

Abstract
RNA viruses are responsible for major human diseases such as flu, bronchitis, dengue, Hepatitis C or measles. They also represent an emerging threat because of increased worldwide exchanges and human populations penetrating more and more natural ecosystems. A good example of such an emerging situation is chikungunya virus epidemics of 2005-2006 in the Indian Ocean. Recent progresses in our understanding of cellular pathways controlling viral replication suggest that compounds targeting host cell functions, rather than the virus itself, could inhibit a large panel of RNA viruses. Some broad-spectrum antiviral compounds have been identified with host target-oriented assays. However, measuring the inhibition of viral replication in cell cultures using reduction of cytopathic effects as a readout still represents a paramount screening strategy. Such functional screens have been greatly improved by the development of recombinant viruses expressing reporter enzymes capable of bioluminescence such as luciferase. In the present report, we detail a high-throughput screening pipeline, which combines recombinant measles and chikungunya viruses with cellular viability assays, to identify compounds with a broad-spectrum antiviral profile.
Keywords: Immunology, Issue 87, Viral infections, high-throughput screening assays, broad-spectrum antivirals, chikungunya virus, measles virus, luciferase reporter, chemical libraries
Introduction
RNA viruses are responsible for a large variety of human infections, and have a huge impact on populations worldwide both in terms of public health and economical cost. Efficient vaccines have been developed against several human RNA viruses, and are widely used as prophylactic treatments. However, there is still a critical lack of therapeutic drugs against RNA virus infections. Indeed, efficient vaccines are not available against major human pathogens such as dengue virus, Hepatitis C virus or human respiratory syncytial virus (hRSV). Besides, RNA viruses are responsible for a majority of emerging diseases, which have increased in frequency because of global exchanges and human impact on ecological systems. Against this threat that represent RNA viruses, our therapeutic arsenal is extremely limited and relatively inefficient1-3. Current therapies are essentially based on recombinant type I interferons (IFN-α/β) to stimulate innate immunity, or the administration of ribavirin. Although the mode of action of this ribonucleoside analog is controversial and probably relies on various mechanisms, the inhibition of cellular IMPDH (inosine monophosphate dehydrogenase), which depletes intracellular GTP pools, is clearly essential4. Ribavirin, in combination with pegylated IFN-α, is the main treatment against Hepatitis C virus. However, IFN-α/β and ribavirin treatments are of relatively poor efficacy in vivo against most RNA viruses as they efficiently blunt IFN-α/β signaling through expression of virulence factors5 and often escape ribavirin3. This added to the fact that ribavirin treatment is raising important toxicity issues, although it was recently approved against severe hRSV disease with controversial benefits6. More recently, some virus-specific treatments have been marketed, in particular against influenza virus with the development of neuraminidase inhibitors3. However, the large diversity and permanent emergence of RNA viruses precludes the development of specific treatments against each one of them in a relatively close future. Altogether, this stresses the need for efficient strategies to identify and develop potent antiviral molecules in the near future.
It is trivial to say that a broad-spectrum inhibitor active against a large panel of RNA viruses would solve this problem. Although such a molecule is still a virologist's dream, our better understanding of cellular defense mechanisms and innate immune system suggest that some possibilities exist7,8. Several academic and industrial laboratories are now seeking molecules that stimulate specific facets of cellular defense mechanisms or metabolic pathways to blunt viral replication. Although such compounds will probably show significant side effects, treatments against acute viral infections will be administered for a relatively short time, making them acceptable despite some potential toxicity on the long term. Various strategies have been developed to identify such broad-spectrum antiviral molecules. Some research programs aim at finding molecules that target specific defense or metabolic pathways. This includes, for example, pathogen recognition receptors to elicit antiviral gene expression9 and activate antiviral factors such as RNaseL10, autophagy machinery to promote virus degradation11, nucleoside synthesis pathways12,13, or apoptotic cascades to precipitate death of virus-infected cells14. Other groups have developed phenotypic screens that are not target-based13,15-17. In that case, antiviral molecules are simply identified by their capacity to block viral replication in a given cellular system. The general assumption is that a compound inhibiting 2-3 unrelated RNA viruses would have a suitable profile for a broad-spectrum antiviral molecule. The mode of action of hit compounds selected with such an empirical approach is only determined in a second time and eventually, may lead to the identification of novel cellular targets for antivirals. Interestingly, a retrospective analysis of new drugs approved by the US Food and Drug Administration between 1999 and 2008 has shown that in general, such phenotypic screenings tend to perform better than target-based approaches to discover first-in-class small-molecule drugs18.
