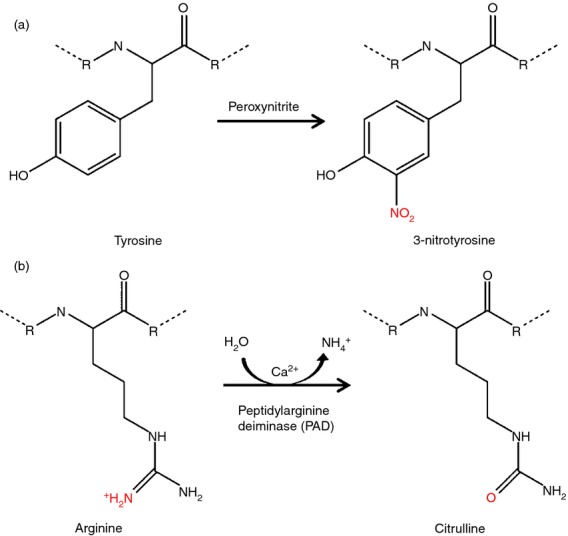
Post-translational modifications to the amino acids tyrosine and arginine. (a) Nitration of tyrosine by peroxynitrite results in the formation of 3-nitrotyrosine. This reaction can occur to both peptidyl and free tyrosine residues. (b) Citrullination is the deimination of arginine to citrulline by peptidylarginine deiminase. The substrate for this enzyme is peptidylarginine, the reaction does not occur on free arginine. Production of free citrulline, however, can occur as a by-product of nitric oxide synthase (NOS) or the urea cycle. Both of these modifications are not just passive, they can alter protein function, for example chemokines.
Figure 3.
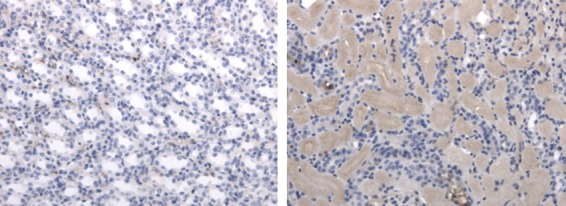
Protein nitration in ischaemia–reperfusion injury. Kidney sections from a murine model of ischaemia–reperfusion injury were stained for 3-nitrotyrosine (brown) and counterstained with haematoxylin (blue) by immunohistochemistry. The right kidney was ischaemic for 45 min and organs were harvested 24 hr after reperfusion, the left kidney is the contralateral control. Magnification ×20.
Nitration is not a silent modification and can have profound effects on protein function. Effects include attenuation of antioxidants, creating a positive feedback loop further increasing oxidative stress. Peroxynitrite inactivation of copper/zinc superoxide dismutase is by modification of a histidine48 residue in the active site. Manganese superoxide dismutase inactivation, however, is by nitration of a specific tyrosine residue in the active site, which prevents catalysis. This has been observed in human renal allografts undergoing chronic rejection,49 contributing to the positive feedback loop and worsening inflammation.
As discussed earlier, chemokines are present at increased levels during periods of oxidative stress and so could be targets of modification. Several chemokines have been found to be targets of peroxynitrite modification including CCL250,51 and CCL5.52 Generally it is thought that modification of pro-inflammatory cytokines by peroxynitrite abrogates function, but this is dependent on numerous factors, including cell type. One study shows that peroxynitrite-treated CCL2 loses its ability to recruit CD8+ T cells but the recruitment of myeloid-derived suppressor cells is unaltered.50 An earlier study, however, shows that CCL2 nitration impairs monocyte recruitment.51 No mechanisms of action have been defined in these papers, but as yet unpublished data from our group suggest that nitration of CCL2 decreases its chemotactic ability by both decreasing binding to the receptor CCR2, and by abrogating its GAG binding ability.
Although not able to nitrate chemokines, the ROS hydrogen peroxide can modify chemokines by causing oxidative cross-linking of tyrosine residues in CCL5.53 This causes multimer formation of the chemokine but does not alter T-cell chemotaxis or receptor usage. Interestingly, GAG binding prevents this modification from occurring.
