

Abstract
Viewing multiple sclerosis (MS) as both neuroinflammation and neurodegeneration has major implications for therapy, with neuroprotection and neurorepair needed in addition to controlling neuroinflammation in the central nervous system (CNS). While Fasudil, an inhibitor of Rho kinase (ROCK), is known to suppress experimental autoimmune encephalomyelitis (EAE), an animal model of MS, it relies on multiple, short-term injections, with a narrow safety window. In this study, we explored the therapeutic effect of a novel ROCK inhibitor FSD-C10, a Fasudil derivative, on EAE. An important advantage of this derivative is that it can be used via non-injection routes; intranasal delivery is the preferred route because of its efficient CNS delivery and the much lower dose compared with oral delivery. Our results showed that intranasal delivery of FSD-C10 effectively ameliorated the clinical severity of EAE and CNS inflammatory infiltration and promoted neuroprotection. FSD-C10 effectively induced CNS production of the immunoregulatory cytokine interleukin-10 and boosted expression of nerve growth factor and brain-derived neurotrophic factor proteins, while inhibiting activation of p-nuclear factor-κB/p65 on astrocytes and production of multiple pro-inflammatory cytokines. In addition, FSD-C10 treatment effectively induced CD4+ CD25+, CD4+ FOXP3+ regulatory T cells. Together, our results demonstrate that intranasal delivery of the novel ROCK inhibitor FSD-C10 has therapeutic potential in EAE, through mechanisms that possibly involve both inhibiting CNS inflammation and promoting neuroprotection.
Keywords: anti-inflammatory, experimental autoimmune encephalomyelitis, FSD-C10-Fasudil derivative, intranasal route, neuroprotection
Introduction
Multiple sclerosis (MS) is an immune-mediated chronic inflammatory demyelinating disease of the central nervous system (CNS), which affects over 2 million people worldwide, often resulting in serious disabilities.1 The pathological hallmarks of MS are demyelination, multifocal inflammation, axonal degeneration and oligodendrocyte loss. Despite intensive investigation, the exact aetiology of MS remains elusive. Our understanding of the immunological processes potentially triggering MS has been greatly aided by the use of animal models such as experimental autoimmune encephalomyelitis (EAE). Studies on the pathogenesis of MS and EAE provide increasing evidence that the activation of autoreactive T cells and a breakdown of the blood–brain barrier cause inflammatory cells to infiltrate the CNS. Subsequently, local re-activation triggers proliferation of various immune cell subtypes and initiation of the inflammatory cascade, leading to demyelination and axonal damage.2–4
Various therapeutic strategies are available for treatment of MS including immunosuppressants, immunomodulators and monoclonal antibodies.5 The complexity of inflammatory demyelination in MS also comprises neurodegenerative processes that may occur within acute phases of inflammation, but that are also temporally independent and outside inflammatory lesions, or even in so-called normal white matter. Multimodal MS therapeutics and further elucidation of neuroprotective pathways within the autoimmune process will be useful to deepen our insight into the complexity of the disease and to improve therapy, especially as regards long-term disability and cognitive decline.6 Rho kinase (ROCK) is expressed both centrally and peripherally and is involved in various basic cellular biological activities including migration, proliferation and apoptosis/survival.7,8 A series of studies have demonstrated that it is crucially responsible for the lack of axonal regeneration in the CNS.9,10 ROCK inhibitor Y-27632 reduced leucocyte infiltration in several models of inflammation, such as ischaemic heart injury, and endotoxic liver and lung injury.11–13 Fasudil (1-[5-isoquinolinesulphonyl]-homopiperazine), a ROCK inhibitor, has been widely used clinically since 1995 for the treatment of subarachnoid haemorrhage in Japan.14 Evaluating the role of ROCK signalling in an optic nerve lesion model showed that pharmacological inhibition of ROCK significantly enhanced regeneration of optic nerve axons after lesion.15 Interestingly, ROCK inhibition has yielded neuroprotective effects and has prevented apoptosis of retinal ganglion cells after optic nerve axotomy.16,17 Our group and others have also found that inhibition of ROCK by Fasudil ameliorated the development of EAE, possibly by blocking inflammatory responses in the CNS.18,19
Currently, clinical application of Fasudil is mainly limited to short-term treatment of frequent injections (twice a day) for acute onset of CNS diseases (≤ 2 weeks). It is, therefore, not practical as a long-term MS therapy. Further, we have observed that Fasudil's security window is narrow (data not shown). Given the complex pathology of MS, it is necessary to develop multi-functional ROCK inhibitor derivatives that are effective, safe, easily delivered and more effective in the combat of MS. Traditionally, neurological disorders have been treated through peripheral administration (predominantly oral administration). However, because of the blood–brain barrier, which keeps foreign materials out, many molecules, particularly large and/or charged ones, do not easily enter the brain from the bloodstream. Additionally, the bioavailability of orally administered drugs can be greatly reduced by first-pass metabolism, which prevents all but a small amount of active drug from ultimately reaching the brain.20 Another concern with systemic administration is the production of undesirable, peripherally induced adverse effects. Although intra-cerebroventricular injection can deliver drugs directly to the brain, it is highly invasive and so not feasible for clinical applications. Intranasal administration, in contrast, has been shown to directly achieve CNS delivery of a variety of compounds;21,22 it rapidly increases CNS levels of these compounds, improves bioavailability, and has no peripherally induced adverse effects. It may, therefore, represent the most promising, novel, non-invasive method for delivering therapeutic substances directly to the CNS for the treatment of CNS disorders, such as MS, Alzheimer's disease, Parkinson's disease and Huntington's disease.23
In this study, we used a novel Fasudil derivative, FSD-C10, via direct intranasal delivery, and explored its therapeutic effect and systemic response, as well as possible mechanisms in the treatment of EAE.