Viral replication in high-throughput cell-based assays is usually determined from virus cytopathic effects. Cells are infected and cultured in 96- or 384-well plates in the presence of tested compounds. After few days, cellular layers are fixed and stained with dyes such as crystal violet. Finally, absorbance is determined with a plate reader and compounds inhibiting viral replication are identified by their capacity to preserve cellular layers from virus-induced cytopathic effect. Alternatively, virally-mediated cytopathic effects are assessed using standard viability assays such as MTS reduction. Such assays are highly tractable and cost-effective, but suffer from three major limitations. First, they require a virus-cell combination where viral replication is cytopathic in only few days but this is not always possible, thus calling for alternative approaches19. Second, they are poorly quantitative since they are based on an indirect measure of viral replication. Finally, toxic compounds can be scored as positive hits, and therefore must be eliminated with a counter screen measuring cellular viability. To overcome some of these hurdles, recombinant viruses or replicons have been engineered by reverse genetics to express reporter proteins, such as EGFP or luciferase, from an additional transcription unit or in frame with viral protein genes (few examples are 20-23). When these viruses replicate, reporter proteins are produced together with viral proteins themselves. This provides a very quantitative assay to measure viral replication and evaluate the inhibitory activity of candidate molecules. This is particularly true for recombinant viruses expressing luciferase (or other enzymes capable of bioluminescence) since this reporter system exhibits a wide dynamic range with a high sensitivity and virtually no background. Furthermore, there is no excitation light source, thus preventing interference with compound fluorescence24.
Here, we detail a high-throughput protocol to screen chemical libraries for broad-spectrum inhibitors of RNA viruses. Compounds are tested first on human cells infected with a recombinant measles virus (MV) expressing firefly luciferase25 (rMV2/Luc, Figure 1, primary screen). MV belongs to Mononegavirales order, and is often considered as a prototypical member of negative-strand RNA viruses. As such, MV genome is used as a template by the viral polymerase to synthesize mRNA molecules encoding for viral proteins. In the recombinant MV strain called rMV2/Luc, luciferase expression is expressed from an additional transcription unit inserted between P and M genes (Figure 2A). In parallel, compounds are tested for their toxicity on human cells using a commercial luciferase-based reagent that evaluates, by ATP quantification, the number of metabolically active cells in culture (Figure 1, primary screen). Entire chemical libraries can be easily screened with these two assays in order to select compounds that are not toxic and efficiently block MV replication. Then, hits are retested for dose-response inhibition of MV replication, the lack of toxicity as well as for their capacity to impair chikungunya virus (CHIKV) replication (Figure 1, secondary screen). CHIKV is a member of Togaviridae family and its genome is a positive single-strand RNA molecule. As such, it is completely unrelated to measles and compounds inhibiting both MV and CHIKV stand a great chance to inhibit a large panel of RNA viruses. CHIKV nonstructural proteins are directly translated from the viral genome, whereas structural proteins are encoded by transcription and translation of a subgenomic mRNA molecule. Our in vitro replication assay for CHIKV is based on a recombinant strain called CHIKV/Ren, which expresses Renilla luciferase enzyme as a cleaved part of the nonstructural polyprotein through an insertion of the reporter gene between nsP3 and nsP4 sequences26 (Figure 2B). The measure of Renilla luciferase activity allows the monitoring of viral replication at the early stage of CHIKV life cycle.
This high-throughput protocol was used to quickly identify compounds with a suitable profile for broad-spectrum antivirals in a commercial library of 10,000 molecules enriched for chemical diversity. Compounds were essentially following Lipinski's rule of five, with molecular weights ranging from 250 to 600 daltons, and log D values below 5. Most of these molecules were new chemical entities not available in other commercial libraries.
Protocol
1. Preparation of 96-well Daughter Plates with Compounds
Move 96-well mother plates containing 10 mM stock solutions of chemical compounds in DMSO from -20 °C to room temperature. Dilute compounds 5x in DMSO to obtain intermediate 96-well dilution plates at 2 mM. Dilution is achieved by pipetting 5 μl into 20 μl of DMSO and mixing.
Pipette 1 μl from dilution plates into dry wells of white, bar-coded tissue culture 96-well plates. This first set of daughter plates (D1) will be used to evaluate the toxicity of compounds at a final concentration of 20 μM. Store daughter plates at -20 °C until use.
Dilute compounds again 1:10 by pipetting 4 μl from dilution plates into 36 μl of DMSO and mixing. Pipette 1 μl into dry wells of white, flat bottom, bar-coded tissue culture 96-wells plates. This second set of daughter plates (D2) will be used to evaluate the inhibition of MV replication by compounds at a final concentration of 2 μM. Store daughter plates at -20 °C until use.
2. Preparation of Cell Cultures to Determine Compound Toxicity
Grow HEK-293T cells at 37 °C and 5% CO2 in Dulbecco's modified Eagle's medium (DMEM) with stabilized L-glutamine and supplemented with 10% fetal calf serum (FCS), streptomycin (100 μg/ml) and penicillin (100 IU/ml). Cells are passaged by trypsinization every 4-5 days, and diluted 1:10 into a fresh flask. Cells should be in log phase for experiments.
On the day of the experiment, recover cells by trypsinization and perform cell counting. Pellet cells by centrifugation, and resuspend them at 3 x 105 cells/ml in culture medium. Consider that 12.5 ml of cell suspension are required for each screening plate.
Transfer cells to a trough, and dispense 100 μl of cell suspension in D1 plates containing spiked chemical compounds. Cell suspension within the trough is regularly agitated to avoid sedimentation.
Spike control wells A1, C1, E1, G1, A12, C12, E12 and G12 with 1 μl of DMSO. Corresponding wells will be used as reference for living cells (no toxicity). Spike control wells B1, D1, F1, H1, B12, D12, F12 and H12 with 1 μl of DMSO and 2.5 μl of a 0.5% IGEPAL solution to kill cells. Corresponding wells will be used as a reference for dead cells (high toxicity).