The ability of factors involved in oxidative stress to both induce the production of chemokines and to down-regulate their function, promoting the resolution of inflammation, further adds to the complexity of chemokine regulation.
Molon et al.50 reported that nitrated CCL2 cannot be detected by commercially available antibodies because the modification causes epitope loss. This has important implications for studies using antibody-based techniques to study chemokine levels in a stressful and inflammatory environment, as discussed later.
Citrullination
Enzymatic residue modification of chemokines includes citrullination. This is the formation of citrulline by the deimination of arginine, by peptidylarginine deiminase (PAD). This modification only occurs post-translationally as the enzyme substrate is peptidylarginine not free arginine.
As shown in Fig. 2(b), citrullination gives a 1-Da mass reduction per arginine residue modified but more profound functional consequences are likely to result from loss of the basic charge. This is especially likely for chemokines that strongly rely on their cationic residues for GAG interactions. Arginine residues often play an important role in the structural integrity of proteins because of ionic interactions with negatively charged residues and cofactors, and by forming hydrogen bonds. Citrullination could logically therefore lead to changes in protein structure and hence function.
Five human PAD isoforms have been identified and their roles in pathology and chemokine interactions have been reviewed by Moelants et al.54 The isoforms have differing tissue expression patterns, and substrate specificity is also exhibited, For example only PAD2 citrullinates β-actin, and only PAD4 citrullinates histone H3.55 PADs are expressed by the epidermis, granulocytes, lymphocytes and some epithelial cells, with increased expression associated with a range of inflammatory diseases including rheumatoid arthritis and multiple sclerosis. Stress-activated pathways involving p53 both regulate and are regulated by PAD4 citrullination,56 with autocitrullination further regulating enzyme activity.57
All human PAD isoforms are calcium binding and calcium levels are crucial in regulating enzyme activity.54 Normal intracellular calcium levels are insufficient for function, but oxidative stress is known to cause an influx of cytoplasmic calcium, suggesting that PAD will be active in situations such as ischaemia–reperfusion and inflammation. Serum PAD has increased activity in rheumatoid arthritis patients.58
Citrullination of multiple protein families has been identified, including the structural proteins actin and cytokeratin. Histone citrullination alters chromatin remodelling and so affects protein expression. Citrullination is also needed for neutrophil extracellular trap formation and so is heavily involved in immune regulation.54
Chemokines can be citrullinated and, as with nitration, this is not a silent modification, but can affect function. All modifications found so far show citrullination to be an anti-inflammatory modification, decreasing in vivo chemokine function. Citrullination of both CXCL10 and CXCL11 does not alter CXCR3 binding but does result in a decrease in calcium signalling, chemotactic activity and heparin binding.59 Studies of CXCL12 show that PAD decreases function, with an increase in functional impairment correlated with an increased number of citrullinated arginines.60 Such modified chemokines may occur in, for example, Crohn's disease, where co-expression of CXCL12 and PAD has been found.60 CXCL8 has increased CXCR1 binding once citrullinated on specific arginines but decreased heparin binding does not induce neutrophil chemotaxis in vivo.61 These modifications highlight important differences between in vivo and in vitro chemokine function.
Bacterial PAD have also been identified, produced by P. gingivalis although unlike the human isoforms these can modify both free and peptidyl-arginine and are not calcium dependent. As yet, chemokine modification by these enzymes is unconfirmed.47
As well as modifying GAG binding, citrullination may also have a role in preventing enzymatic cleavage. Citrullination of CXCL8 prevents cleavage by gingipains to more active forms of the chemokine, so potentially dampening inflammation.47
Implication for biomarkers and detection
There are an increasing number of studies of stress-induced proteomic changes following organ transplantation. Chemokines, due to their role in inflammation, are logical candidates and are often studied.62 However, measurement of chemokine levels alone, as candidate biomarkers of rejection, is not providing the whole picture.27,63 As discussed in this review, it is becoming clear that measurement of the function of post-translationally modified proteins may be more important than absolute protein levels; this is particularly relevant for proteins in stressful situations due to the increased potential for modification.