Materials and methods
Mice
Female C57BL/6 mice (8–10 weeks and 18–20 g) were purchased from Vital River Laboratory Animal Technology Co. Ltd. (Beijing, China). All experiments were conducted in accordance with the International Council for Laboratory Animal Science guidelines. The study was approved by the Council for Laboratory and Ethics Committee of Shanxi Datong University, Datong, China. All mice were housed under pathogen-free conditions and kept in a reversed 12 : 12-hr light/dark cycle in a temperature-controlled room (25 ± 2°) for 1 week before experimental manipulation.
Induction of chronic EAE
Mouse myelin oligodendrocyte glycoprotein peptide35–55 (MOG35–55, MEVGWYRSPFSRVVHLYRNGK) was produced in an automatic synthesizer (CL Bio-Scientific. Company, Xi'an, China). Purity of the peptide was > 95% as determined by HPLC.
Chronic EAE was induced by subcutaneous immunization on the upper dorsal flanks with 300 μg of MOG35–55 in Freund's complete adjuvant (Sigma, St Louis, MO) supplemented with 3 mg/ml of Mycobacterium tuberculosis H37Ra (BD Difco, Franklin Lakes, NJ) (400 μg/mice). Mice were injected with 750 ng of pertussis toxin (Enzo Life Sciences, Farmingdale, NY) into the abdominal cavity at the same time as immunization and again 48 hr later. Animals were weighed and evaluated for clinical score daily [from day 8 to day 28 post-immunization (p.i.)] in a blinded fashion by at least two investigators. Clinical score of EAE was graded according to the following criteria: 0, healthy; 1, limp tail; 2, ataxia and/or paresis of hind limbs; 3, paralysis of hind limbs and/or paresis of forelimbs; 4, tetraparalysis; and 5, moribund or death. Once the clinical score of EAE reached 3, we provided special care, i.e. softening the food with water in a dish, adding nutrients such as egg, and putting the dish at the bottom of the cage, making it easy for mice to obtain food, water and nutrition.
Administration of Fasudil derivative FSD-C10
Mice were divided into two groups: FSD-C10 experimental group (FSD-C10-treated group) and ddH2O control group (EAE control group) (n = 12 each group). FSD-C10 is a Fasudil derivative that is easily soluble in ethanol, and suitably water-soluble. FSD-C10 (from Tianjin Chase Sun Pharmaceutical Co., Ltd, Tianjin, China) was dissolved in sterile ddH2O. For intranasal administration, mice were lightly anaesthetized with diethylether, and then received 10 μl/nostril (5 μg/μl) at the entrance of the nostrils on day 3 p.i. until day 27 p.i., allowing the animal to sniff the solution into the upper nasal cavity. Mice that received the same volume of ddH2O nasally served as untreated EAE controls.
Histopathology and immunohistochemistry
On day 28 p.i., mice were perfused with saline and 4% buffered paraformaldehyde. Brains and spinal cords (lower thoracic–lumbar) were sliced (10 μm), and pathological changes were detected by haematoxylin & eosin staining and Luxol Fast Blue staining. For immunohistochemistry, non-specific binding was blocked with 1% BSA (Sigma), and permeabilized with 0·3% Triton X-100 in 1% BSA-PBS for 30 min. The sections were incubated at 4° overnight with anti-CD4 (Sigma), anti-CD68 (Serotec, Kidlington, UK), anti-CD11b (eBiosence, San Diego, CA), anti-GFAP (Millipore, Billericay, MA), and anti-p-nuclear factor-κB (NF-κB)/p65 (Cell Signaling Technology, Danvers, MA), anti-neuron specific nuclear protein (anti-NeuN; Millipore), anti-microtubule associated protein-2 (anti-MAP2; Millipore), anti-forkhead box p3 (anti-FOXP3; Millipore), anti- brain-derived neurotrophic factor (anti-BDNF; Promega, Madison, WI) and anti-nerve growth factor (anti-NGF; Promega), then incubated with the corresponding secondary antibodies at room temperature for 2 hr. As a negative control, additional sections were treated similarly, but the primary antibodies were omitted. Results were visualized under fluorescence microscopy by image-pro plus software (Media Cybernetics, Inc., Rockville, MD) in a blinded fashion. Quantification was performed on three sections per mouse, and four mice per group were analysed in a blinded fashion.
Cytokine measurement by ELISA
On day 28 p.i., mice were killed, and brains and spinal cords were homogenized on ice with an extraction kit supplemented with protease inhibitors (Millipore) in a microcontent motor-operated tissue homogenizer (Kimble Kontes Glass Co., Vineland, NJ). Lysates were centrifuged at 10 000 g for 20 min at 4°, and protein concentration was determined by a bicinchoninic acid protein assay kit (Pierce Biotechnology, Rockford, IL). Levels of interleukin-β (IL-1β), IL-6, tumour necrosis factor-α (TNF-α), interferon-γ (IFN-γ), IL-17 and IL-10 (Peprotech, Rocky Hill, NJ) were measured using sandwich ELISA kits in accordance with the manufacturer's instructions. Determinations were performed in duplicate and results were expressed as pg/10 mg protein.
Flow cytometry analysis
Splenocytes were prepared on day 28 p.i. by pushing spleen trough a sterile 70-μm nylon cell strainer (BD, Franklin Lakes, NJ) to generate a single-cell suspension. Mononuclear cells were stained for 20 min at room temperature in 1% BSA-PBS buffer with the following panel of antibodies: allophycocyanin-conjugated anti-CD4 (eBioscience) and phycoerythrin-conjugated anti-CD25 (eBioscience). Data were acquired on a FACSCalibur and analysed using cellquest software (Becton Dickinson, Bedford, MA).