Incubate cells for 24 hr at 37 °C and 5% CO2.
3. Preparation of Cell Cultures Infected by rMV2/Luc
Grow and recover cells as described in 2.1 and 2.2. Again, resuspend cells at 3 x 105 cells/ml in culture medium. Consider that 12.5 ml of cell suspension are required for each screening plate.
Optional: supplement culture medium with uridine at 20 μg/ml. Note: When screening chemical libraries for viral replication inhibitors, it seems that compounds targeting early steps of pyrimidine biosynthesis pathway are frequently isolated13,16,27,28. The addition of uridine to culture medium is used to filter out such antiviral molecules. Indeed, this efficiently restores viral replication when upstream enzymes of pyrimidine biosynthesis pathway are blocked.
Save 1:10 of cell suspension for control wells with non-infected cells, and proceed to infection with remaining volume.
Thaw the appropriate volume of rMV2/Luc stock solution. MV stock solutions are usually at 106-108 infectious particles per ml. Culture must be infected with 0.1 infectious particles of rMV2/Luc per target cells, which corresponds to 0.1 multiplicity of infection (MOI). Note: rMV2/Luc is derived from MV vaccine (Schwarz strain) and is manipulated in a BSL2 environment.
Add the virus to cell suspension and mix gently. Transfer infected cells to a trough, and dispense 100 μl of infected cell suspension in columns 2 to 11 of D2 plates containing spiked chemical compounds. Cell suspension within the trough is regularly mixed gently.
In columns 1 and 12, dispense 100 μl of non-infected cells a well on two. Infected cells are dispensed in the others wells.
Incubate cells for 24 hr at 37 °C and 5% CO2.
4. Luciferase Activity Measures and Data Analysis
Remove D1 plates from the incubator, and determine cellular viability by adding 50 μl of luciferase-based viability reagent directly to culture wells. Mix and incubate for 10 min at room temperature. Read plates with a luminometer. Integration time is set at 100 msec per well.
Remove D2 plates from the incubator, and determine luciferase activity by adding 50 μl of luciferase substrate directly to culture wells. Incubate for 6 min at room temperature, and read plates with a luminometer as described above.
Calculate a Z'-factor for each D1 plate of the toxicity assay to warrant the quality of the screen29. Z' = 1-3*(σ+ + σ-)/(µ+ - µ-), where µ+ and σ+ correspond to means of luminescence and standard deviations for HEK-293T cells with DMSO alone (no toxicity), and µ- and σ- are means of luminescence and standard deviations for culture wells treated with IGEPAL to kill cells. Z' is expected to be above 0.5, or the corresponding plate is discarded.
Set toxicity threshold at µ+/2. Discard compounds with luminescence values below this cut-off (Figure 3A).
Calculate a Z'-factor for each D2 plate of the antiviral assay. Here, µ+ and σ+ correspond to means of luminescence and standard deviations for infected cells cultured with DMSO alone, and µ- and σ- are means of luminescence and standard deviations for non-infected cells. Again, plates with Z' values below 0.5 are discarded.
Calculate the inhibition of viral replication as follow: percentage of inhibition = (µ+ - luciferase activity for compound X)/(µ+ - µ-)*100. Set inhibition threshold at 75%, and select compounds that both reduce luminescence values below this cut-off (Figure 3B) and are not toxic according to criteria described in 4.4.
5. Secondary Screen for Toxicity and Broad-spectrum Antiviral Activity
Make 1:2 serial dilutions for each hit compound in DMSO, starting from 500 µM to 4 µM (8 dilutions). Fill white, bar-coded tissue culture plates with 50 µl of culture medium. Add 1 µl of each compound dilution in culture wells. Repeat twice to have 3 culture plates with duplicate serial dilutions for each hit compound.
Prepare 37.5 ml of a HEK-293T cell suspension at 6 x 105 cells/ml in culture medium as described above (in 2.1 and 2.2). Dispense 2x 12.5 ml in falcon tubes and infect cells with either rMV2/Luc at MOI = 0.1 or recombinant CHIKV expressing Renilla luciferase (CHIKV/Ren) at MOI = 0.2. Mix by inverting. Note: CHIKV/Ren is derived from a wild-type strain of CHIKV and therefore should be manipulated in a BSL3 environment. Plates can be moved from BSL3 to BSL1 once Renilla substrate has been added to culture wells (see below).
Dispense 50 µl of uninfected, rMV2/Luc-infected, or CHIKV/Ren-infected cells in culture plates containing serial dilutions of hit compounds. Incubate 24 hr at 37 °C.
Add 50 µl of firefly luciferase substrate to determine firefly luciferase activity in rMV2/Luc-infected wells. Add 50 µl of Renilla luciferase substrate to determine Renilla luciferase activity in CHIKV/Ren-infected wells. Finally, add 50 µl of luciferase-based viability assay reagent to uninfected cells to determine cellular viability.
Plot data (Figure 4) and determine hit compound concentrations that inhibit MV and CHIKV replication by 50%. Disregard compound showing some toxicity in this assay.
