

Abstract
Mounting evidence points to roles for miRNA gene regulation in promoting development, function, and cell survival in the mammalian retina. However, little is known regarding which retinal genes are targets of miRNAs. Here, we employed a systematic, nonbiased, biochemical approach to identify targets of miRNA gene regulation in the bovine retina, a common model species for vision research. Using Argonaute high-throughput sequencing of RNAs isolated by cross-linking immunoprecipitation analysis, we identified 348 high-confidence miRNA target sites within 261 genes. This list was enriched in rod and cone photoreceptor genes and included 28 retinal disease genes, providing further evidence of a role of miRNAs in the pathology of blinding diseases.
Substantial progress has been made recently in assessing the role of miRNAs in the mammalian visual system. Loss of DICER1 (and hence most mature miRNAs) in the developing mouse neuroretina led to aberrant differentiation of neuroretinal cell types or apoptosis of retinal progenitors.1,2 More recently, it was demonstrated that a loss of activity of the photoreceptor-enriched miR-183 cluster led to enhanced sensitivity to light damage- or age-induced retinal degeneration, suggesting a role for this cluster in promoting photoreceptor cell survival during cellular stress.3,4 Significant retinal degeneration was also reported in mature rod photoreceptor-specific Dicer1 conditional knockout mice, indicating that additional miRNAs are also important for the survival of rods.5 In addition, changes in miRNA expression have been demonstrated in models of experimental autoimmune uveoretinitis, diabetic retinopathy, retinitis pigmentosa, and retinoblastoma, suggesting a link between miRNAs and retinal degenerative diseases.2,6,7
Identification of physiologically relevant direct targets of miRNAs is of paramount importance to understanding their functional significance. Until recently, the use of target prediction algorithms followed by validation of direct miRNA targeting of selected 3′-UTRs in a cell culture transfection assay was the primary way to establish miRNA target genes. However, this approach has significant limitations. Target prediction algorithms exhibit a high rate of false positives and ignore several types of physiologically relevant targets. Factors other than seed region complementarity (i.e., expression of other miRNAs, differential expression of miRNA-induced silencing complex components, presence of other mRNA-binding factors, target mRNAs, target site-containing noncoding RNAs, etc.) can also regulate miRNA–target interactions.2 Therefore, it is vital to biochemically identify miRNA targets directly in a tissue of interest.
Combining the newly developed technologies of next-generation DNA sequencing (NGS) and cross-linking immunoprecipitation (CLIP) now makes it possible to systematically identify transcripts directly targeted by miRNAs. This technique, known as Argonaute high-throughput sequencing of RNAs isolated by crosslinking immunoprecipitation (HITS-CLIP), provides a “snapshot” of miRNA–target interactions in living cells from a given tissue.8 Argonaute family proteins directly interact with miRNAs to form the core of the miRNA-induced silencing complex that effects miRNA-mediated repression of gene expression. Argonaute family proteins can be cross-linked both to bound miRNAs and to the target mRNAs with which they interact in vivo, allowing high-stringency co-immunoprecipitation of target transcripts and their identification by NGS technology. As RNAs associate with Argonaute proteins postlysis, cross-linking is critical for detecting only invivo-relevant interactions.9 This approach constitutes a definitive biochemical method for studying miRNA–target interactions directly in a tissue of interest. However, no retinal miRNA–target interactions have yet been biochemically validated.
