

Abstract
Painful venoms are used to deter predators. Pain itself, however, can signal damage and thus serves an important adaptive function. Evolution to reduce general pain responses, although valuable for preying on venomous species, is rare, likely because it comes with the risk of reduced response to tissue damage. Bark scorpions capitalize on the protective pain pathway of predators by inflicting intensely painful stings. However, grasshopper mice regularly attack and consume bark scorpions, grooming only briefly when stung. Bark scorpion venom induces pain in many mammals (house mice, rats, humans) by activating the voltage-gated Na+ channel Nav1.7, but has no effect on Nav1.8. Grasshopper mice Nav1.8 has amino acid variants that bind bark scorpion toxins and inhibit Na+ currents, blocking action potential propagation and inducing analgesia. Thus, grasshopper mice have solved the predator-pain problem by using a toxin bound to a nontarget channel to block transmission of the pain signals the venom itself is initiating.
Pain is adaptive in that it warns of tissue damage. Thus, pain sensitivity is essential for survival. Humans who lack pain sensitivity often suffer through regular injury (1). Many animals (e.g., jellyfish, ants, wasps, spiders, scorpions, vipers, stonefish, platypus) capitalize on the critical role of the sensory pain-pathway by producing venoms that induce extreme pain, likely as a mechanism for deterring predators (2). Immediate, intense pain may stun a predator, providing prey with the opportunity to escape. Longer term, the predator may avoid future encounters with known painful prey. While counter-selection on the predator might be expected, evolving sensory neurons that have a higher pain threshold may put animals at risk for injury and death from other environmental insults. Thus, the lack of examples documenting predators that are resistant to painful prey is not surprising.
Bark scorpions (Centruroides spp., Family Buthidae) are well known for inflicting intensely painful, potentially lethal stings (3–7). Grasshopper mice (Onychomys spp.), desert-dwelling rodents that prey on scorpions, are resistant to Centruroides’ lethal toxins (8). In this report, we examine whether the mice have also evolved resistance to the pain-inducing components in bark scorpion venom. Observations from a staged feeding study showed that southern grasshopper mice (O. torridus) sympatric with Arizona bark scorpions (C. sculpturatus, formerly known as C. exilicauda) responded to the scorpion’s sting by grooming for only a few seconds before pressing their attack, voraciously killing and consuming the scorpion [(9), see also movie S1].
The mice’s response was surprisingly brief given that anecdotal reports describe C. sculpturatus’ stings as producing an immediate burning sensation followed by prolonged throbbing pain that can last for hours. Our observations suggested that O. torridus have evolved reduced sensitivity to this painful venom. We here confirm that O. torridus are less sensitive than house mice (Mus musculus) to C. sculpturatus’ pain-inducing toxins. We show that, in O. torridus, the channel (Nav1.8) responsible for transmitting pain signals to their central nervous system (CNS) has amino acid variants that bind venom peptides and inhibit channel current, paradoxically blocking pain signals instead of transmitting them.
Effects of Venom and Formalin on Sensory-Pain Behavior in Mice
We injected venom into the hind paws of O. torridus and M. musculus, using physiological saline as a control, and recorded the amount of time the mice spent licking their paws over a 15-min period. To select a venom dose that induces pain in the control species without lethal effects, we evaluated the response of M. musculus to three doses of venom having concentrations less than the median lethal dose (LD50) (10) (fig. S1). M. musculus substantially increased their paw licking in response to venom as compared to saline, whereas O. torridus responded to venom by licking their paws only briefly (Fig. 1A). Indeed, O. torridus licked their paws less in response to venom than to saline.
Fig. 1. Grasshopper mice (O. torridus) do not show pain responses to bark scorpion (C. sculpturatus) venom, but do to formalin.
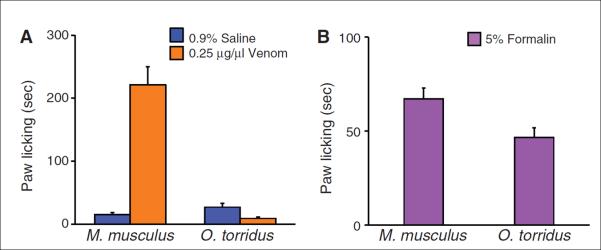
All data are shown as the mean + 1 SE. (A) O. torridus and M. musculus differed significantly in their response to venom. M. musculus increased their paw licking in response to venom as compared to saline (saline = 15.30 + 3.22 s, venom = 221.63 + 28.46 s, n = 8 mice). In contrast, O. torridus decreased their paw licking in response to venom as compared to saline (saline = 26.86 + 5.78 s, n = 6 mice, venom = 8.98 + 2.48 s, n = 8 mice). Two-way ANOVA species by treatment interaction, F (1,26) = 51.02, P < 0.0001. (B) Both M. musculus and O. torridus licked their paws in response to formalin (M. musculus: 67.26 + 5.65 s; O. torridus: 46.67 + 5.21 s; each species, n = 8 mice). However, O. torridus licked their paws significantly less than M. musculus in response to the treatment [oneway ANOVA, species by treatment interaction, F (1,14) = 7.18, P = 0.02].