Representative Results
This screening pipeline relies first on the selection of compounds that inhibit MV replication and do not show any significant cellular toxicity (Figure 1, primary screen). It takes advantage of luciferase-based assays to determine cellular toxicity and the inhibition of viral replication. All pipetting steps and data acquisition can be performed in a high-throughput setting with a robotic platform.
Cellular toxicity of compounds is assessed using a luciferase-based viability assay that evaluates the number of living cells in culture based on quantification of the ATP present, which corresponds to metabolically active cells. Because luciferin oxidation by luciferase is ATP dependent, luminescence signal is directly proportional to cellular ATP provided by living cells. Figure 3A is showing results for one representative screening plate. As expected, very distinct luciferase activity values were measured in control wells that contained either living cells or dead cells killed with IGEPAL. Threshold is empirically set at µ+/2 to filter out compounds showing some cellular toxicity and in the present case, 21 compounds from this plate were discarded. In parallel, antiviral activity is measured using rMV2/Luc, a recombinant strain of MV expressing luciferase (Figure 2). Human HEK-293T cells are used because they are highly sensitive to MV infection. As a result, luciferase activity in infected culture wells is very high (from 200 to 300 x 103 luciferase activity units in control wells). This provides a large dynamic range that facilitates the selection of MV replication inhibitors. Figure 3B is showing representative results obtained for one screening plate. Four compounds inhibited viral replication by more than 75% in this example and were selected.
Finally, compounds that are not toxic and scored positive in the viral replication assay are selected (Figure 3A and 3B). Their half maximal inhibitory concentration (IC50) is determined on both MV and CHIKV, which are two unrelated RNA viruses (Figure 1, secondary screen). Antiviral activity of hit compounds is determined using recombinant virus strains expressing either firefly (rMV2/Luc) or Renilla (CHIKV/Ren) luciferase. Figure 4A and 4B show representative inhibition curves for MV and CHIKV replication, respectively. These curves are used to determine IC50s of hit compounds. In parallel, the lack of cellular toxicity of selected compounds is confirmed in a dose-response experiment using the luciferase-based viability assay described above (Figure 4C). Table 1 is showing IC50 values for MV and CHIKV replication on a set of 13 non-toxic compounds identified from a chemical library of 10,000 molecules. These compounds are novel both in terms of chemical structure and biological activity. Some of these molecules shared structural similarities, and can be gathered into three distinct chemical families. As a reference, IC50 values obtained with brequinar are also shown in Table 1. Brequinar is an inhibitor of dihydroorotate dehydrogenase, the fourth enzyme of pyrimidine biosynthesis pathway, with a potent antiviral activity in vitro30,31. When looking at IC50 values, less than one order of magnitude separated this reference molecule from most active hits identified by screening. This established the strong antiviral activity of selected compounds.
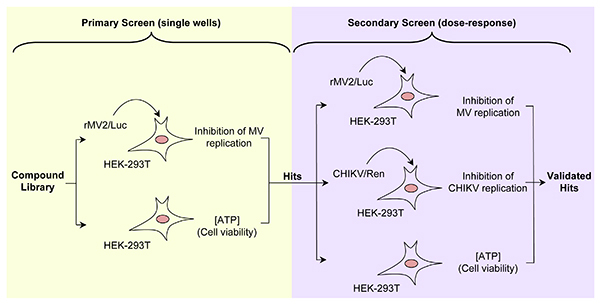 Figure 1. Summary of the screening pipeline. Left panel is showing the primary screen where compounds are selected for the lack of toxicity and capacity to block MV replication. Right panel is showing the secondary screen where the antiviral activity of selected compounds is confirmed, and their capacity to block CHIKV replication is determined. Compounds that efficiently block MV and CHIKV without any significant toxicity are considered as potential broad-spectrum inhibitors of RNA viruses. Click here to view larger image.
Figure 1. Summary of the screening pipeline. Left panel is showing the primary screen where compounds are selected for the lack of toxicity and capacity to block MV replication. Right panel is showing the secondary screen where the antiviral activity of selected compounds is confirmed, and their capacity to block CHIKV replication is determined. Compounds that efficiently block MV and CHIKV without any significant toxicity are considered as potential broad-spectrum inhibitors of RNA viruses. Click here to view larger image.
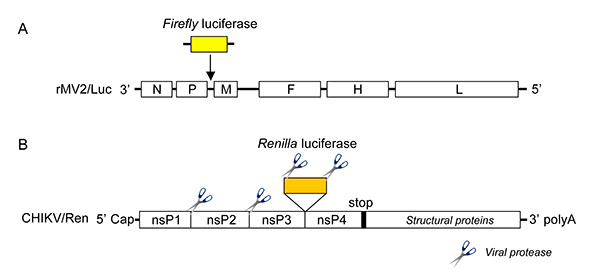 Figure 2. Genomic organization of recombinant RNA viruses carrying luciferase gene. (A) rMV2/Luc: the MV negative-sense RNA genome is displayed with its 3' end on the left, with the six genes indicated in capital letters and depicted as white rectangles. The additional transcriptional units encoding for the reporter gene is inserted between P and M genes and depicted as a yellow rectangle. (B) CHIKV/Ren: the CHIKV positive-sense RNA genome is displayed with its 5'-capped end on the left, with the open reading frame encoding the four non-structural proteins (nsP1-4). Click here to view larger image.