Post-translational modifications alter the function of chemokines, both enhancing and abrogating their potency on a case-by-case basis. Furthermore, in some instances, such as CCL2, nitration can prevent detection by antibodies, potentially limiting the biological relevance of some immunochemical and proteomic biomarkers. Not all modifications affect function and not all alter our ability to detect protein presence, but it is this heterogeneity that makes understanding chemokines in the inflammatory environment so important. In the future, more emphasis should perhaps be placed on measurements of chemokine function, rather than mere presence. Furthermore, new strategies to measure levels of modified chemokines, using antibodies specific to modified forms of chemokine, or mass spectrometry, should be further developed for more widespread use.
Conclusions
Current knowledge of the post-translational modification of chemokines is, on the whole, anecdotal. To further our understanding a systematic approach needs to be adopted to determine which chemokines can be modified and by what mechanisms. Binding to conventional receptors, immunoregulatory atypical receptors and GAGs must be assessed on a case-by-case basis to ascertain true biological potency. Determining both the functional and antigenic effects of these modifications is crucial to establish chemokine levels in vivo and the function of these proteins. It is in stressful and inflammatory situations that this is most crucial due to both increased chemokine levels and increased opportunities for modification.
Acknowledgments
This work was supported by the MRC and the NIHR Biomedical Research Centre for Ageing and Age-related Disease. Murine kidney tissue was kindly provided by Prof. Neil Sheerin and Christopher Fox.
Glossary
- GAG
glycosaminoglycan
- MMP
matrix metalloproteinase
- NOS
nitric oxide synthase
- PAD
peptidylarginine deiminase
- PPAD
peptidylarginine deiminase from Porphyromonas gingivalis
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- SpyCEP
Streptococcus pyogenes cell envelope protease
Disclosures
The authors of this manuscript have no conflicts of interest to disclose.
References
- 1.Eltzschig HK, Eckle T. Ischemia and reperfusion – from mechanism to translation. Nat Med. 2011;17:1391–401. doi: 10.1038/nm.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ďuračková Z. Some current insights into oxidative stress. Physiol Res. 2010;59:459–69. doi: 10.33549/physiolres.931844. [DOI] [PubMed] [Google Scholar]
- 3.Papadopoulos D, Siempis T, Theodorakou E, Tsoulfas G. Hepatic ischemia and reperfusion injury and trauma: current concepts. Arch Trauma Res. 2013;2:63–70. doi: 10.5812/atr.12501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bokov A, Chaudhuri A, Richardson A. The role of oxidative damage and stress in aging. Mech Ageing Dev. 2004;125(10–11 SPEC. ISS):811–26. doi: 10.1016/j.mad.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 5.Dröge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 6.Weston CJ, Adams DH. Hepatic consequences of vascular adhesion protein-1 expression. J Neural Transm. 2011;118:1055–64. doi: 10.1007/s00702-011-0647-0. [DOI] [PubMed] [Google Scholar]