To determine whether this response is specific to C. sculpturatus venom or represents a general insensitivity to painful stimuli, we injected formalin (11, 12) into the hind paws of O. torridus and M. musculus and recorded their paw licking for 15 min. Although O. torridus licked their paws less than M. musculus in response to formalin, the duration of licking was longer than their response to saline, demonstrating that they are sensitive to alternative painful stimuli (Fig. 1B). Although M. musculus found the venom to be substantially more irritating than formalin, a post hoc comparison showed that O. torridus found the injections of formalin to be more irritating than saline, and the saline to be more irritating than the venom (fig. S2).
Effects of Venom on Dorsal Root Ganglion–Expressed TTX-R Na+ Current
In mammals, acute pain (nociception) is transmitted to the CNS mainly by two voltage-gated sodium (Na+) channels, tetrodotoxin-sensitive (TTX-S) Nav1.7 and tetrodotoxin-resistant (TTX R) Nav1.8, that are expressed in small-diameter dorsal root ganglion (DRG) neurons (nociceptors) (13). Nociceptors also express Nav1.9, a second TTX-R Na+ current involved in diabetic neuropathy and inflammatory pain (13–17). Venoms from Old and New World Buthidae scorpions initiate acute pain in sensitive mammals (e.g., house mice, rats, humans) by activating Nav1.7, but have no effect on Nav1.8 (18–21). We used TTX to block Nav1.7 and confirmed that C. sculpturatus venom does not affect DRG-expressed TTX-R Na+ current in M. musculus (this TTX-R current is primarily, if not exclusively, due to Nav1.8 as Nav1.9 loses activity rapidly in dissociated neurons) (Fig. 2, A and B). However, venom inhibited TTX-R Na+ currents recorded from dissociated DRG neurons in O. torridus (Fig. 2, A and B). Venom did not significantly affect the voltage dependence of channel activation or steady-state inactivation in either species (Fig. 2C). Increasing venom concentrations had no effect on M. musculus but decreased O. torridus TTX-R Na+ currents in a dose-dependent manner (Fig. 2D).
Fig. 2. C. sculpturatus venom inhibits O. torridus, but not M. musculus TTX-R Na+ current.
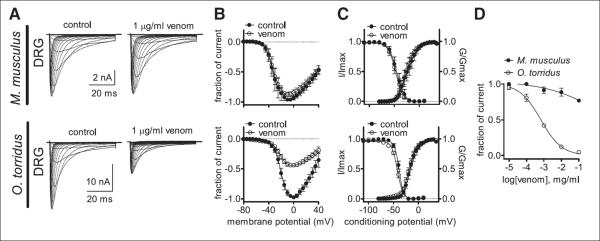
(A) Current traces and (B) the current-voltage relation demonstrate that venom inhibits O. torridus TTX-R Na+ currents recorded from native channels expressed in small-diameter DRG neurons but has no effect on M. musculus TTX-R currents. Currents were elicited by 50-ms depolarizing steps to voltages ranging from –80 to 40 mV in 5-mV increments. All currents induced before (control) and after the application of venom were normalized to the maximum amplitude of control peak current. (C) Venom did not significantly affect the voltage dependence of channel activation or steady-state inactivation in either species. The voltage dependence of steady-state inactivation was estimated with a standard double-pulse protocol in which Na+ currents were induced by a 20-ms depolarizing potential of 0 mV following a 500-ms prepulse at voltages ranging from –110 to 10 mV. Currents were plotted as a fraction of the maximum peak current. Data points were fitted with the Boltzmann equation [(M. musculus V1/2 for activation: control = –45.0 ± 1.4 mV, venom = –47.0 ± 1.2 mV; V1/2 for steady-state inactivation: control = –24.3 ± 1.5 mV, venom = –26.0 ± 1.1 mV), (O. torridus V1/2 for activation: control = –38.7 ± 0.4 mV, venom = –42.2 ± 0.5 mV; V1/2 for steady-state inactivation: control = –17.1 ± 0.7 mV, venom = –18.1 ± 0.6 mV)]. (D) Venom inhibited O. torridus’ DRG-expressed TTX-R Na+ current in a concentration-dependent manner (IC50 = 0.7 μg/ml), whereas high concentrations of venom had little effect on M. musculus TTX-R current (IC50 = 5.7 mg/ml). The inhibitory response was assessed with a single pulse protocol in which current was elicited every 5 s by a 50-ms depolarizing potential of 0 mV. Data points were fit with the Hill logistic equation: Y = bottom + (top – bottom)/(1 + 10((logEC 50−X) × Hill slope)), where EC50 is median effective concentration. For (A) to (D), dissociated DRG cells were pretreated with 500 nM TTX to block TTX-S Na+ channels and held at −80 mV (n = 4 or 5 cells obtained from two different animals of each species). For (B) to (D), data are shown as the mean ± 1 SE.
Effects of Venom on Nociceptor Excitability and Action Potential Generation
Empirical and modeling studies show that whereas Nav1.7 initiates action potentials in nociceptors, Nav1.8 Na+ currents are necessary for their sustained firing and propagation (13, 22–24). Thus, we predicted that venom-induced inhibition of Nav1.8 Na+ currents would decrease membrane excitability and block action potential propagation in O. torridus nociceptors. To test this, we examined the effects of venom on action potential firing in small-diameter DRG neurons. We recorded baseline firing by injecting ramp currents into dissociated DRG neurons to mimic the small, slow depolarization of membranes elicited by noxious stimuli. O. torridus and M. musculus differed in DRG neuron current threshold, with O. torridus neurons requiring significantly more current than M. musculus neurons to reach baseline excitability (fig. S3). After DRG neurons in both species reached an equivalent baseline level of firing, we measured the effect of venom on nociceptor excitability. Venom significantly increased membrane excitability and action potential firing in M. musculus neurons, but had the opposite affect on O. torridus neurons, in which venom significantly decreased membrane excitability and blocked action potential firing (Fig. 3, A and B). Instead of inducing pain in O. torridus, C. sculpturatus venom paradoxically blocks pain signaling to the CNS. By blocking signals to the CNS, it follows that venom should also reduce O. torridus’ response to subsequent painful stimuli; that is, venom may induce analgesia in O. torridus.