Figure 2. Genomic organization of recombinant RNA viruses carrying luciferase gene. (A) rMV2/Luc: the MV negative-sense RNA genome is displayed with its 3' end on the left, with the six genes indicated in capital letters and depicted as white rectangles. The additional transcriptional units encoding for the reporter gene is inserted between P and M genes and depicted as a yellow rectangle. (B) CHIKV/Ren: the CHIKV positive-sense RNA genome is displayed with its 5'-capped end on the left, with the open reading frame encoding the four non-structural proteins (nsP1-4). Click here to view larger image.
 Figure 3. Representative results for one 96-well screening plate. (A) Luciferase values obtained with the luciferase-based viability assay. Columns 1 and 12 correspond to alternate controls with DMSO alone ("+"; no toxicity) or IGEPAL ("-"; high toxicity). Blue color is highlighting compounds that are considered not toxic according to ATP concentrations detected in culture wells, whereas white wells correspond to toxic compounds (luciferase values < 12,203 x 103). (B) Luciferase values obtained with rMV2/Luc-infected cells. Columns 1 and 12 correspond to alternate controls, i.e. non-infected HEK-293T cells ("-") or rMV2/Luc-infected cells treated with DMSO alone ("+"). For each tested compound, the figure is showing both the luciferase luminescence signal and the percentage of inhibition relative to control wells. Compounds inhibiting luminescence signal by more than 75% are highlighted in yellow or green depending if they were considered toxic or not as determined in (A).Thus, green color is showing hit compounds that inhibit MV replication and do not affect cellular viability. Click here to view larger image.
Figure 3. Representative results for one 96-well screening plate. (A) Luciferase values obtained with the luciferase-based viability assay. Columns 1 and 12 correspond to alternate controls with DMSO alone ("+"; no toxicity) or IGEPAL ("-"; high toxicity). Blue color is highlighting compounds that are considered not toxic according to ATP concentrations detected in culture wells, whereas white wells correspond to toxic compounds (luciferase values < 12,203 x 103). (B) Luciferase values obtained with rMV2/Luc-infected cells. Columns 1 and 12 correspond to alternate controls, i.e. non-infected HEK-293T cells ("-") or rMV2/Luc-infected cells treated with DMSO alone ("+"). For each tested compound, the figure is showing both the luciferase luminescence signal and the percentage of inhibition relative to control wells. Compounds inhibiting luminescence signal by more than 75% are highlighted in yellow or green depending if they were considered toxic or not as determined in (A).Thus, green color is showing hit compounds that inhibit MV replication and do not affect cellular viability. Click here to view larger image.
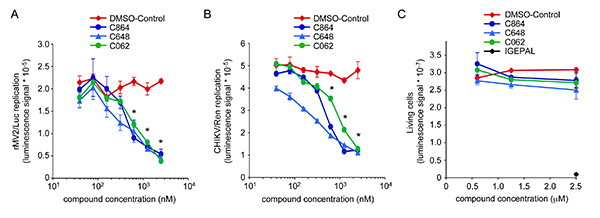 Figure 4. Dose-response antiviral activity and toxicity of compounds C864, C648 and C062. (A) HEK-293T cells were infected with rMV2/Luc strain of MV expressing luciferase (MOI=0.1), and incubated with increasing doses of C864, C648, C062 or DMSO alone. After 24 hr, luciferase expression was determined. * indicates that observed luciferase inhibitions were statistically significant with all three compounds (p-values < 0.05). (B) HEK-293T cells were infected with CHIKV/Ren strain of CHIKV expressing Renilla luciferase (MOI=0.2), and incubated with increasing doses of C864, C648, C062 or DMSO alone. After 24 hr, Renilla luciferase expression was determined. * indicates statistically significant inhibitions with all three compounds (p-values < 0.05). (C) HEK-293T cells were incubated with increasing doses of C864, C648, C062 or DMSO alone. As a control for toxicity, cell cultures were supplemented with 2.5 μl of a 0.5% IGEPAL solution. After 24 hr, the number of living cells was determined using the luciferase-based viability assay. Click here to view larger image.
Figure 4. Dose-response antiviral activity and toxicity of compounds C864, C648 and C062. (A) HEK-293T cells were infected with rMV2/Luc strain of MV expressing luciferase (MOI=0.1), and incubated with increasing doses of C864, C648, C062 or DMSO alone. After 24 hr, luciferase expression was determined. * indicates that observed luciferase inhibitions were statistically significant with all three compounds (p-values < 0.05). (B) HEK-293T cells were infected with CHIKV/Ren strain of CHIKV expressing Renilla luciferase (MOI=0.2), and incubated with increasing doses of C864, C648, C062 or DMSO alone. After 24 hr, Renilla luciferase expression was determined. * indicates statistically significant inhibitions with all three compounds (p-values < 0.05). (C) HEK-293T cells were incubated with increasing doses of C864, C648, C062 or DMSO alone. As a control for toxicity, cell cultures were supplemented with 2.5 μl of a 0.5% IGEPAL solution. After 24 hr, the number of living cells was determined using the luciferase-based viability assay. Click here to view larger image.