- 7.Urtasun R, de la Rosa LC, Nieto N. Oxidative and nitrosative stress and fibrogenic response. Clin Liver Dis. 2008;12:769–90. doi: 10.1016/j.cld.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xia Y, Zweier JL. Superoxide and peroxynitrite generation from inducible nitric oxide synthase in macrophages. Proc Natl Acad Sci USA. 1997;94:6954–8. doi: 10.1073/pnas.94.13.6954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Woodbine L, Brunton H, Goodarzi AA, Shibata A, Jeggo PA. Endogenously induced DNA double strand breaks arise in heterochromatic DNA regions and require ataxia telangiectasia mutated and Artemis for their repair. Nucleic Acids Res. 2011;39:6986–97. doi: 10.1093/nar/gkr331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rai P. Oxidation in the nucleotide pool, the DNA damage response and cellular senescence: defective bricks build a defective house. Mutat Res. 2010;703:71–81. doi: 10.1016/j.mrgentox.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 11.Szabo C, Ischiropoulos H, Radi R. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discovery. 2007;6:662–80. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- 12.Dong A, Shen J, Zeng M, Campochiaro PA. Vascular cell-adhesion molecule-1 plays a central role in the proangiogenic effects of oxidative stress. Proc Natl Acad Sci U S A. 2011;108:14614–9. doi: 10.1073/pnas.1012859108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cole NB, Daniels MP, Levine RL, Kim G. Oxidative stress causes reversible changes in mitochondrial permeability and structure. Exp Gerontol. 2010;45:596–602. doi: 10.1016/j.exger.2010.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Acosta JC, O'Loghlen A, Banito A, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133:1006–18. doi: 10.1016/j.cell.2008.03.038. [DOI] [PubMed] [Google Scholar]
- 15.Sasaki M, Miyakoshi M, Sato Y, Nakanuma Y. Modulation of the microenvironment by senescent biliary epithelial cells may be involved in the pathogenesis of primary biliary cirrhosis. J Hepatol. 2010;53:318–25. doi: 10.1016/j.jhep.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 16.Brain JG, Robertson H, Thompson E, et al. Biliary epithelial senescence and plasticity in acute cellular rejection. Am J Transplant. 2013;13:1688–702. doi: 10.1111/ajt.12271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.El-Sawy T, Fahmy NM, Fairchild RL. Chemokines: directing leukocyte infiltration into allografts. Curr Opin Immunol. 2002;14:562–8. doi: 10.1016/s0952-7915(02)00382-5. [DOI] [PubMed] [Google Scholar]
- 18.Zlotnik A, Yoshie O. Chemokines: a new classification system and their role in immunity. Immunity. 2000;12:121–7. doi: 10.1016/s1074-7613(00)80165-x. [DOI] [PubMed] [Google Scholar]
- 19.Bachelerie F, Ben-Baruch A, Burkhardt AM, et al. International Union of Pharmacology LXXXIX. Update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptors. Pharmacol Rev. 2014;66:1–79. doi: 10.1124/pr.113.007724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Øynebråten I, Barois N, Hagelsteen K, Johansen FE, Bakke O, Haraldsen G. Characterization of a novel chemokine-containing storage granule in endothelial cells: evidence for preferential exocytosis mediated by protein kinase A and diacylglycerol. J Immunol. 2005;175:5358–69. doi: 10.4049/jimmunol.175.8.5358. [DOI] [PubMed] [Google Scholar]
- 21.O'Boyle G, Ali S, Kirby JA. Chemokines in transplantation: what can atypical receptors teach us about anti-inflammatory therapy? Transplant Rev (Orlando) 2011;25:136–44. doi: 10.1016/j.trre.2010.10.005. [DOI] [PubMed] [Google Scholar]
- 22.Weber M, Hauschild R, Schwarz J, et al. Interstitial dendritic cell guidance by haptotactic chemokine gradients. Science. 2013;339:328–32. doi: 10.1126/science.1228456. [DOI] [PubMed] [Google Scholar]
- 23.Ali S, Malik G, Burns A, Robertson H, Kirby JA. Renal transplantation: examination of the regulation of chemokine binding during acute rejection. Transplantation. 2005;79:672–9. doi: 10.1097/01.tp.0000155961.57664.db. [DOI] [PubMed] [Google Scholar]
- 24.Halloran PF, Fairchild RL. The puzzling role of CXCR3 and its ligands in organ allograft rejection. Am J Transplant. 2008;8:1578–9. doi: 10.1111/j.1600-6143.2008.02323.x. [DOI] [PubMed] [Google Scholar]