Assay of ROCK activity
ROCK activity was measured by The CycLex Research Product ROCK assay kit, which is used to determine ROCK activity in tissue cytosols and cell extracts (cyLex Co., Ltd, Nagano, Japan). On day 28 p.i., mice were killed, and brain, spinal cord and spleen were homogenized on ice in four volumes of an appropriate extraction buffer (50 mm Tris–HCl, pH 8·0, 0·1% Triton X-100, 1 mm EDTA, 1 mm EGTA, 0·5 mm PMSF, 10 mm NaF, 10 mm β-mercaptoethanol), and centrifuged for 30 min at 12 000 g to pellet the insoluble membrane/organelle fraction. ROCK activity in the supernatant fraction was determined following the manufacturer's instructions. Results were expressed as optical density at 450 nm (OD450).
Statistical analysis
All experiments were repeated two or three times and graphpad prism software (Cabit Information Technology Co., Ltd., Shanghai, China) was used for statistical analysis. For clinical mean score, non-parametric Kruskal–Wallis test was performed to determine whether an overall statistically significant change existed before using the Mann–Whitney U-test to analyse the difference between any two groups. Student's t-test was used to compare demyelination, inflammation and in vitro experiments. Statistical significance for the other results was analysed using two-way analysis of variance with multiple comparison post test (Bonferroni). Significance level was set at P < 0·05.
Results
Intranasal administration of FSD-C10 suppresses the activity of ROCK
We first explored whether intranasal administration of FSD-C10 had an inhibitory effect on ROCK activity in EAE. As shown in Fig. 1, while certain levels of ROCK activity were exhibited in spinal cord, brain and spleen of untreated EAE mice, this activity was significantly inhibited in spinal cord and spleen, but not in brain, of mice treated by intranasal administration of FSD-C10 (P < 0·01 for both spinal cord and spleen). These results show that FSD-C10, delivered via intranasal route, has a potent inhibitory effect on ROCK activity in vivo.
Figure 1.
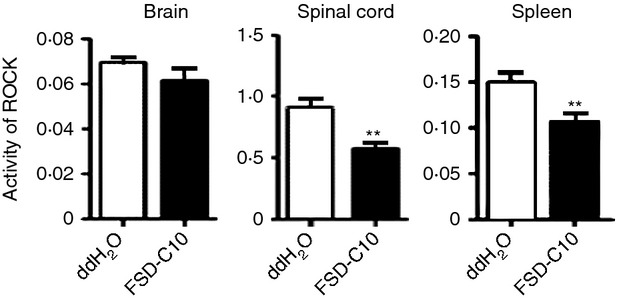
FSD-C10 inhibits Rho kinase (ROCK) activity. Brain, spinal cord and spleen of mice with experimental autoimmune encephalomyelitis (EAE) treated with nasal FSD-C10 or saline were harvested at day 28 post-immunization (n = 12 each group). These tissues were homogenized, and ROCK activity in the supernatant fraction was measured by commercial kit. **P < 0·01.
Intranasal FSD-C10 attenuates the severity of EAE and improves demyelination
In the present study, mice were immunized with MOG35-55 peptide to develop an EAE model that closely imitates many characteristics of MS. Starting on day 3 p.i., mice received, daily, a 20-μl drop containing 5 μg/μl FSD-C10 or ddH2O by bilateral intranasal instillation. Clinical score and bodyweight were then monitored from day 8 to 28 p.i. As shown in Fig. 2(a) and Table 1, the incidence of disease (33·3%) in FSD-C10-treated mice was significantly decreased compared with EAE control mice (100%). In the EAE control group, the mean onset date was at day 14·63 p.i., and the mean maximum score was 2·81. Intranasal administration of FSD-C10 delayed onset (mean onset date = 20·67, P < 0·05 versus EAE control) and reduced maximum clinical score (mean maximum score = 0·8, P < 0·01 versus EAE control), showing significantly attenuated development of EAE in FSD-C10-treated mice. It is known that there is a relationship between loss of body weight and severity of clinical score during the course of EAE. We also observed that intranasal treatment of FSD-C10 reduced body weight loss as compared with that of EAE control mice (Fig. 2a).
Figure 2.
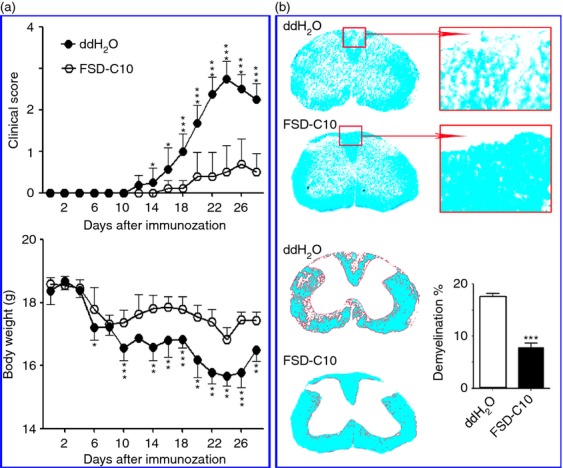
FSD-C10 ameliorates the severity of experimental autoimmune encephalomyelitis (EAE) and improves demyelination. FSD-C10 was given by nasal instillation and saline was set up as control (n = 12 each group) in a similar manner. (a) Clinical score of EAE mice and mean bodyweight, and (b) representative microphotographs for demyelination (Luxol Fast Blue staining) and quantitative analysis of spinal cord lesions. Data represent mean standard error (SEM) of demyelination score from seven or eight mice in each group. *P < 0·05, **P < 0·01, ***P < 0·001.