| Chemical Family | Compound | MV (IC50 μM) | CHIKV (IC50 μM) | CC50 (μM) |
| 1 | C864 | 0.6 | 0.6 | > 5 |
| 1 | C877 | 0.2 | 0.2 | > 5 |
| 1 | C957 | 1.2 | 1.2 | > 5 |
| 1 | C963 | 1.2 | 0.9 | > 5 |
| 1 | C967 | 1.7 | 1.6 | > 5 |
| 1 | C348 | 0.25 | 0.3 | > 5 |
| 1 | C265 | 0.25 | 0.3 | > 5 |
| 1 | C270 | 0.5 | 0.5 | > 5 |
| 1 | C350 | 0.4 | 0.5 | > 5 |
| 2 | C646 | 0.16 | 0.15 | > 5 |
| 2 | C814 | 1.9 | 1.1 | > 5 |
| 2 | C648 | 0.7 | 0.4 | > 5 |
| 3 | C062 | 1 | 1.2 | > 5 |
| Brequinar | 0.04 | 0.04 | > 5 |
Table 1. Antiviral activity and cytotoxicity of the 13 compounds selected from the original chemical library. Results are expressed as IC50 values for the inhibition of MV or CHIKV replication. CC50 values were found >5 μM for all compounds in dose-response experiments from the secondary screen. But the scoring filter threshold for cytoxicity from the primary screen suggest that CC50 values are even >20 μM.
Discussion
The screening pipeline described here aims at selecting compounds with a suitable profile as broad-spectrum inhibitors of RNA viruses. When a library of 10,000 compounds was screened with this protocol and aforementioned filtering criteria were applied, i.e. inhibition of MV replication superior to 75%, 40 compounds (0.4%) scored positive. Besides, about half of them showed some toxicity in the luciferase-based viability assay, and were disregarded for this reason. Finally, a dozen of hits readily available in larger amount were retested. This small set was easily retested for both cellular toxicity and the inhibition of viral replication against MV and CHIKV in dose-response experiments (Figure 4). The antiviral activity of all selected compounds was confirmed, and the overall identification hit rate for this screening pipeline was 1.3‰. This score is very similar to results obtained by other groups with different high-throughput replication-based screening assays for antivirals27,32, and falls in the expected range for cell-based phenotypic assays in general (1-5‰33). However, a hit rate is highly dependent on both the compound library that is screened and the scoring filter thresholds, which can be adjusted depending on the profile of collected data. Here, selected thresholds for the inhibition of MV replication and cellular toxicity were empirically determined, and trade off was set to obtain a tractable number of hits that can be easily retested in the secondary screen against CHIKV.
In the primary screen, compounds were tested for both their capacity to block MV replication and the lack of cellular toxicity. Antiviral activity was determined at a concentration of 2 μM on rMV2/Luc-infected cells whereas cellular toxicity was determined at 20 μM. These experimental settings were found valuable to directly select for compounds with a high antiviral activity relative to cellular toxicity. In addition, the primary screen was performed in the presence of uridine. Indeed, recent publications suggest that compounds targeting early steps of pyrimidine biosynthesis pathway are frequently isolated when looking for inhibitors of RNA viruses13,16,27,28. In particular, research groups looking for antiviral molecules have found inhibitors of dihydroorotate dehydrogenase, the fourth enzyme of this metabolic pathway31. Uridine in culture medium efficiently trans-complements for the inhibition of this enzyme, and thus restores viral replication. In this way, compounds that prevent MV replication through inhibition of pyrimidine biosynthesis pathway are directly excluded from the antiviral hits. Finally, the Z'-factor was found to be systematically above 0.5, thus demonstrating the high robustness and quality of the assay.
In parallel, cellular toxicity was determined using a high-throughput luciferase-based assay that determines the number of metabolically active cells in culture based on quantification of the ATP. In each screening plate, IGEPAL was added in control wells for toxicity. This non-denaturing, non-ionic detergent has the advantage of killing efficiently all cell types non-specifically without affecting luciferase enzyme activity. Z'-factor was systematically above 0.5 over 125 plates, thus demonstrating the high robustness and quality of the assay. One limitation of this cellular toxicity assay is its relatively high cost. As an alternative assay to determine cellular viability, some groups have used cell lines constitutively expressing luciferase from a stable transgene34. In such assays, toxic compounds are expected to decrease or even suppress luciferase expression when affecting cellular viability. We recently developed a stable cell line expressing luciferase upon IFN-β stimulation35, and this cell line was used to test a training set of 80 compounds for cellular toxicity. Surprisingly, the correlation coefficient (CC) between this assay and the luciferase-based viability assay described above was relatively low (CC = 0.67). Indeed, several compounds impaired luciferase expression from the transgene, whereas ATP metabolism was unaffected. Some broad-spectrum antiviral compounds are likely to affect cellular functions such as cell signaling, gene transcription or protein translation, which will also impair cellular gene expression. Although there is no perfect screening system for cellular viability, it is at risk that filtering toxic compounds based on cellular gene expression will potentially lead to exclude bona fide antiviral molecules.