Fig. 3. C. sculpturatus venom decreases membrane excitability and action potential firing in O. torridus’ small-diameter DRG neurons, reducing their sensitivity to formalin.
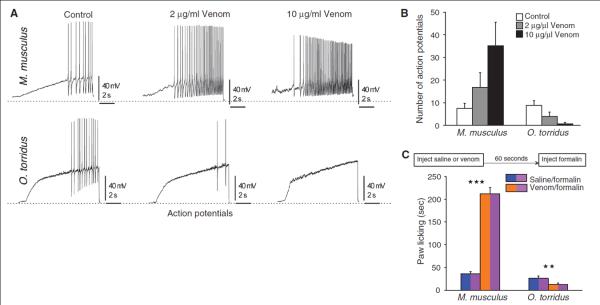
(A) Ramp currents were injected into DRG neurons to produce small, slow depolarization, similar to that evoked by pain-inducing stimuli. Whereas venom increased membrane excitability and action potential (AP) firing in M. musculus DRG neurons, it decreased excitability and blocked AP firing in O. torridus DRG neurons. (B) O. torridus and M. musculus differed significantly in their response to increasing doses of venom. Venom increased the number of APs generated by M. musculus DRG neurons (control = 7.5 + 2.2 APs, 2 μg/ml venom = 16.83 + 6.45 APs, 10 μg/ml venom = 35.2 + 10.33 APs, n = 6 cells from 2 different animals), but decreased the number of APs generated by O. torridus DRG neurons (control = 8.85 + 2.05 APs, 2 μg/ml venom = 3.85 + 1.96 APs, 10 μg/ml venom = 0.66 + 0.66 APs, n = 7 cells from 3 different animals). Data are shown as the mean number of APs (+ 1 SE), multivariate ANOVA with repeated measures, species by treatment interaction, F (2, 8) = 7.52, P = 0.01. (C) Preinjecting M. musculus with 0.25 μg/μl venom significantly increased their paw licking in response to an injection of 0.1% formalin as compared to preinjecting with 0.9% saline (saline/formalin = 36.35 + 4.84 s, venom/formalin = 212.25 + 13.16 s, n = 16 mice, paired t test, ***P < 0.0001). In contrast, preinjecting O. torridus with 0.25 μg/μl venom significantly decreased their paw licking in response to an injection of 0.5% formalin as compared to preinjecting with 0.9% saline (saline/formalin = 26.14 + 4.19 s, venom/formalin = 12.47 + 3.03 s, n = 16 mice, paired t test, **P = 0.01). Data are shown as the mean duration (+ 1 SE) of paw licking recorded for 20 min after the second injection in each treatment. Two-way ANOVA with repeated measures, species by treatment interaction, F (1, 30) = 145.42, P < 0.0001. For each species, one group of 16 mice was used for both treatments (i.e., each mouse served as its own control). Each mouse received one set of injections to a right or left hind paw and 1 week later received the second set of injections in the alternate paw (order of injections into the right or left hind paw was counterbalanced).
Effects of Venom on Response to Alternative Painful Stimuli
To determine whether venom induces analgesia in O. torridus, we examined paw-licking responses to 5% formalin injected into a hind paw after an injection of venom. Formalin concentrations >1% induce a biphasic response in rodents; an immediate, brief response mediated, in part, by the nociceptor-expressed transient receptor potential cation channel (TRPA1), and a delayed, persistent response mediated by inflammatory agents and changes to the CNS (11, 12, 25, 26). To minimize responses due to tissue inflammation, we measured paw licking during the 5-min period immediately after formalin injections. A two-way analysis of variance (ANOVA) revealed a significant species-by-treatment interaction. M. musculus licked their paws more in response to formalin after pretreatment with venom as compared to formalin without venom pretreatment (fig. S4). However, O. torridus decreased their paw licking in response to formalin when pretreated with venom (fig. S4). We replicated this study using lower concentrations of formalin (<1%) to elicit nociceptor-mediated paw licking and further minimize inflammation-induced licking. We accomplished this by selecting doses of formalin that produced paw licking within the first 10 min after formalin injections, but that produced little or no licking after 10 min (fig. S5). The results of the replicated study confirmed that pretreating with venom before formalin injections significantly increased paw licking in M. musculus but significantly decreased paw licking in O. torridus (Fig. 3C).