- 25.Sakihama H, Masunaga T, Yamashita K, Hashimoto T, Inobe M, Todo S, Uede T. Stromal cell-derived factor-1 and CXCR4 interaction is critical for development of transplant arteriosclerosis. Circulation. 2004;110:2924–30. doi: 10.1161/01.CIR.0000146890.93172.6C. [DOI] [PubMed] [Google Scholar]
- 26.Belperio JA, Ardehali A. Chemokines and transplant vasculopathy. Circ Res. 2008;103:454–66. doi: 10.1161/CIRCRESAHA.108.182865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berres ML, Trautwein C, Schmeding M, et al. Serum chemokine CXC ligand 10 (CXCL10) predicts fibrosis progression after liver transplantation for hepatitis C infection. Hepatology. 2011;53:596–603. doi: 10.1002/hep.24098. [DOI] [PubMed] [Google Scholar]
- 28.Mortier A, Van Damme J, Proost P. Regulation of chemokine activity by posttranslational modification. Pharmacol Ther. 2008;120:197–217. doi: 10.1016/j.pharmthera.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 29.Falk V, Soccal PM, Grünenfelder J, Hoyt G, Walther T, Robbins RC. Regulation of matrix metalloproteinases and effect of MMP-inhibition in heart transplant related reperfusion injury. Eur J Cardiothorac Surg. 2002;22:53–8. doi: 10.1016/s1010-7940(02)00207-5. [DOI] [PubMed] [Google Scholar]
- 30.Kuyvenhoven JP, Molenaar IQ, Verspaget HW, et al. Plasma MMP-2 and MMP-9 and their inhibitors TIMP-1 and TIMP-2 during human orthotopic liver transplantation. The effect of aprotinin and the relation to ischemia/reperfusion injury. Thromb Haemost. 2004;91:506–13. doi: 10.1160/TH03-05-0272. [DOI] [PubMed] [Google Scholar]
- 31.Dean RA, Cox JH, Bellac CL, Doucet A, Starr AE, Overall CM. Macrophage-specific metalloelastase (MMP-12) truncates and inactivates ELR+ CXC chemokines and generates CCL2, -7, -8, and -13 antagonists: potential role of the macrophage in terminating polymorphonuclear leukocyte influx. Blood. 2008;112:3455–64. doi: 10.1182/blood-2007-12-129080. [DOI] [PubMed] [Google Scholar]
- 32.Urtasun R, Cubero FJ, Vera M, Nieto N. Reactive nitrogen species switch on early extracellular matrix remodeling via induction of MMP1 and TNFa. Gastroenterology. 2009;136:1410–22.e4. doi: 10.1053/j.gastro.2008.12.065. [DOI] [PubMed] [Google Scholar]
- 33.Tester AM, Cox JH, Connor AR, Starr AE, Dean RA, Puente XS, López-Otín C, Overall CM. LPS responsiveness and neutrophil chemotaxis in vivo require PMN MMP-8 activity. PLoS ONE. 2007;2:e312. doi: 10.1371/journal.pone.0000312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Van Den Steen PE, Proost P, Wuyts A, Van Damme J, Opdenakker G. Neutrophil gelatinase B potentiates interleukin-8 tenfold by aminoterminal processing, whereas it degrades CTAP-III, PF-4, and GRO-α and leaves RANTES and MCP-2 intact. Blood. 2000;96:2673–81. [PubMed] [Google Scholar]
- 35.Van Den Steen PE, Wuyts A, Husson SJ, Proost P, Van Damme J, Opdenakker G. Gelatinase B/MMP-9 and neutrophil collagenase/MMP-8 process the chemokines human GCP-2/CXCL6, ENA-78/CXCL5 and mouse GCP-2/LIX and modulate their physiological activities. Eur J Biochem. 2003;270:3739–49. doi: 10.1046/j.1432-1033.2003.03760.x. [DOI] [PubMed] [Google Scholar]
- 36.McQuibban GA, Butler GS, Gong JH, Bendall L, Power C, Clark-Lewis I, Overall CM. Matrix metalloproteinase activity inactivates the CXC chemokine stromal cell-derived factor-1. J Biol Chem. 2001;276:43503–8. doi: 10.1074/jbc.M107736200. [DOI] [PubMed] [Google Scholar]