The secondary screen aims at establishing the antiviral activity of selected compounds against two completely unrelated RNA viruses, while determining precisely their IC50s. Interestingly, it should be noticed that luciferase enzyme inhibitors could be wrongly scored as positives in the rMV2/Luc assay but such false-positives will be filtered-out by the secondary screen when tested for the inhibition of CHIKV/Ren. Indeed, rMV2/Luc and CHIKV/Ren express firefly and Renilla luciferase, respectively, and these two enzymes are unrelated and catalyze different chemical reactions. Compounds that inhibit firefly luciferase reporter from rMV2/Luc in the primary screen will not show any activity against CHIKV/Ren and will be discarded, thus preventing their erroneous selection as antivirals. Unexpectedly, all 13 primary hits blocking MV replication also inhibited CHIKV in this secondary screen. This is not always the case as few MV-specific inhibitors were isolated when screening other libraries (data not shown). Furthermore, selected compounds share structural similarities and can be grouped into only three chemical families (Table 1). Compounds within a chemical family probably have the same mode of action, and therefore can be considered as analogs of the same chemical entity. This puts into perspective the fact that all 13 selected compounds inhibited both MV and CHIKV replication. Nevertheless, these results could also suggest that broad-spectrum antivirals are more frequently identified than virus-specific inhibitors. The rational behind this hypothesis is that, despite obvious specificities, all RNA viruses rely on the same basic cellular functions to replicate, such as ribosomal machinery, cytoskeleton or mitochondria as a source of energy. These functional modules are complex molecular systems, which provide a relatively large panel of potential targets for broad-spectrum antivirals. In contrast, there is only a limited set of viral or cellular factors that represent appropriate targets to raise virus-specific inhibitors. In the future, new panels of antivirals identified by phenotypic screening should provide necessary data to confirm or invalidate this hypothesis.
As aforementioned, isolated compounds had a suitable profile for broad-spectrum inhibitors of RNA viruses with several showing IC50 values below 1 μM in both MV and CHIKV replication assays. This will need to be confirmed with more conventional viral replication assays, including measures of viral progeny in culture supernatants. Antiviral activity of selected compounds will also have to be confirmed on a much larger panel of RNA viruses. In conclusion, these compounds represent excellent starting points for their pharmaceutical development, with the demonstration of their antiviral activity in animal infection models as a primary goal.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
We thank Dr. Yves L. Janin for his fruitful comments and suggestions. We would like to thank H.H. Gad for CHIK/Ren and C. Combredet for her technical support. This work was supported by the Institut Pasteur, the Centre National de la Recherche Scientifique (CNRS), the Institut National de la Santé Et de la Recherche Médicale (INSERM), the Institut Carnot - Pasteur Maladies Infectieuses (Programme STING to P.O.V and H.M.-L), the Agence Nationale pour la Recherche (ANR-RPIB, Programme STING 2.0 to P.O.V), and the "Conseil Régional d'Ile-de-France" (Chemical Library Project, grants n° I 06-222/R and I 09-1739/R to H.M.-L.). The work on CHIKV/Ren was supported by the project ArbOAS (ANR grant 2010-INTB-1601-02).
References
- De Clercq E. Antiviral drugs in current clinical use. J Clin Virol. 2004;30:115–133. doi: 10.1016/j.jcv.2004.02.009. [DOI] [PubMed] [Google Scholar]
- Debing Y, Jochmans D, Neyts J. Intervention strategies for emerging viruses: use of antivirals. Curr Opin Virol. 2013. [DOI] [PMC free article] [PubMed]
- Leyssen P, De Clercq E, Neyts J. Molecular strategies to inhibit the replication of RNA viruses. Antiviral Res. 2008;78:9–25. doi: 10.1016/j.antiviral.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyssen P, Balzarini J, De Clercq E, Neyts J. The predominant mechanism by which ribavirin exerts its antiviral activity in vitro against flaviviruses and paramyxoviruses is mediated by inhibition of IMP dehydrogenase. J Virol. 2005;79:1943–1947. doi: 10.1128/JVI.79.3.1943-1947.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang BX, Fish EN. The yin and yang of viruses and interferons. Trends Immunol. 2012;33:190–197. doi: 10.1016/j.it.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Empey KM, Peebles RS, Kolls JK. Pharmacologic advances in the treatment and prevention of respiratory syncytial virus. Clin Infect Dis. 2010;50:1258–1267. doi: 10.1086/651603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan SL, Ganji G, Paeper B, Proll S, Katze MG. Systems biology and the host response to viral infection. Nat Biotechnol. 2007;25:1383–1389. doi: 10.1038/nbt1207-1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prussia A, Thepchatri P, Snyder JP, Plemper RK. Systematic Approaches towards the Development of Host-Directed Antiviral Therapeutics. Int J Mol Sci. 2011;12:4027–4052. doi: 10.3390/ijms12064027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly DJ, O'Neill LA. New developments in Toll-like receptor targeted therapeutics. Curr Opin Pharmacol. 2012. [DOI] [PubMed]