Nav1.8 Molecular Structure
Voltage-gated Na+ channels are membrane-spanning proteins constructed of four homologous domains (DI to DIV), with six transmembrane segments (S1 to S6) composing each domain (27–29) (fig. S6A). Positively charged residues in S4 of each domain serve as sensors that detect changes in membrane voltage to regulate channel gating. The reentrant loop between S5 and S6 in each domain join to form the channel pore. Venoms of buthid scorpions such as C. sculpturatus consist of multiple, low–molecular weight peptides that bind either to the amino acids in the extracellular loops connecting the transmembrane segments or to the residues lining the pore of the channel (10, 30–34). Because C. sculpturatus venom inhibits TTX-R Na+ current in O. torridus Nav1.8 but has no effect on M. musculus, we predicted that the structure of Nav1.8 would differ between the two species. To identify structural differences, we cloned and sequenced the gene (Scn10a) that encodes Nav1.8 from O. torridus and compared it to orthologous sequence from M. musculus. We identified multiple amino acid variants in O. torridus Nav1.8 (fig. S6, A and B). Many of these variants were distributed throughout the extracellular loops and pore region of the channel where they would be accessible to venom peptides.
Nav1.8 Chimeras
To identify the region of the channel critical for venom-induced inhibition of Na+ current, we made constructs of Nav1.8 for O. torridus (otNav1.8) and M. musculus (mNav1.8) by inserting Scn10a from each species into expression vectors (Fig. 4A). We confirmed that C. sculpturatus venom had no effect on mNav1.8 Na+ current but dose dependently blocked otNav1.8 (Fig. 4, B and C). Using otNav1.8 as the framework, we made four chimeras in which each domain of otNav1.8 was replaced with the corresponding domain from mNav1.8. We tested venom on the chimeras and found that replacing domain I in otNav1.8 did not affect venom activity, and replacing domains III and IV only slightly reduced venom activity (Fig. 4D). However, replacing domain II in otNav1.8 with the corresponding domain from mNav1.8 abolished the effects of the venom on the channel (Fig. 4D). Moreover, reversing the chimera by using mNav1.8 as the framework and replacing domain II with the corresponding domain from otNav1.8, imparted venom sensitivity to mNav1.8 (Fig. 4E). C. sculpturatus venom dose dependently inhibited mNav1.8 Na+ current after exchanging domain II with otNav1.8. Thus, domain II is the major contributor to venom sensitivity in otNav1.8.
Fig. 4. Domain II is the major contributor to the sensitivity of O. torridus Nav1.8 to C. sculpturatus venom.

(A) Schematic diagram of sodium channel α subunit Nav1.8 from O. torridus (otNav1.8) and M. musculus (mNav1.8). OI to OIV and MI to MIV represent four different domains of otNav1.8 and mNav1.8, respectively. (B) C. sculpturatus venom inhibited otNav1.8 TTX-resistant Na+ current, but not mNav1.8, expressed by ND7/23 cells. Cells were pretreated with 500 nM TTX to block TTX-sensitive (i.e., Nav1.7) currents and held at –80 mV. Na+ currents were induced by 50-ms depolarizing steps to various potentials ranging from –80 to +60 mV in 5-mV increments. All currents elicited before (control) and after venom treatment were normalized to the maximum amplitude of control peak current. (C) Concentration-response inhibitory curves for C. sculpturatus venom on wild-type otNav1.8 (OIOIIOIIIOIV, filled circles) and mNav1.8 (MIMIIMIIIMIV, open circles). The IC50 for otNav1.8 was 2.2 μg/ml. (D) Effects of C. sculpturatus venom on four otNav1.8/mNav1.8 chimeras. Each of the four domains in otNav1.8 was replaced by the corresponding domain from mNav1.8. Venom inhibited Na+ current expressed by chimeras where domains I, III, and IV were exchanged (IC50 values: MIOIIOIIIOIV = 1.5 μg/ml; OIOIIMIIIOIV = 3.0 μg/ml; OIOIIOIIIMIV = 2.4 μg/ml). Venom had no effect on the domain II chimera (IC50 value: OIMIIOIIIOIV > 524.6 μg/ml). (E) The reverse replacement of otNav1.8-DII significantly increased the sensitivity of mNav1.8 to C. sculpturatus venom (IC50 value: MIOIIMIIIMIV = 3.2 μg/ml). Each data point is shown as the mean ± SE. Data for (C) to (E) were obtained from 3 to 7 separate cells expressing each channel construct.
Site-Directed Mutagenesis
To identify the amino acid(s) in domain II that impart venom sensitivity to otNav1.8, we used site-directed mutagenesis to replace amino acid variants in otNav1.8 domain II with residues corresponding to the same positions in mNav1.8. Initially we focused on two amino acid variants expressed in O. torridus domain II S3-S4 because scorpion peptides known to target domain II in other Na+ channels bind to amino acids in this loop (31). We replaced two hydrophobic amino acids (I746 and A747) in the otNav1.8 domain II S3-S4 with hydrophilic residues (T746 and S747) that correspond to the same positions in mNav1.8 (Fig. 5A). However, C. sculpturatus venom dose dependently blocked Na+ current in both the wild-type construct (otNav1.8) and the mutant (otNav1.8 I746T/A747S) (Fig. 5, B and C), demonstrating that these variants do not contribute to venom sensitivity in O. torridus.
Fig. 5. An acidic residue (E862) in domain II near the pore region (SS2-S6 linker) plays a critical role in the sensitivity of otNav1.8 to C. sculpturatus venom.