Table 1.
The clinical evaluation of mice with experimental autoimmune encephalomyelitis
| Group | n | Incidence (%) | Mean onset date | Mean score of maximal symptom |
|---|---|---|---|---|
| ddH2O treatment | 12 | 100 | 14·63 ± 2·39 | 2·81 ± 0·59 |
| FSD-C10 treatment | 12 | 33·3* | 20·67 ± 5·03* | 0·8 ± 0·75** |
FSD-C10 was administrated by bilateral intranasal instillation at 5 mg/kg/day daily starting from day 3 post-immunization.
P < 0·05,
P < 0·01.
At the end of the experiment on day 28 p.i., we evaluated the effect of intranasal FSD-C10 on CNS demyelination using Luxol Fast Blue staining. Consistent with clinical observation, although extensive demyelination was observed in the white matter of spinal cords of EAE control mice, foci of demyelination were rare in FSD-C10-treated mice, and the difference was significant (P < 0·001, Fig. 2b).
Intranasal FSD-C10 increases expression of NeuN protein, MAP2,BDNF and NGF in EAE
We explored the effect of nasally administered FSD-C10 on CNS neuroprotection in EAE mice. Immunocytochemical analysis revealed a particularly evident immunoreactivity of MAP2 and NeuN in the entire spinal cord of all the mice. While reactivity of these markers was not significantly different in the grey matter, their density in the white matter was considerably enhanced after administration of FSD-C10 (Fig. 3a). We also determined expression levels of neurotrophic factors in the spinal cord, given the endogenous neuroprotective role of these factors, and found that BDNF is mainly in the grey matter, while NGF is mainly in the white matter. FSD-C10 promotes the expression of BDNF and NGF protein in the spinal cord (Fig. 3b). Expression of neural and neurotrophin markers in brain, including MAP2, NeuN, NGF and BDNF, was not significantly different between FSD-C10-treated and untreated groups (data not shown).
Figure 3.

Intranasal FSD-C10 improves expression of neural markers brain-derived neurotrophic factor (BDNF) and nerve growth factor (NGF) protein in experimental autoimmune encephalomyelitis (EAE). Lumbar regions of spinal cords were harvested for immunostaining of NeuN, MAP2, NGF and BDNF. (a) Representative microphotographs and quantitative analysis for expression of NeuN and MAP2 in spinal cords, (b) representative microphotographs and quantitative analysis for the expression of BDNF and NGF in spinal cords. BDNF is mainly in the grey matter, while NGF is mainly in the white matter. Quantitative analyses represent mean standard error (SEM) from seven or eight mice in each group. ***P < 0·001.
Intranasal FSD-C10 inhibits inflammatory infiltration into the CNS
We then evaluated the effect of FSD-C10 nasal administration on infiltration of inflammatory cells into the CNS of EAE mice. As shown in Fig. 4, extensive CNS inflammatory infiltration was found in control EAE mice by haematoxylin and eosin staining, while intranasal administration of FSD-C10 obviously reduced inflammatory infiltration foci (Fig. 4a). When cell types of CNS inflammatory infiltration were determined by immunostaining, we found that spinal cord and brain of EAE control mice contained high numbers of CD4+ T cells (Fig. 4b) and CD68+ macrophages (Fig. 4c), and the numbers of these cells was significantly reduced in mice treated with nasal FSD-C10 (all P < 0·001).
Figure 4.

FSD-C10 suppresses central nervous system inflammation. Brains and lumbar regions of spinal cords were harvested for haematoxylin & eosin staining and immunostaining. (a) Representative microphotographs for inflammatory haematoxylin eosin (H & E) response of spinal cords and quantitative analysis of spinal cord lesions; (b) infiltration of CD4 T cells in brains and spinal cords (green) and quantitative analysis of brains and spinal cords; and (c) infiltration of CD68 macrophages in brains and spinal cords (red) and quantitative analysis of brains and spinal cords. Data represent mean standard error (SEM) from seven or eight mice in each group. ***P < 0·001.
Intranasal FSD-C10 inhibits NF-κB-mediated inflammatory responses in the CNS
It was considered likely that an increased number of infiltrating immune cells would produce pro-inflammatory mediators, so creating an inflammatory microenvironment in the CNS, and that FSD-C10 would switch this into an immunomodulatory one. To test this hypothesis, we measured levels of inflammatory cytokines such as IFN-γ, IL-17, IL-1β, IL-6 and TNF-α in brain and spinal cord. As expected, levels of IL-1β, IL-6, IL-17 and TNF-α were significantly suppressed, and the level of IL-10 was significantly enhanced in mice treated with intranasal administration of FSD-C10 compared with control EAE mice (Fig. 5, P < 0·01–0·001). The level of IFN-γ production was also increased in FSD-C10-treated mice compared with EAE control mice (P < 0·001). These results indicate that intranasal administration of FSD-C10 not only suppressed CNS infiltration, but also changed the CNS microenvironment from pro-inflammatory to immunomodulatory.
Figure 5.