- Thakur CS, et al. Small-molecule activators of RNase L with broad-spectrum antiviral activity. Proc Natl Acad Sci U S A. 2007;104:9585–9590. doi: 10.1073/pnas.0700590104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoji-Kawata S, et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature. 2013;494:201–206. doi: 10.1038/nature11866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman ES, Smith RE, Renaud RC. From the analyst's couch: TLR-targeted therapeutics. Nat Rev Drug Discov. 2005;4:879–880. doi: 10.1038/nrd1880. [DOI] [PubMed] [Google Scholar]
- Bonavia A, et al. Identification of broad-spectrum antiviral compounds and assessment of the druggability of their target for efficacy against respiratory syncytial virus (RSV) Proc Natl Acad Sci U S A. 2011;108:6739–6744. doi: 10.1073/pnas.1017142108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rider TH, et al. Broad-spectrum antiviral therapeutics. PLoS One. 2011;6 doi: 10.1371/journal.pone.0022572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krumm SA, et al. Potent host-directed small-molecule inhibitors of myxovirus RNA-dependent RNA-polymerases. PLoS One. 2011. [DOI] [PMC free article] [PubMed]
- Hoffmann HH, Kunz A, Simon VA, Palese P, Shaw ML. Broad-spectrum antiviral that interferes with de novo pyrimidine biosynthesis. Proc Natl Acad Sci U S A. 2011;108:5777–5782. doi: 10.1073/pnas.1101143108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey R, et al. GSK983: a novel compound with broad-spectrum antiviral activity. Antiviral Res. 2009;82:1–11. doi: 10.1016/j.antiviral.2008.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinney DC, Anthony J. How were new medicines discovered. Nat Rev Drug Discov. 2011;10:507–519. doi: 10.1038/nrd3480. [DOI] [PubMed] [Google Scholar]
- Jochmans D, Leyssen P, Neyts J. A novel method for high-throughput screening to quantify antiviral activity against viruses that induce limited CPE. J Virol Methods. 2012;183:176–179. doi: 10.1016/j.jviromet.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig-Basagoiti F, et al. High-throughput assays using a luciferase-expressing replicon, virus-like particles, and full-length virus for West Nile virus drug discovery. Antimicrob Agents Chemother. 2005;49:4980–4988. doi: 10.1128/AAC.49.12.4980-4988.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panchal RG, et al. Development of high-content imaging assays for lethal viral pathogens. J Biomol Screen. 2010;15:755–765. doi: 10.1177/1087057110374357. [DOI] [PubMed] [Google Scholar]
- Pohjala L, et al. Inhibitors of alphavirus entry and replication identified with a stable Chikungunya replicon cell line and virus-based assays. PLoS One. 2011;6 doi: 10.1371/journal.pone.0028923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou G, Xu HY, Qing M, Wang QY, Shi PY. Development and characterization of a stable luciferase dengue virus for high-throughput screening. Antiviral Res. 2011;91:11–19. doi: 10.1016/j.antiviral.2011.05.001. [DOI] [PubMed] [Google Scholar]
- Thorne N, Inglese J, Auld DS. Illuminating insights into firefly luciferase and other bioluminescent reporters used in chemical biology. Chem Biol. 2010;17:646–657. doi: 10.1016/j.chembiol.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komarova AV, et al. Proteomic analysis of virus-host interactions in an infectious context using recombinant viruses. Mol Cell Proteomics. 2011. [DOI] [PMC free article] [PubMed]
- Henrik Gad H, et al. The E2-E166K substitution restores Chikungunya virus growth in OAS3 expressing cells by acting on viral entry. Virology. 2012;434:27–37. doi: 10.1016/j.virol.2012.07.019. [DOI] [PubMed] [Google Scholar]
- Wang QY, et al. Inhibition of dengue virus through suppression of host pyrimidine biosynthesis. J Virol. 2011;85:6548–6556. doi: 10.1128/JVI.02510-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smee DF, Hurst BL, Day CW. D282, a non-nucleoside inhibitor of influenza virus infection that interferes with de novo pyrimidine biosynthesis. Antivir Chem Chemother. 2012;22:263–272. doi: 10.3851/IMP2105. [DOI] [PubMed] [Google Scholar]
- Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- Qing M, et al. Characterization of dengue virus resistance to brequinar in cell culture. Antimicrob Agents Chemother. 2010;54:3686–3695. doi: 10.1128/AAC.00561-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munier-Lehmann H, Vidalain PO, Tangy F, Janin YL. On dihydroorotate dehydrogenases and their inhibitors and uses. J Med Chem. 2013;56:3148–3167. doi: 10.1021/jm301848w. [DOI] [PubMed] [Google Scholar]
- Yoon JJ, et al. High-throughput screening-based identification of paramyxovirus inhibitors. J Biomol Screen. 2008;13:591–608. doi: 10.1177/1087057108321089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maréchal E, Roy S, Lafanechère L. Chemogenomics and Chemical Genetics: A User's Introduction for Biologists, Chemists and Informaticians. Springer; 2011. [Google Scholar]
- Gentzsch J, et al. Hepatitis C virus complete life cycle screen for identification of small molecules with pro- or antiviral activity. Antiviral Res. 2011;89:136–148. doi: 10.1016/j.antiviral.2010.12.005. [DOI] [PubMed] [Google Scholar]
- Caignard G, et al. The V protein of Tioman virus is incapable of blocking type I interferon signaling in human cells. PLoS One. 2013;8 doi: 10.1371/journal.pone.0053881. [DOI] [PMC free article] [PubMed] [Google Scholar]