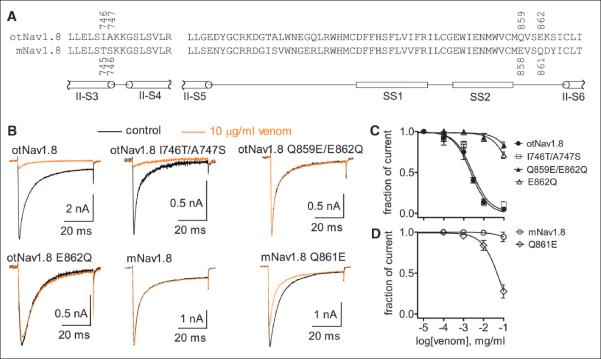
(A) The amino acid sequence representing domain II S3-S4 and S5-S6 linkers from otNav1.8 is aligned with the corresponding sequence from mNav1.8. The position of each amino acid residue of interest is designated with a number. (B) Effects of C. sculpturatus venom on wild-type and mutant Nav1.8 channels expressed by ND7/23 cells. Cells were pretreated with 500 nM TTX and held at –80 mV. Representative current traces before (control, black) and after 10 μg/ml venom treatment (orange) were elicited by a 50-ms depolarizing potential of +20 mV. (C) Concentration-response inhibitory curves show the effect of C. sculpturatus venom on wild-type and mutant otNav1.8 channels. C. sculpturatus venom IC50 values were estimated to be >251.4 μg/ml on the single-mutant E862Q and >531.5 μg/ml on the double-mutant Q859E/E862Q, respectively. (D) The reverse mutation Q861E in mNav1.8 increased the sensitivity of the channel to C. sculpturatus venom. The IC50 value was 66.9 μg/ml for the mutant Q861E. Each data point is shown as the mean ± SE. Data for (C) and (D) were obtained from 3 to 7 separate cells expressing each channel construct. Abbreviations for the amino acid residues are as follows: A, Ala; C, Cys; D, Asp; E, Glu; F, Phe; G, Gly; H, His; I, Ile; K, Lys; L, Leu; M, Met; N, Asn; P, Pro; Q, Gln; R, Arg; S, Ser; T, Thr; V, Val; W, Trp; and Y, Tyr.
We then focused on two amino acid variants expressed in O. torridus domain II SS2-S6 linker, a structure that is adjacent to the channel pore. In O. torridus SS2-S6 loop, the positions of a hydrophilic (Q859) and an acidic (E862) residue are reversed as compared to corresponding residues in M. musculus (Fig. 5A). We made a construct in which we changed the hydrophilic (Q859) and acidic (E862) residues in OtNav1.8 to acidic (E859) and hydrophilic (Q862) residues, respectively. C. sculpturatus venom had no effect on the double mutant (otNav1.8 Q859E/E862Q), suggesting not only that an acidic residue is important for the activity of the venom, but also that the position of the acidic residue is critical for venom sensitivity (Fig. 5, B and C). To confirm this, we replaced the acidic residue (E862) with the hydrophilic (Q862) residue and found that C. sculpturatus venom had almost no effect on Na+ current expressed by the single mutant (otNav1.8 E862Q) (Fig. 5, B and C). These results demonstrate that glutamic acid (E862) expressed adjacent to the pore at position 862 is critical for imparting venom sensitivity to otNav1.8. Further, changing the hydrophilic residue (Q861) to glutamic acid (E861) imparted venom sensitivity to the M. musculus construct as C. sculpturatus venom inhibited Na+ current in the single mutant (mNav1.8 Q861E) (Fig. 5, B and D). However, whereas replacement of domain II in mNav1.8 with domain II from otNav1.8 inhibited most of the Na+ current in the mutant with a median inhibitory concentration (IC50) similar to that of the otNav1.8 wild type, the Q861E replacement in mNav1.8 did not completely block Na+ current. These results suggest that glutamic acid (E862) is necessary for TTX-R Na+ current inhibition in O. torridus Nav1.8, but that additional amino acid variants in domain II may contribute to venom sensitivity.
Amino Acid Variants Critical for Venom-Pain Insensitivity Occur Frequently in Rodent and Nonrodent Mammals
To determine whether the amino acids critical for venom-pain insensitivity are unique to O. torridus, we compared orthologous sequence from 18 species, including rodents and nonrodents from 15 mammalian families (Fig. 6). The sequence alignment revealed that all 18 species express either glutamic acid (E) or glutamine (Q) at positions 859 and 862. The amino acid variants (Q859 and E862) observed in O. torridus occurred frequently in both closely and distantly related rodents as well as nonrodent mammals. The glutamic acid (E862) that is critical for blocking Na+ current in O. torridus Nav1.8 was expressed in 7 of the 10 rodent species and one primate. Two of these rodents (Mesocricetus auratus, Cavia porcellus), as well as the primate (Otolemur garnettii), also expressed the glutamine (Q859) residue. Conservation of positions 859 and 862 for expression of either glutamic acid or glutamine across a diverse group of mammals suggests that these amino acids play a critical role in the structure and function of the Nav1.8 domain II SS2-S6 linker. However, positions 859 and 862 are not conserved by the particular order in which the amino acids are expressed. All four possible permutations for the two amino acids are expressed across the species shown in the alignment. Thus, although E862 most likely evolved in a mammalian ancestor under selection pressures that were not associated with scorpion venom, O. torridus can use this negatively charged amino acid to reduce their sensitivity to venom-induced pain.
Fig. 6. O. torridus Nav1.8 DII SS2-S6 linker amino acid variants critical for venom insensitivity are expressed frequently in rodent and non-rodent mammals.