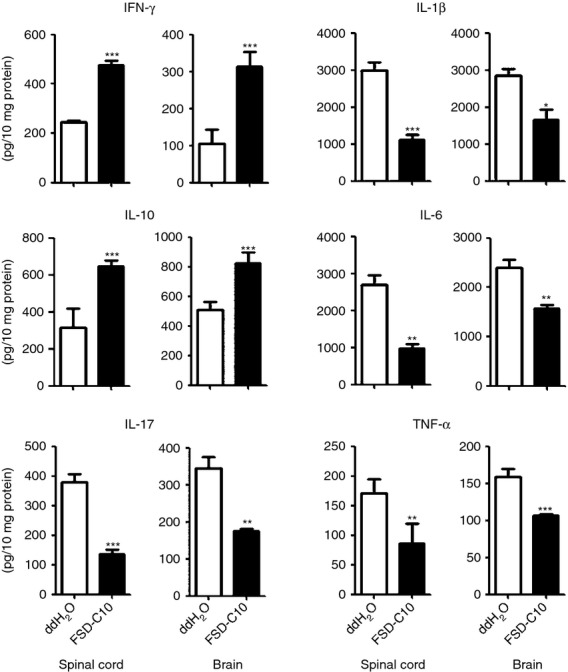
FSD-C10 reduces inflammatory responses in the central nervous system. Brains and spinal cords were collected, and protein extracts were obtained. Production of interferon-γ (IFN-γ), interleukin-10 (IL-10), IL-17, IL-1β, IL-6 and tumour necrosis factor-α (TNF-α) was measured using ELISA kits. Quantitative analysis represents mean standard error (SEM) form seven or eight mice in each group.*P < 0·05; **P < 0·01, ***P < 0·001.
To further define the molecular mechanism underlying the anti-inflammatory effect of intranasal administration of FSD-C10, we determined NF-κB activation in CNS tissue, given the accumulating evidence that the NF-κB signalling pathway contributes to inflammatory responses. Indeed, brain and spinal cord of EAE control mice exhibited a high level of p-NF-κB/p65 expression, and this expression was significantly inhibited after FSD-C10 treatment (Fig. 6a,b, P < 0·001).
Figure 6.
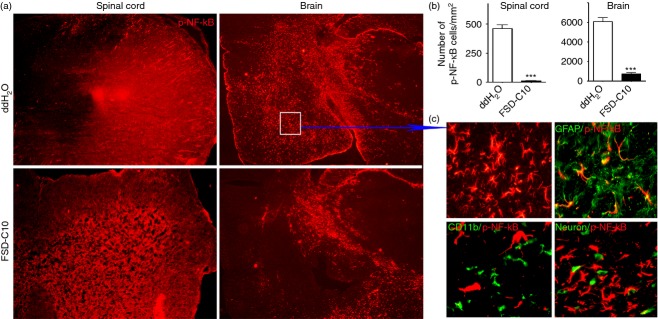
FSD-C10 inhibits expression of p-nuclear factor-κB (NF-κB)/p65 in the central nervous system. Brains and lumbar regions of spinal cords were harvested for immunostaining of p-NF-κB/p65. (a) Representative microphotographs for expression of p-NF-κB/p65 in brains and spinal cords; (b) quantitative analysis of p-NF-κ B/p65 positive cells in brains and spinal cords; and (c) location of p-NF-κ B/p65 in GFAP+ astrocytes, but not in microglia and neurons. Quantitative analyses represent mean standard error (SEM) from seven or eight mice in each group. ***P < 0·001.
When the cellular localization of p-NF-κB/p65 expression in brain was addressed, we found that p-NF-κB/p65 was extensively observed throughout the whole brain. Double immunohistochemistry showed that astrocytes, but not microglia or neurons, overlapped with p-NF-κB/p65 (Fig. 6c). These results suggest that astrocytes are the main cell source of p-NF-κB/p65 activation in EAE, and that intranasal administration of FSD-C10 inhibited activation of p-NF-κB/p65 on astrocytes.
Taken together, our results demonstrate that intranasal administration of FSD-C10 ameliorates disease progression in EAE, possibly by targeting the NF-κB-mediated inflammatory pathway.
Intranasal FSD-C10 modulates systemic immune responses in EAE
Although intranasal delivery of drugs is directly targeted to the CNS, it has been reported that this type of drug administration results in significantly higher bioavailability and higher plasma drug levels than the oral pathway.24–26 In this study, we explored whether intranasal administration of FSD-C10 could modulate systemic immune responses in EAE mice. As shown in Fig. 7, percentages and numbers of CD4+ CD25+, CD4+ FOXP3+ regulatory T cells were increased in splenocytes of mice treated with FSD-C10 compared with EAE control mice (P < 0·001), indicating an induction of regulatory T cells.
Figure 7.
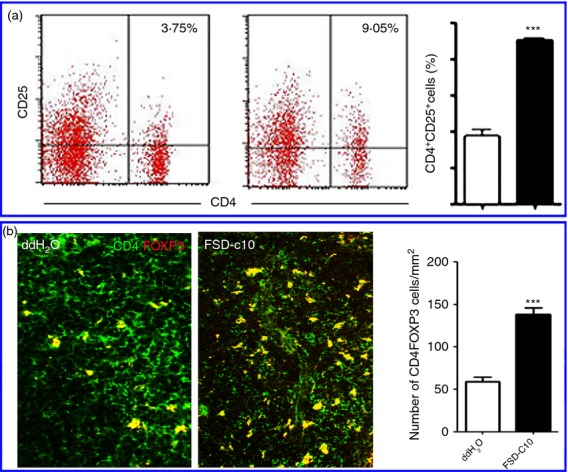
FSD-C10 modulates peripheral immune responses in spleen. (a) Splenic mononuclear cells (MNCs) were prepared, stained with T-cell marker CD4 and CD25, and percentages of CD4+ CD25+ regulatory T cells were measured by flow cytometry. Representative dot plots of CD4+ CD25+ regulatory T cells from one of three experiments with similar results and quantitative analysis of CD4+ CD25+ regulatory T cells; (b) immunostaining of CD4 (green) and FOXP3 (red) in spleen; yellow cells indicate merging of CD4+ with FOXP3+ cells. *P < 0·05, ***P < 0·001.