- 37.Ohnuma K, Dang NH, Morimoto C. Revisiting an old acquaintance: CD26 and its molecular mechanisms in T cell function. Trends Immunol. 2008;29:295–301. doi: 10.1016/j.it.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 38.Ludwig A, Schiemann F, Mentlein R, Lindner B, Brandt E. Dipeptidyl peptidase IV (CD26) on T cells cleaves the CXC chemokine CXCL11 (I-TAC) and abolishes the stimulating but not the desensitizing potential of the chemokine. J Leukoc Biol. 2002;72:183–91. [PubMed] [Google Scholar]
- 39.Oravecz T, Pall M, Roderiquez G, et al. Regulation of the receptor specificity and function of the chemokine RANTES (regulated on activation, normal T cell expressed and secreted) by dipeptidyl peptidase IV (CD26)-mediated cleavage. J Exp Med. 1997;186:1865–72. doi: 10.1084/jem.186.11.1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Struyf S, De Meester I, Scharpé S, Lenaerts JP, Menten P, Wang JM, Proost P, Van Damme J. Natural truncation of RANTES abolishes signaling through the CC chemokine receptors CCR1 and CCR3, impairs its chemotactic potency and generates a CC chemokine inhibitor. Eur J Immunol. 1998;28:1262–71. doi: 10.1002/(SICI)1521-4141(199804)28:04<1262::AID-IMMU1262>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 41.Guan E, Wang J, Roderiquez G, Norcross MA. Natural truncation of the chemokine MIP-b/CCL4 affects receptor specificity but not anti-HIV-1 activity. J Biol Chem. 2002;277:32348–52. doi: 10.1074/jbc.M203077200. [DOI] [PubMed] [Google Scholar]
- 42.Sadir R, Imberty A, Baleux F, Lortat-Jacob H. Heparan sulfate/heparin oligosaccharides protect stromal cell-derived factor-1 (SDF-1)/CXCL12 against proteolysis induced by CD26/dipeptidyl peptidase IV. J Biol Chem. 2004;279:43854–60. doi: 10.1074/jbc.M405392200. [DOI] [PubMed] [Google Scholar]
- 43.Ellyard JI, Simson L, Bezos A, Johnston K, Freeman C, Parish CR. Eotaxin selectively binds heparin: an interaction that protects eotaxin from proteolysis and potentiates chemotactic activity in vivo. J Biol Chem. 2007;282:15238–47. doi: 10.1074/jbc.M608046200. [DOI] [PubMed] [Google Scholar]
- 44.Edwards RJ, Taylor GW, Ferguson M, et al. Specific C-terminal cleavage and inactivation of interleukin-8 by invasive disease isolates of Streptococcus pyogenes. J Infect Dis. 2005;192:783–90. doi: 10.1086/432485. [DOI] [PubMed] [Google Scholar]
- 45.Kurupati P, Turner CE, Tziona I, et al. Chemokine-cleaving Streptococcus pyogenes protease SpyCEP is necessary and sufficient for bacterial dissemination within soft tissues and the respiratory tract. Mol Microbiol. 2010;76:1387–97. doi: 10.1111/j.1365-2958.2010.07065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mikolajczyk-Pawlinska J, Travis J, Potempa J. Modulation of interleukin-8 activity by gingipains from Porphyromonas gingivalis: implications for pathogenicity of periodontal disease. FEBS Lett. 1998;440:282–6. doi: 10.1016/s0014-5793(98)01461-6. [DOI] [PubMed] [Google Scholar]
- 47.Moelants EA, Loozen G, Mortier A, et al. Citrullination and proteolytic processing of chemokines by Porphyromonas gingivalis. Infect Immun. 2014;82:2511–9. doi: 10.1128/IAI.01624-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alvarez B, Demicheli V, Durán R, Trujillo M, Cerveñansky C, Freeman BA, Radi R. Inactivation of human Cu, Zn superoxide dismutase by peroxynitrite and formation of histidinyl radical. Free Radical Biol Med. 2004;37:813–22. doi: 10.1016/j.freeradbiomed.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 49.Macmillan-Crow LA, Crow JP, Kerby JD, Beckman JS, Thompson JA. Nitration and inactivation of manganese superoxide dismutase in chronic rejection of human renal allografts. Proc Natl Acad Sci USA. 1996;93:11853–8. doi: 10.1073/pnas.93.21.11853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Molon B, Ugel S, Del Pozzo F, et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J Exp Med. 2011;208:1949–62. doi: 10.1084/jem.20101956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sato E, Simpson KL, Grisham MB, Koyama S, Robbins RA. Effects of reactive oxygen and nitrogen metabolites on MCP-1-induced monocyte chemotactic activity in vitro. Am J Physiol Lung Cell Mol Physiol. 1999;277:L543–9. doi: 10.1152/ajplung.1999.277.3.L543. [DOI] [PubMed] [Google Scholar]