(A) Schematic diagram of domain II from a typical voltage-gated Na+ channel α subunit designating the DII SS2-S6 linker (red star) where O. torridus expresses amino acid variants critical for venom-pain insensitivity. (B) An alignment of O. torridus Nav1.8 DII with orthologous sequence from rodent and nonrodent mammals shows that the extracellular SS2-S6 linker is more variable than flanking intracellular regions (DII pore loop and DII S6 segment). However, all species shown express either glutamic acid (E) or glutamine (Q) at positions 859 and 862 (yellow background), suggesting that these sites are conserved. Ten species, including rodents and nonrodent mammals, express either one or both of the amino acid variants (site 859, Q shown in red; site 862, E shown in red) observed in O. torridus that are critical for venom insensitivity. M. auratus, C. porcellus, and O. garnettii express Q at site 859 and E at site 862. M. ochrogaster, J. jaculus, H. glaber, C. lanigera, and O. degus express E at site 862. C. cristata and S. araneus express Q at site 859. Sequences were downloaded from the NCBI and aligned with the Clustal format (MAFFT L-INS-1, v6.850b). Sequences were arranged in descending order according to taxonomic family and relatedness to O. torridus. Identical amino acids are shown as (*), highly conserved amino acid variants as (:), similar amino acid variants as (.), and dissimilar amino acid variants or gaps as a blank (). Rodents [Cricetidae: O. torridus (southern grasshopper mouse), Microtus ochrogaster (prairie vole, XM_005348098), Mesocricetus auratus (golden hamster, XM_005348098); Muridae: Mus musculus (house mouse, NM_001205321), Rattus norvegicus (Norway rat, NM_017247); Dipodidae: Jaculus jaculus (lesser Egyptian jerboa, XM_004662101); Sciuridae: Ictidomys tridecemlineatus (thirteen-lined ground squirrel, XM_005317385); Bathyergidae: Heterocephalus glaber (naked mole-rat, JF_912495); Caviidae: Cavia porcellus (guinea pig, XM_003464141); Chinchillidae: Chinchilla lanigera (long-tailed chinchilla, XM_005386574); Octodontidae: Octodon degus (degu, XM_004642215)]. Nonrodent mammals [Galagidae: Otolemur garnettii (small-eared galago, XM_003794468); Cercopithecidae: Papio anubis (olive baboon, XM_003894661); Hominidae: Homo sapiens (human, NM_006514); Talpidae: Condylura cristata (star-nosed mole, XM_004676631); Soricidae: Sorex araneus (European shrew, XM_004614563); Felidae: Felis catus (domestic cat, XM_003992249); Bovidae: Bos taurus (cattle, XM_002696916); Elephantidae: Loxodonta africana (African elephant, XM_003415747)].
Discussion
Pain sensitivity is critical for survival. Thus, many animals use painful venom to deter predators. Because voltage-gated Na+ channels play a major role in transmitting sensory-pain signals to the CNS, they are often the targets of pain-inducing venoms. C. sculpturatus venom induces pain by activating the channel (Nav1.7) responsible for initiating pain signaling in nociceptors. Although pain-inducing venom could impose strong selection on the receiver, counteradaptation may be constrained by the risks associated with reduced pain sensitivity. Given the important role of Na+ channels in pain signaling, counteradaptation may also be constrained by the conservation of Na+ channel structure and function. However, O. torridus can sustain multiple stings during predatory attacks on C. sculpturatus. In O. torridus, the channel (Nav1.8) responsible for transmitting pain signals to their CNS has amino acid variants that bind venom peptides and inhibit channel current, blocking pain signals instead of transmitting them. This mechanism represents a unique evolutionary strategy as many examples of resistance to deadly toxins involve structural modifications to the target channel or receptor that interfere with toxin binding [e.g., resistance to cobra toxin (35, 36), resistance to tetrodotoxin (37–40)]. In O. torridus, however, a channel (Nav1.8) that is not the target of the venom has amino acid variants that facilitate venom binding. Moreover, scorpion peptides that target Na+ channels typically activate the channel or block inactivation, prolonging channel activity and increasing neuron excitability (31, 32). In contrast, scorpion peptides inhibit O. torridus Nav1.8 Na+ current and decrease neuron excitability, blocking neuronal signaling and inducing analgesia.
A fascinating parallel to the grasshopper mouse–bark scorpion case involves African naked mole rats that are insensitive to acid-induced pain (41, 42). Naked mole rats live in subterranean colonies where they are exposed to high concentrations of carbon dioxide (CO2). Increased CO2 environments induce pain by activating proton-gated acid sensors expressed in nociceptors. Instead of modifications to the proton-gated acid sensors, naked mole rats evolved amino acid variants in Nav1.7, the channel responsible for initiating pain signals in nociceptors. In naked mole rats, protons bind to the pore region of Nav1.7 and block Na+ current, inhibiting action potential initiation and preventing pain signaling. Thus, in naked mole rats, a nontarget Na+ channel expressed in the sensory pain pathway evolved structural variations that facilitate proton binding, ultimately blocking the pain signals that the protons are initiating.