Discussion
The major finding of this study is that intranasal administration of FSD-C10 delays the onset of EAE, attenuates clinical severity, suppresses inflammation and demyelination, and promotes neuroprotection.
Our previous study showed that Fasudil is effective in the treatment of EAE, but that its security window is narrow as regards neurotoxicity. It is necessary to further develop multifunctional Fasudil derivatives for an effective, safe and easily delivered approach (e.g. oral or nasal administration) as a long-term MS therapy, given the life-long disease course of MS. Our data show a significant reduction in demyelination areas in mice treated with FSD-C10, which is consistent with previous findings about Fasudil. Immunofluoresence labelling indicated that the number of infiltrated inflammatory cells also decreased after intranasal administration of FSD-C10. Simultaneously, compared with control, FSD-C10-treated mice exhibited neuroprotection in the spinal cord. These data support the hypothesis that intranasal FSD-C10 affects autoimmune responses in the periphery and in inflammatory foci of the CNS and plays a neuroprotective role in EAE. To our knowledge, this is the first time that the effect of intranasal FSD-C10 on anti-inflammation and neuroprotection has been evaluated.
Pioneering studies demonstrated that inhibition of ROCK can reduce inflammatory responses, promote synapse formation and mobilize neural stem cells to differentiate into oligodendrocytes,the myelin-forming cells.9 Intraperitoneal injection of ROCK inhibitor Fasudil ameliorated clinical severity of EAE, accompanied by a decrease in demyelination and suppression of inflammatory cells.27 Whether FSD-C10 also plays an anti-inflammatory and neuroprotective role is not known. In our current study, intranasal delivery of FSD-C10 suppressed EAE, with a significant reduction in the production of pro-inflammatory cytokines IL-1β, IL-6, IL-17 and TNF-α, as well as decreased NF-κB activation. In contrast, production of immunoregulatory cytokine IL-10 was significantly increased. Further, FSD-C10 treatment effectively increased the percentage of CD4+ CD25+ T cells, as well as Foxp3 expression, indicating a regulatory T-cell phenotype. Collectively, these data support an immunoregulatory effect of FSD-C10.
Although IFN-γ serves as a hallmark of T helper type 1 (Th1) cells, the effect of this cytokine on EAE is questionable. Interferon-γ-deficient mice were susceptible to EAE, showed massive inflammatory infiltrates, and had poorer clinical outcomes than control mice.28 This result was further confirmed by using antibodies to neutralize IFN-γ, which showed that monoclonal anti-IFN-γ antibodies exacerbated the clinical symptoms of EAE.29 Our previous and present studies also showed that Fasudil and FSD-C10 increase the production of IFN-γ and attenuate the clinical score of EAE. These results suggest that IFN-γ can provide a certain degree of immunomodulation against disease progression. Interferon-γ signalling to astrocytes limited demyelination and down-regulated inflammation during acute EAE.30 Exogenous administration of IFN-γ decreased Th17 cells in EAE, orchestrating the number and function of Th17 cells.31
Interestingly, Fasudil and FSD-C10 have been found to possess a potentially neuroprotective function; however, the mechanism underlying this effect is not clear. We have observed in the present study the effect of intranasal FSD-C10 treatment on neurons. It was found that after treatment, the density of NeuN and MAP2 proteins in the FSD-C10 group was markedly higher than in the control group, and that NGF and BDNF protein expression of mice in the FSD-C10 group was statistically significantly higher than in the control group. NGF and BDNF are important neurotrophic factors, whose capacity to promote and maintain neurons against excitotoxicity and to promote the development, differentiation, growth and regeneration of a variety of nerve cells including neurons has been proved.32 By inducing these neurotrophic factors, FSD-C10 treatment could therefore be an important mechanism underlying its neuroprotective effect in EAE. The intranasal route allows rapid drug delivery directly from nasal mucosa to the brain via extracellular and intracellular mechanisms.21,22 Extracellular transport along peri-neuronal and peri-vascular channels is rapid and delivers drugs to the brain within minutes, whereas intracellular axonal delivery is a slow process, requiring internalization of the drug within the neurons and lasting from several hours to several days. The nasal route for drug delivery is a rapidly growing therapy, and there are currently more than 300 clinical trials under way in the USA alone using intranasal administration (http://www.clinical trials.gov). Nasal drug delivery is also being widely explored by pharmaceutical companies. Low-molecular-weight and lipophilic drugs are more effectively absorbed by the intranasal route for efficacious brain targeting, with a bioavailability close to 90%.33,34 In our previous study, the dosage of autoantigen by the nasal route was much smaller than by the oral route, yet induced immune tolerance to a similar extent.35–37 In our pilot studies, the dosage of intraperitoneal injection of Fasudil and FSD-C10 that attenuated clinical scores in EAE was 800 μg/mouse/day (data not shown). However, in this study, we used only 100 μg of FSD-C10 to treat EAE mice by the intranasal route, and this dosage also had a significant therapeutic effect and pathogenic improvement. These results indicate that intranasal delivery of a low dose of the small lipophilic molecule FSD-C10 holds great therapeutic potential. Unlike intranasal administration of hydrophilic compounds, which typically target only the brain, with low or no systemic exposure, intranasal delivery of a small lipophilic molecule may have systemic exposure, and studies on the efficacy of this approach are lacking.38
In conclusion, our results demonstrate that, unlike Fasudil, which has a narrow safety window and cannot be used for frequent injections, its derivative, FSD-C10, can easily be delivered by nasal administration and it effectively suppresses EAE by inhibiting neuroinflammation in the CNS and also by regulating immune responses in the periphery and promoting neuroprotection. Thus, our present study using intranasal delivery of the novel ROCK inhibitor represents a cutting-edge approach for the rapid, easy and non-invasive delivery of drugs into the CNS for treatment of EAE, eventually for MS and probably other diseases of the CNS.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (Nos. 81272163, 81070957 and 81371414), the Natural Science Foundation of Shanxi (2012021034-2), and by grants from the Department of Science and Technology, Shanxi Province of China (No. 2013081058) and from Shanxi University of Traditional Chinese Medicine (No. 2011PY-1). We thank Katherine Regan for editorial assistance.