Here, we employed Argonaute HITS-CLIP to identify miRNA targets in the bovine retina, an animal model used extensively in vision research. Through an arrangement with a local slaughterhouse, we were able to isolate retinas and perform UV-induced cross-linking within 1 h of the death of an animal, minimizing the impact of post-mortem changes in gene expression on the identified miRNA target genes. The tissue was lysed, subjected to limited RNase digestion, and immunoprecipitated with an antibody specific to Argonaute family proteins.10 Cross-linked protein–RNA complexes were then radiolabeled by ligation of a 32P-labeled RNA linker oligonucleotide to the 3′ end of the RNA. These protein RNA complexes were resolved by electrophoresis and subjected to autoradiography (Figure 1a). Autoradiograms revealed a single, Argonaute-specific antibody-dependent band slightly larger than the size of bovine Argonaute family proteins (97–99 kDa), with a smear of higher-molecular weight (MW) complexes above it. This region of the membrane was excised and digested with protease; co-immunoprecipitated RNAs were eluted, and another RNA linker oligonucleotide was ligated to the 5′ end. cDNA was generated and amplified by polymerase chain reaction (PCR), revealing a major band corresponding to the length of the 3′ and 5′ linkers (41 nucleotides) plus the size of ∼22-nucleotide miRNAs (Figure 1b,c). Sequencing of cDNAs derived from this band revealed mainly miRNAs (not shown). In addition to this band, a smear of higher-MW complexes appeared at later cycle numbers, corresponding to fragments of co-immunoprecipitated mRNAs. Libraries were prepared for Illumina-based sequencing of these higher-MW cDNAs according to Chi et al.8 In parallel, libraries were prepared for NGS-based transcriptome analysis from the same bovine retina tissue samples. Libraries for transcriptome analysis and HITS-CLIP were prepared from three biological replicates of bovine retinal tissue. Enrichment analysis was performed, comparing the HITS-CLIP reads to the transcriptome reads using model-based analysis of ChIPseq (MACS) software.11 Enrichment analysis ensures that analysis of Argonaute-interacting RNAs is not biased by nonspecific interactions of Argonaute family proteins with the most abundant cellular RNAs. Peaks with a P value for enrichment of ≤1 × 10–20 present in all three biological replicates were considered high-confidence miRNA target sites. Panels d and e of Figure 1 show examples of how enrichment analysis along with the requirement for reproducibility in binding sites across three biological replicates selected high-confidence miRNA target sites.
Figure 1.
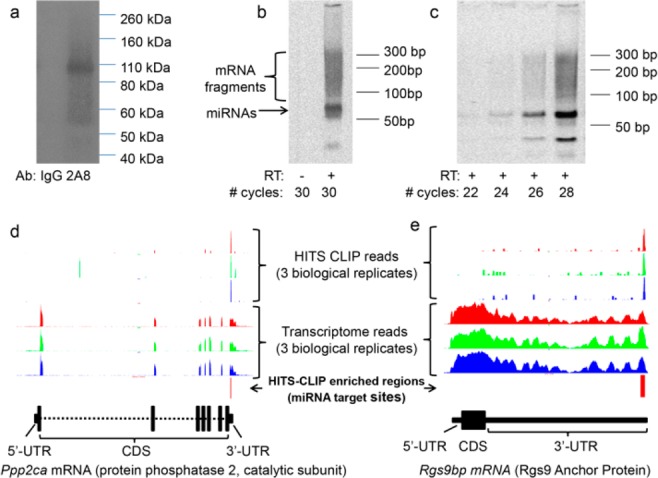
Bovine retina HITS-CLIP library production and analysis. (a) Autoradiograms of labeled protein–RNA complexes generated by CLIP using either normal IgG or the Argonaute-specific 2A8 mouse monoclonal antibody. (b and c) PCR amplification of cDNA derived from Argonaute CLIP on bovine retina. (d and e) Bovine retina Argonaute HITS-CLIP and transcriptome sequencing results for the miRNA target genes Ppp2ca and Rgs9bp were mapped onto the UCSC genome browser. High-stringency enrichment analysis requiring high-confidence peaks in all three biological replicates identified a single high-confidence miRNA target site in each transcript.
We identified 348 high-confidence miRNA target sites in the bovine retina that mapped to 261 genes (Table S1 of the Supporting Information). With more than 9600 genes expressed in the bovine retina at one read per kilobase of transcript per 1 million mapped reads (RPKM) or higher, this represents a precise high-confidence list. It should be noted, however, that as miRNAs have been implicated in responding to cellular stress, as well as conferring robustness to gene networks by buffering gene expression against potentially deleterious fluctuations, many miRNA–target mRNA interactions are likely to be transient in nature.12,13 Therefore, our list of retinal miRNA target genes is meant to represent a snapshot of retinal miRNA gene regulation and not a complete list of retinal miRNA-targeted genes.