The pain-reducing mechanisms observed in southern grasshopper mice and African naked mole rats are exciting because they demonstrate that although Na+ channels are structurally conserved, variation exists among species. Moreover, slight variations in Na+ channel structure can produce substantial physiological effects. Reversing the positions of a hydrophilic and an acidic amino acid in Nav1.8 is critical for imparting venom-pain insensitivity to O. torridus, and converting a positively charged amino acid motif to a negatively charged motif in Nav1.7 renders naked mole rats insensitive to acid-induced pain. Because the amino acid variants in O. torridus Nav1.8 and naked mole rat Nav1.7 are each directed against the source of the pain (venom peptides and protons, respectively), O. torridus can exploit a biochemically protected food resource and naked mole rats can live in subterranean colonies while circumventing the constraints associated with evolving generally desensitized nociceptors. Future studies should examine whether there has been counterselection on C. sculpturatus, perhaps resulting in peptides modified to overcome the mice’s resistance to this scorpion’s painful sting. Given that venom toxins and their ion-channel targets are both products of gene families, C. sculpturatus and O. torridus provide an excellent model for investigating coevolution and arms races at the molecular and biochemical levels.
C. sculpturatus and O. torridus also provide a unique model for analgesia studies. Nav1.7 is considered a target for analgesic development because of its role in human pain disorders (13). However, our results demonstrate the key role Nav1.8 plays in pain signaling and its potential to serve as an analgesic target. Moreover, given that few toxins have been identified that bind Nav1.8, and none that bind selectively (43), the molecular and biochemical interactions between venom peptides and Nav1.8 could serve as the basis for designing highly selective, nonaddictive analgesics.
Supplementary Material
Acknowledgments
We thank M. Heitlinger, the Santa Rita Experimental Range (Univ. of Arizona), and the Arizona Game and Fish Department for assistance with animal collections. We also thank Y. Lu and M. Wu for technical assistance with Nav1.8 sequencing and construct assembly. This work was supported, in part or in whole, by NSF IOS Award 1122115 (to A.H.R., M.P.R., and H.H.Z.), Department of the Army grants W911NF-06-1-0213 and W911NF-09-1-0355 from the Army Research Office Life Sciences Division (to A.H.R. and H.H.Z.), NIH grant NS 053422 from the National Institute of Neurological Disorders and Stroke (to T.R.C. and Y.X.), and a Faculty Research Grant from the Office of Research and Sponsored Projects, Sam Houston State University (to M.P.R.). The Department of Biological Sciences at Sam Houston State University provided logistical support for fieldwork.
Footnotes
We declare no conflict of interest. The data are included in the main article and in the supplementary materials. The gene (Scn10a) encoding O. torridus Nav1.8 has been deposited in NCBI’s GenBank gene database. Information about the GenBank accession numbers can be found in the supplementary materials.
References and Notes
- 1.Cox JJ, et al. Nature. 2006;444:894–898. doi: 10.1038/nature05413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schmidt JO. In: Insect Defenses: Adaptive Mechanisms and Strategies of Prey and Predators. Evans DL, editor. State University of New York Press; Albany, NY: 1990. pp. 383–417. [Google Scholar]
- 3.Curry SC, Vance MV, Ryan PJ, Kunkel DB, Northey WT. J. Toxicol. Clin. Toxicol. 1983;21:417–449. doi: 10.3109/15563658308990433. [DOI] [PubMed] [Google Scholar]
- 4.Fet V, Lowe G. In: Catalog of the Scorpions of the World (1758-1998) Fet V, Sissom WD, Lowe G, Braunwalder ME, editors. Vol. 1. New York Entomological Society; New York: 2000. pp. 54–286. [Google Scholar]
- 5.LoVecchio F, McBride C, Toxicol J. Clin. Toxicol. 2003;41:937–940. doi: 10.1081/clt-120026514. [DOI] [PubMed] [Google Scholar]
- 6.Boyer LV, et al. N. Engl. J. Med. 2009;360:2090–2098. doi: 10.1056/NEJMoa0808455. [DOI] [PubMed] [Google Scholar]
- 7.Skolnik AB, Ewald MB. Pediatr. Emerg. Care. 2013;29:98–103. doi: 10.1097/PEC.0b013e31827b5733. quiz 104–105. [DOI] [PubMed] [Google Scholar]
- 8.Rowe AH, Rowe MP. Toxicon. 2008;52:597–605. doi: 10.1016/j.toxicon.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 9.Rowe AH, Rowe MP. Anim. Behav. 2006;71:725–734. [Google Scholar]
- 10.Simard JM, Meves H, Watt DD. In: Natural Toxins: Toxicolgy, Chemistry and Safety. Keeler RF, Mandava NB, Tu AT, editors. Vol. 1. Alaken; Fort Collins, CO: 1992. pp. 236–263. [Google Scholar]