Glossary
- CNS
central nervous system
- EAE
experimental allergic encephalomyelitis
- MOG35–55
myelin oligodendrocyte glycoprotein peptide35–55
- MS
multiple sclerosis
- ROCK
Rho kinase
Disclosures
None of the authors has any conflict of interest related to this manuscript.
References
- 1.Patani R, Chandran S. Experimental and therapeutic opportunities for stem cells in multiple sclerosis. Int J Mol Sci. 2012;13:14470–91. doi: 10.3390/ijms131114470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang J, Yan Y, Ciric B, et al. Evaluation of bone marrow- and brain-derived neural stem cells in therapy of central nervous system autoimmunity. Am J Pathol. 2010;177:1989–2001. doi: 10.2353/ajpath.2010.091203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stamatovic SM, Keep RF, Kunkel SL, et al. Potential role of MCP-1 in endothelial cell tight junction ‘opening’: signaling via Rho and Rho kinase. J Cell Sci. 2003;116:4615–28. doi: 10.1242/jcs.00755. [DOI] [PubMed] [Google Scholar]
- 4.Sagar D, Lamontagne A, Foss CA, et al. Dendritic cell CNS recruitment correlates with disease severity in EAE via CCL2 chemotaxis at the blood–brain barrier through paracellular transmigration and ERK activation. J Neuroinflammation. 2012;9:245. doi: 10.1186/1742-2094-9-245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Filippini G, Del Giovane C, Vacchi L, et al. Immunomodulators and immunosuppressants for multiple sclerosis: a network meta-analysis. Cochrane Database Syst Rev. 2013;6:CD008933. doi: 10.1002/14651858.CD008933.pub2. [DOI] [PubMed] [Google Scholar]
- 6.Stroet A, Linker RA, Gold R. Advancing therapeutic options in multiple sclerosis with neuroprotective properties. J Neural Transm. 2013;120(Suppl. 1):S49–53. doi: 10.1007/s00702-013-1037-6. [DOI] [PubMed] [Google Scholar]
- 7.Street CA, Bryan BA. Rho kinase proteins–pleiotropic modulators of cell survival and apoptosis. Anticancer Res. 2011;31:3645–57. [PMC free article] [PubMed] [Google Scholar]
- 8.Guilluy C, Garcia-Mata R, Burridge K. Rho protein crosstalk: another social network? Trends Cell Biol. 2011;21:718–26. doi: 10.1016/j.tcb.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mueller BK, Mack H, Teusch N. Rho kinase, a promising drug target for neurological disorders. Nat Rev Drug Discov. 2005;4:387–98. doi: 10.1038/nrd1719. [DOI] [PubMed] [Google Scholar]
- 10.Tönges L, Koch JC, Bähr M, et al. ROCKing regeneration: Rho kinase inhibition as molecular target for neurorestoration. Front Mol Neurosci. 2011;4:39. doi: 10.3389/fnmol.2011.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bao W, Hu E, Tao L, et al. Inhibition of Rho kinase protects the heart against ischemia/reperfusion injury. Cardiovasc Res. 2004;61:548–58. doi: 10.1016/j.cardiores.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 12.Slotta JE, Laschke MW, Menger MD, et al. Rho-kinase signalling mediates endotoxin hypersensitivity after partial hepatectomy. Br J Surg. 2008;95:976–84. doi: 10.1002/bjs.6082. [DOI] [PubMed] [Google Scholar]
- 13.Ding RY, Zhao DM, Zhang ZD, et al. Pretreatment of Rho kinase inhibitor inhibits systemic inflammation and prevents endotoxin-induced acute lung injury in mice. J Surg Res. 2011;171:e209–14. doi: 10.1016/j.jss.2011.08.009. [DOI] [PubMed] [Google Scholar]
- 14.Zhao J, Zhou D, Guo J, et al. Effect of fasudil hydrochloride, a protein kinase inhibitor, on cerebral vasospasm and delayed cerebral ischemic symptoms after aneurysmal subarachnoid hemorrhage. Neurol Med Chir. 2006;46:421–8. doi: 10.2176/nmc.46.421. [DOI] [PubMed] [Google Scholar]
- 15.Lingor P, Teusch N, Schwarz K, et al. Inhibition of Rho kinase (ROCK) increases neurite outgrowth on chondroitin sulphate proteoglycan in vitro and axonal regeneration in the adult optic nerve in vivo. J Neurochem. 2007;103:181–9. doi: 10.1111/j.1471-4159.2007.04756.x. [DOI] [PubMed] [Google Scholar]
- 16.Lingor P, Tönges L, Pieper N, et al. ROCK inhibition and CNTF interact on intrinsic signalling pathways and differentially regulate survival and regeneration in retinal ganglion cells. Brain. 2008;131:250–63. doi: 10.1093/brain/awm284. [DOI] [PubMed] [Google Scholar]
- 17.Planchamp V, Bermel C, Tönges L, et al. BAG1 promotes axonal outgrowth and regeneration in vivo via Raf-1 and reduction of ROCK activity. Brain. 2008;131:2606–19. doi: 10.1093/brain/awn196. [DOI] [PubMed] [Google Scholar]
- 18.Sun X, Minohara M, Kikuchi H, et al. The selective Rho-kinase inhibitor Fasudil is protective and therapeutic in experimental autoimmune encephalomyelitis. J Neuroimmunol. 2006;180:126–34. doi: 10.1016/j.jneuroim.2006.06.027. [DOI] [PubMed] [Google Scholar]