The localization of miRNA targets is consistent with the biology of miRNA gene regulation with the bulk of the target sites (56%) located within the mRNA 3′-UTR and a smaller fraction (20%) residing within the coding sequence (Figure 2a). Several additional high-confidence miRNA target sites were localized to long noncoding RNAs (lncRNAs), suggesting roles for miRNAs in the regulation of lncRNAs or a function for lncRNAs as miRNA sponges within the retina.14 MEME-based nonbiased analysis of 6–8mer sequence motifs enriched within the identified target sites identified perfect seed region complementarity to miR-124 family miRNAs and miR-183 cluster miRNAs as the second and third most statistically significantly enriched motifs, respectively (Figure 2b). Intriguingly, the most significantly enriched motif also matched the abundant miR-183 cluster miRNAs if one allows for one or two G-U wobble base pairs, suggesting that seed region G-U wobble base pairing could be an important component of miRNA target recognition in the retina.
Figure 2.
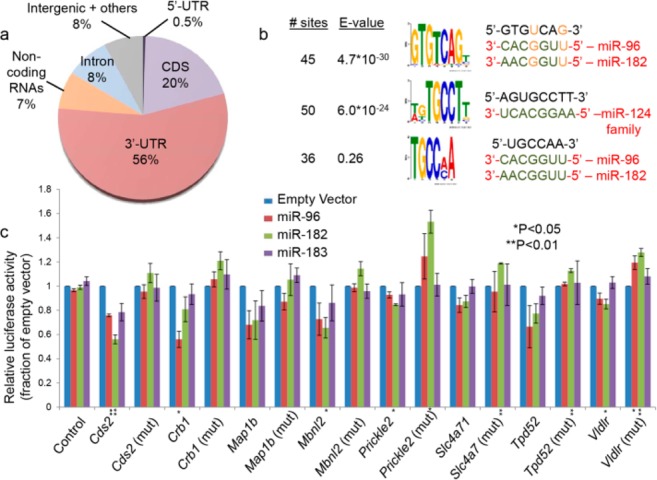
Retinal miRNA targets identified by Argonaute HITS-CLIP. (a) Pie chart showing the localization of identified miRNA target sites within retinal mRNAs. (b) A MEME-based, nonbiased search for 6–8mer sequence motifs enriched in retinal miRNA target sites identified seed region matches to abundant retinal miRNAs. (c) 293T cell culture-based luciferase assays show the effect of overexpressed miR-183 cluster miRNAs on expression of firefly luciferase reporter constructs containing identified miRNA target sites exhibiting perfect seed region complementarity to miR-183 cluster miRNAs. Constructs containing the same target gene fragment with the miR-183 cluster seed region match(es) mutated were also tested. Data represent means ± the standard deviation based on three independent experiments.
We used a 293T cell-based dual luciferase assay to validate repression by miR-183 cluster miRNAs of identified target sites bearing seed region matches. The ∼1 kb fragments surrounding the identified target sites were cloned into the 3′-UTR of the firefly luciferase gene in the pmiRGLO vector (Promega, Madison, WI). Eight of 10 constructs tested showed some level of repression upon cotransfection of plasmids driving overexpression of miR-183 cluster miRNAs, and this repression was relieved by deletion of three nucleotides in the center of the miRNA seed region complementary sequence (Figure 2C). Consistent with the notion that miRNAs “fine-tune” gene expression, repression was generally modest. miRs-96, -182, and -183 produced variable levels of repression upon being cotransfected with reporter vectors. While some overlap in miRNA target genes is expected given the seed region similarity among miR-183 cluster miRNAs, this result suggests that these miRNAs repress different sets of target genes. Taken together, these results provide validation for the identified high-confidence mammalian retinal miRNA target sites.
Mammalian retinal miRNA target genes conduct diverse functions, with genes involved in gene regulation, metabolism, signal transduction, and transport being the most abundant (Table S1 of the Supporting Information). Several recent reports have implicated rod and cone photoreceptors as cell types within the retina where miRNA gene regulation is particularly critical to survival and function.3−5,15,16 Consistent with this notion, 102 of 261 (39%) of the identified miRNA target genes were statistically significantly downregulated in rhodopsin knockout mice, suggesting enrichment in rod and cone photoreceptors (data are available in the GEO database). More compelling is the finding that 28 of 261 (11%) of the high-confidence retinal miRNA target genes were linked with retinal degenerative disorders in humans (Table S1 of the Supporting Information), suggesting a role for retinal miRNAs in the pathology of retinal degenerative diseases.