- 11.Tjølsen A, Berge OG, Hunskaar S, Rosland JH, Hole K. Pain. 1992;51:5–17. doi: 10.1016/0304-3959(92)90003-T. [DOI] [PubMed] [Google Scholar]
- 12.Sufka KJ, Watson GS, Nothdurft RE, Mogil JS. Eur. J. Pain. 1998;2:351–358. doi: 10.1016/s1090-3801(98)90033-7. [DOI] [PubMed] [Google Scholar]
- 13.Dib-Hajj SD, Cummins TR, Black JA, Waxman SG. Annu. Rev. Neurosci. 2010;33:325–347. doi: 10.1146/annurev-neuro-060909-153234. [DOI] [PubMed] [Google Scholar]
- 14.Cummins TR, et al. J. Neurosci. 1999;19:RC43. doi: 10.1523/JNEUROSCI.19-24-j0001.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dib-Hajj S, Black JA, Cummins TR, Waxman SG. Trends Neurosci. 2002;25:253–259. doi: 10.1016/s0166-2236(02)02150-1. [DOI] [PubMed] [Google Scholar]
- 16.Lolignier S, et al. PLOS ONE. 2011;6:e23083. doi: 10.1371/journal.pone.0023083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leo S, D’Hooge R, Meert T. Behav. Brain Res. 2010;208:149–157. doi: 10.1016/j.bbr.2009.11.023. [DOI] [PubMed] [Google Scholar]
- 18.Saab CY, Cummins TR, Dib-Hajj SD, Waxman SG. Neurosci. Lett. 2002;331:79–82. doi: 10.1016/s0304-3940(02)00860-1. [DOI] [PubMed] [Google Scholar]
- 19.Maertens C, et al. Mol. Pharmacol. 2006;70:405–414. doi: 10.1124/mol.106.022970. [DOI] [PubMed] [Google Scholar]
- 20.Moraes ER, Kalapothakis E, Naves LA, Kushmerick C. Neurotox. Res. 2011;19:102–114. doi: 10.1007/s12640-009-9144-8. [DOI] [PubMed] [Google Scholar]
- 21.Rowe AH, et al. PLoS ONE. 2011;6:e23520. doi: 10.1371/journal.pone.0023520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Renganathan M, Cummins TR, Waxman SG. J. Neurophysiol. 2001;86:629–640. doi: 10.1152/jn.2001.86.2.629. [DOI] [PubMed] [Google Scholar]
- 23.Blair NT, Bean BP. J. Neurosci. 2002;22:10277–10290. doi: 10.1523/JNEUROSCI.22-23-10277.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choi J-S, Waxman SG. J. Neurophysiol. 2011;106:3173–3184. doi: 10.1152/jn.00100.2011. [DOI] [PubMed] [Google Scholar]
- 25.McNamara CR, et al. Proc. Natl. Acad. Sci. U.S.A. 2007;104:13525–13530. doi: 10.1073/pnas.0705924104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shields SD, Cavanaugh DJ, Lee H, Anderson DJ, Basbaum AI. Pain. 2010;151:422–429. doi: 10.1016/j.pain.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Catterall WA. Physiol. Rev. 1992;72(suppl.):S15–S48. doi: 10.1152/physrev.1992.72.suppl_4.S15. [DOI] [PubMed] [Google Scholar]
- 28.Catterall WA. Neuron. 2000;26:13–25. doi: 10.1016/s0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- 29.Catterall WA, Goldin AL, Waxman SG. International Union of Pharmacology, Pharmacol. Rev. 2005;57:397–409. doi: 10.1124/pr.57.4.4. [DOI] [PubMed] [Google Scholar]
- 30.Rogers JC, Qu Y, Tanada TN, Scheuer T, Catterall WA. J. Biol. Chem. 1996;271:15950–15962. doi: 10.1074/jbc.271.27.15950. [DOI] [PubMed] [Google Scholar]
- 31.Cestèle S, et al. Neuron. 1998;21:919–931. doi: 10.1016/s0896-6273(00)80606-6. [DOI] [PubMed] [Google Scholar]
- 32.de la Vega R. C. Rodríguez, Possani LD. Toxicon. 2005;46:831–844. doi: 10.1016/j.toxicon.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 33.Zhang JZ, et al. J. Biol. Chem. 2012;287:30719–30728. doi: 10.1074/jbc.M112.370742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gurevitz M. Toxicon. 2012;60:502–511. doi: 10.1016/j.toxicon.2012.03.022. [DOI] [PubMed] [Google Scholar]
- 35.Kreienkamp HJ, Sine SM, Maeda RK, Taylor P. J. Biol. Chem. 1994;269:8108–8114. [PubMed] [Google Scholar]
- 36.Takacs Z, Wilhelmsen KC, Sorota S. Mol. Biol. Evol. 2001;18:1800–1809. doi: 10.1093/oxfordjournals.molbev.a003967. [DOI] [PubMed] [Google Scholar]
- 37.Kaneko Y, Matsumoto G, Hanyu Y. Biochem. Biophys. Res. Commun. 1997;240:651–656. doi: 10.1006/bbrc.1997.7696. [DOI] [PubMed] [Google Scholar]
- 38.Yotsu-Yamashita M, et al. Biochem. Biophys. Res. Commun. 2000;267:403–412. doi: 10.1006/bbrc.1999.1974. [DOI] [PubMed] [Google Scholar]
- 39.Geffeney S, Brodie ED, Jr., Ruben PC, Brodie ED., 3rd Science. 2002;297:1336–1339. doi: 10.1126/science.1074310. [DOI] [PubMed] [Google Scholar]
- 40.Venkatesh B, et al. Curr. Biol. 2005;15:2069–2072. doi: 10.1016/j.cub.2005.10.068. [DOI] [PubMed] [Google Scholar]
- 41.Park TJ, et al. PLOS Biol. 2008;6:e13. doi: 10.1371/journal.pbio.0060013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smith ES, et al. Science. 2011;334:1557–1560. doi: 10.1126/science.1213760. [DOI] [PubMed] [Google Scholar]
- 43.Gilchrist J, Bosmans F. Toxins. 2012;4:620–632. doi: 10.3390/toxins4080620. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.