- 52.Sato E, Simpson KL, Grisham MB, Koyama S, Robbins RA. Effects of reactive oxygen and nitrogen metabolites on RANTES- and IL- 5-induced eosinophil chemotactic activity in vitro. Am J Pathol. 1999;155:591–8. doi: 10.1016/S0002-9440(10)65154-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.MacGregor HJ, Kato Y, Marshall LJ, Nevell TG, Shute JK. A copper-hydrogen peroxide redox system induces dityrosine cross-links and chemokine oligomerisation. Cytokine. 2011;56:669–75. doi: 10.1016/j.cyto.2011.08.025. [DOI] [PubMed] [Google Scholar]
- 54.Moelants EAV, Mortier A, Van Damme J, Proost P, Loos T. Peptidylarginine deiminases: physiological function, interaction with chemokines and role in pathology. Drug Discov Today Technol. 2012;9:e261–80. doi: 10.1016/j.ddtec.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 55.Darrah E, Rosen A, Giles JT, Andrade F. Peptidylarginine deiminase 2, 3 and 4 have distinct specificities against cellular substrates: novel insights into autoantigen selection in rheumatoid arthritis. Ann Rheum Dis. 2012;71:92–8. doi: 10.1136/ard.2011.151712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tanikawa C, Ueda K, Nakagawa H, Yoshida N, Nakamura Y, Matsuda K. Regulation of protein citrullination through p53/PADI4Network in DNA damage response. Cancer Res. 2009;69:8761–9. doi: 10.1158/0008-5472.CAN-09-2280. [DOI] [PubMed] [Google Scholar]
- 57.Méchin MC, Coudane F, Adoue V, et al. Deimination is regulated at multiple levels including auto-deimination of peptidylarginine deiminases. Cell Mol Life Sci. 2010;67:1491–503. doi: 10.1007/s00018-010-0262-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Basu PS, Majhi R, Ghosal S, Batabyal SK. Peptidyl-arginine deiminase: an additional marker of rheumatoid arthritis. Clin Lab. 2011;57:1021–5. [PubMed] [Google Scholar]
- 59.Loos T, Mortier A, Gouwy M, Ronsse I, Put W, Lenaerts JP, Van Damme J, Proost P. Citrullination of CXCL10 and CXCL11 by peptidylarginine deiminase: a naturally occurring posttranslational modification of chemokines and new dimension of immunoregulation. Blood. 2008;112:2648–56. doi: 10.1182/blood-2008-04-149039. [DOI] [PubMed] [Google Scholar]
- 60.Struyf S, Noppen S, Loos T, et al. Citrullination of CXCL12 differentially reduces CXCR4 and CXCR7 binding with loss of inflammatory and anti-HIV-1 activity via CXCR4. J Immunol. 2009;182:666–74. doi: 10.4049/jimmunol.182.1.666. [DOI] [PubMed] [Google Scholar]
- 61.Proost P, Loos T, Mortier A, et al. Citrullination of CXCL8 by peptidylarginine deiminase alters receptor usage, prevents proteolysis, and dampens tissue inflammation. J Exp Med. 2008;205:2085–97. doi: 10.1084/jem.20080305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Friedman BH, Wolf JH, Wang L, Putt ME, Shaked A, Christie JD, Hancock WW, Olthoff KM. Serum cytokine profiles associated with early allograft dysfunction in patients undergoing liver transplantation. Liver Transpl. 2012;18:166–76. doi: 10.1002/lt.22451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sirota JC, Walcher A, Faubel S, Jani A, McFann K, Devarajan P, Davis CL, Edelstein CL. Urine IL-18, NGAL, IL-8 and serum IL-8 are biomarkers of acute kidney injury following liver transplantation. BMC Nephrol. 2013;14:17. doi: 10.1186/1471-2369-14-17. [DOI] [PMC free article] [PubMed] [Google Scholar]