A wealth of emerging evidence links miRNA gene regulation to retinal degeneration and blinding diseases, making miRNAs potentially powerful new therapeutic targets for protecting or restoring vision. Both the isolation and accessibility of the visual system compartment make it ideally suited for gene therapy approaches. As such, miRNA-based gene therapy approaches to preventing or ameliorating retinal degeneration is an attractive option. Clearly, a much more detailed understanding of the physiological impact and molecular mechanism of action of retinal miRNAs is required to pursue therapeutic approaches. This systematic, nonbiased analysis of mammalian retinal miRNA target genes represents an initial step in this process.
Acknowledgments
We thank Zissimos Mourelatos (University of Pennsylvania, Philadelphia, PA) for antibodies and Leslie T. Webster, Jr. (Case Western Reserve University), for critical comments about the manuscript.
Supporting Information Available
Supporting Methods and Table S1. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by funding from the National Eye Institute, National Institutes of Health Grant R01EY022326, and the Arnold and Mabel Beckman Foundation. K.P. is the John H. Hord Professor of Pharmacology.
The authors declare no competing financial interests.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- La Torre A.; Georgi S.; Reh T. A. (2013) Proc. Natl. Acad. Sci. U.S.A. 110, E2362–E2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundermeier T. R.; Palczewski K. (2012) Cell. Mol. Life Sci. 69, 2739–2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q.; Sun W.; Okano K.; Chen Y.; Zhang N.; Maeda T.; Palczewski K. (2011) J. Biol. Chem. 286, 31749–31760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumayag S.; Haldin C. E.; Corbett N. J.; Wahlin K. J.; Cowan C.; Turturro S.; Larsen P. E.; Kovacs B.; Witmer P. D.; Valle D.; Zack D. J.; Nicholson D. A.; Xu S. (2013) Proc. Natl. Acad. Sci. U.S.A. 110, E507–E516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundermeier T. R.; Zhang N.; Vinberg F.; Mustafi D.; Kohno H.; Golczak M.; Bai X.; Maeda A.; Kefalov V. J.; Palczewski K. (2014) FASEB J. 28, 3780–3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genini S.; Guziewicz K. E.; Beltran W. A.; Aguirre G. D. (2014) BMC Genomics 15, 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray A. R.; Chen Q.; Takahashi Y.; Zhou K. K.; Park K.; Ma J. X. (2013) Invest. Ophthalmol. Visual Sci. 54, 1689–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi S. W.; Zang J. B.; Mele A.; Darnell R. B. (2009) Nature 460, 479–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley K. J.; Yario T. A.; Steitz J. A. (2012) RNA 18, 1581–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson P. T.; De Planell-Saguer M.; Lamprinaki S.; Kiriakidou M.; Zhang P.; O’Doherty U.; Mourelatos Z. (2007) RNA 13, 1787–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Liu T.; Meyer C. A.; Eeckhoute J.; Johnson D. S.; Bernstein B. E.; Nusbaum C.; Myers R. M.; Brown M.; Li W.; Liu X. S. (2008) Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelaez N.; Carthew R. W. (2012) Curr. Top. Dev. Biol. 99, 237–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell J. T.; Olson E. N. (2012) Cell 148, 1172–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay Y.; Rinn J.; Pandolfi P. P. (2014) Nature 505, 344–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busskamp V.; Krol J.; Nelidova D.; Daum J.; Szikra T.; Tsuda B.; Juttner J.; Farrow K.; Scherf B. G.; Alvarez C. P.; Genoud C.; Sothilingam V.; Tanimoto N.; Stadler M.; Seeliger M.; Stoffel M.; Filipowicz W.; Roska B. (2014) Neuron 3, 586–600. [DOI] [PubMed] [Google Scholar]
- Sanuki R.; Onishi A.; Koike C.; Muramatsu R.; Watanabe S.; Muranishi Y.; Irie S.; Uneo S.; Koyasu T.; Matsui R.; Cherasse Y.; Urade Y.; Watanabe D.; Kondo M.; Yamashita T.; Furukawa T. (2011) Nat. Neurosci. 14, 1125–1134. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.