- 19.Hou SW, Liu CY, Li YH, et al. Fasudil ameliorates disease progression in experimental autoimmune encephalomyelitis, acting possibly through antiinflammatory effect. CNS Neurosci Ther. 2012;18:909–17. doi: 10.1111/cns.12002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Illum L. Transport of drugs from the nasal cavity to the central nervous system. Eur J Pharm Sci. 2000;11:1–18. doi: 10.1016/s0928-0987(00)00087-7. [DOI] [PubMed] [Google Scholar]
- 21.Bitter C, Suter-Zimmermann K, Surber C. Nasal drug delivery in humans. Curr Probl Dermatol. 2011;40:20–35. doi: 10.1159/000321044. [DOI] [PubMed] [Google Scholar]
- 22.Jiang Y, Zhu J, Xu G, et al. Intranasal delivery of stem cells to the brain. Expert Opin Drug Deliv. 2011;8:623–32. doi: 10.1517/17425247.2011.566267. [DOI] [PubMed] [Google Scholar]
- 23.Danielyan L, Schäfer R, von Ameln-Mayerhofer A, et al. Intranasal delivery of cells to the brain. Eur J Cell Biol. 2009;88:315–24. doi: 10.1016/j.ejcb.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 24.Chapman CD, Frey WHII, Craft S, et al. Intranasal treatment of central nervous system dysfunction in humans. Pharm Res. 2013;30:2475–84. doi: 10.1007/s11095-012-0915-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hanson LR, Fine JM, Svitak AL, et al. Intranasal administration of CNS therapeutics to awake mice. J Vis Exp. 2013;8:e4440. doi: 10.3791/4440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nave R, Schmitt H, Popper L. Faster absorption and higher systemic bioavailability of intranasal fentanyl spray compared to oral transmucosal fentanyl citrate in healthy subjects. Drug Deliv. 2013;20:216–23. doi: 10.3109/10717544.2012.762435. [DOI] [PubMed] [Google Scholar]
- 27.Liu CY, Li YH, Yu JZ, et al. Targeting the shift from M1 to M2 macrophages in experimental autoimmune encephalomyelitis mice treated with fasudil. PLoS One. 2013;8:e54841. doi: 10.1371/journal.pone.0054841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferber IA, Brocke S, Taylor-Edwards C, et al. Mice with a disrupted IFN-γ gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE) J Immunol. 1996;156:5–7. [PubMed] [Google Scholar]
- 29.Lublin FD, Knobler RL, Kalman B, et al. Monoclonal anti-γ interferon antibodies enhance experimental allergic encephalomyelitis. Autoimmunity. 1993;16:267–74. doi: 10.3109/08916939309014645. [DOI] [PubMed] [Google Scholar]
- 30.Hindinger C, Bergmann CC, Hinton DR, et al. IFN-γ signaling to astrocytes protects from autoimmune mediated neurological disability. PLoS One. 2012;7:e42088. doi: 10.1371/journal.pone.0042088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berghmans N, Nuyts A, Uyttenhove C, et al. Interferon-γ orchestrates the number and function of Th17 cells in experimental autoimmune encephalomyelitis. J Interferon Cytokine Res. 2011;31:575–87. doi: 10.1089/jir.2010.0137. [DOI] [PubMed] [Google Scholar]
- 32.Chao MV. Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat Rev Neurosci. 2003;4:299–309. doi: 10.1038/nrn1078. [DOI] [PubMed] [Google Scholar]
- 33.Bahadur S, Pathak K. Physicochemical and physiological considerations for efficient nose-to-brain targeting. Expert Opin Drug Deliv. 2012;9:19–31. doi: 10.1517/17425247.2012.636801. [DOI] [PubMed] [Google Scholar]
- 34.Devillier P, Grassin-Delyle S, Naline E, et al. Intranasal delivery of systemic drugs: a new route for opioid drugs. Therapie. 2010;65:475–81. doi: 10.2515/therapie/2010052. [DOI] [PubMed] [Google Scholar]
- 35.Ma CG, Zhang GX, Xiao BG, et al. Suppression of EAMG by nasal administration of acetylcholine receptor. J Neuroimmunol. 1995;58:51–60. doi: 10.1016/0165-5728(94)00187-s. [DOI] [PubMed] [Google Scholar]
- 36.Shi FD, Bai XF, Xiao BG, et al. Nasal administration of multiple antigens suppresses experimental autoimmune myasthenia gravis, encephalomyelitis and neuritis. J Neurol Sci. 1998;155:1–12. doi: 10.1016/s0022-510x(97)00232-3. [DOI] [PubMed] [Google Scholar]
- 37.Bai XF, Shi FD, Xiao BG, et al. Nasal administration of myelin basic protein prevents relapsing experimental autoimmune encephalomyelitis in DA rats by activating regulatory cells expressing IL-4 and TGF-β mRNA. J Neuroimmunol. 1997;80:65–75. doi: 10.1016/s0165-5728(97)00133-1. [DOI] [PubMed] [Google Scholar]
- 38.Hoekman JD, Ho RJ. Effects of localized hydrophilic mannitol and hydrophobic nelfinavir administration targeted to olfactory epithelium on brain distribution. AAPS PharmSciTech. 2011;12:534–43. doi: 10.1208/s12249-011-9614-1. [DOI] [PMC free article] [PubMed] [Google Scholar]