

Abstract
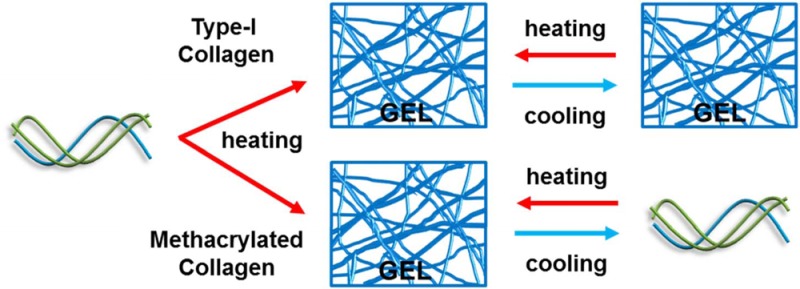
Type-I collagen self-assembles into a fibrillar gel at physiological temperature and pH to provide a cell-adhesive, supportive, structural network. As such, it is an attractive, popular scaffold for in vitro evaluations of cellular behavior and for tissue engineering applications. In this study, type-I collagen is modified to introduce methacrylate groups on the free amines of the lysine residues to create collagen methacrylamide (CMA). CMA retains the properties of collagen such as self-assembly, biodegradability, and natural bioactivity but is also photoactive and can be rapidly cross-linked or functionalized with acrylated molecules when irradiated with ultraviolet light in the presence of a photoinitiator. CMA also demonstrates unique temperature-dependent behavior. For natural type-I collagen, the overall structure of the fiber network remains largely static over time scales of a few hours upon heating and cooling at temperatures below its denaturation point. CMA, however, is rapidly thermoreversible and will oscillate between a liquid macromer suspension and a semisolid fibrillar hydrogel when the temperature is modulated between 10 and 37 °C. Using a series of mechanical, scattering, and spectroscopic methods, we demonstrate that structural reversibility is manifest across multiple scales from the protein topology of the triple helix up through the rheological properties of the CMA hydrogel. Electron microscopy imaging of CMA after various stages of heating and cooling shows that the canonical collagen-like D-periodic banding ultrastructure of the fibers is preserved. A rapidly thermoreversible collagen-based hydrogel is expected to have wide utility in tissue engineering and drug delivery applications as a biofunctional, biocompatible material. Thermal reversibility also makes CMA a powerful model for studying the complex process of hierarchical collagen self-assembly.
Introduction
Thermoreversible hydrogels are of great interest in biomedical research and allow for an extensive array of applications including cell encapsulation, release of drugs, and rapid prototyping.1,2 Ideally, to utilize these gel systems, a sol–gel transition occurs in a temperature range favorable to gel and cell manipulation.3 For example, for in vivo utility, thermoreversible gels should form solutions at cool (<25 °C) or hot temperatures (>50 °C) and transition to a gel at physiological conditions upon injection.1 Thus far, the materials used most extensively are synthetic in nature and lack the natural bioactivity of proteins often found in the extracellular matrix.4,5 Many synthetic polymers used in tissue engineering, such as poly(ethylene glycol) (PEG), poly(ethylene glycol)-diacrylate (PEGDA), and poly(vinyl alcohol) (PVA), are simple cell scaffolds that often require the chemical addition of expensive growth factors or peptides prior to or following gel formation to support cell adhesion.4 Thermoreversible gel systems composed of natural materials, such as gelatin and chitosan, are readily available, but controlling their mechanical properties has proved challenging.4 Our group has developed a collagen-based hydrogel, collagen methacrylamide (CMA), which not only retains the natural properties of collagen but is also photo-cross-linkable and thermoreversible.6
Type-I collagen is a natural, ubiquitous protein comprising approximately 30% of the total protein body content and largely acts as a structural network for tissues such as skin and tendon.7−9 Its inherent properties, including cell attachment and bioactivity, natural biodegradability, amenability to chemical modifications, and/or mechanical cross-linking and self-assembly into a fibrillar gel under physiological conditions, make collagen hydrogels advantageous for tissue engineering and regenerative medicine applications.10,11
The type-I collagen protein fiber is composed of three polypeptide chains that oligomerize into an extended triple helix.12 The constituent triplet for each chain is a Gly-X-Y sequence, where X and Y represent any amino acid but primarily are hydroxyproline and proline, which naturally confer significant strength and rigidity to the protein as well as bioactivity.9,12,13 At physiological temperature and pH, collagen molecules can further associate with other triple-helical monomers to form fibrils and then fibers, where cross-links between adjacent triple helices provide mechanical strength to the growing fiber.13
Type-I collagen can be extracted from connective tissues, typically bovine or porcine, with relative ease, and the resulting protein maintains its ability to self-assemble into a fibrillar hydrogel at physiological pH and temperature, resulting in its widespread use for in vitro and in vivo regenerative medicine applications.10,11 Although collagen gels support cell adhesion and growth and are enzymatically degraded into cell-tolerated products, these scaffolds lack robust control of mechanical properties, which are emerging as important in regulating gross mechanical function and defining the microniche environment of resident cells.14−17
To address this limitation, we developed a protocol to render collagen hydrogels photo-cross-linkable, which allowed spatiotemporal control of mechanical and bioactive properties. Methacrylate groups were covalently bound to the free amines on lysine groups of type-I collagen to create CMA.6 Initial characterization showed that collagen and CMA gels maintained similar properties in secondary structure. Like collagen, CMA self-assembled into a fibrillar gel with similar fibril size and distribution.6 Upon exposure to long-wave UV light (365 nm, 100 mW/cm2) and a photoinitiator in solution, methacrylate groups on collagen formed intermolecular cross-links to stiffen the gel.
As a biomaterial scaffold, collagen is often frozen to generate a highly porous collagen “sponge”, and we were curious if sponges similarly prepared from CMA would retain the ability to be photo-cross-linked.18,19 Subsequently, we discovered that CMA did not freeze like collagen, prompting our interest in studying the temperature-dependent behavior of CMA hydrogels. Herein, to our knowledge, we identify and characterize the first collagen-like protein that can repeatedly thermoreversibly self-assemble into a hydrogel under physiological conditions, as demonstrated on the molecular and supramolecular scales during cooling and reheating. On the basis of these newly discovered properties, CMA can be utilized as a novel model for collagen fibril formation and disassembly as well as a collagen-like thermoreversible hydrogel for tissue engineering applications, including cell encapsulation and cell microenvironment design, drug delivery, and 3D printing.
Materials and Methods
Collagen Methacrylation
The method for collagen methacrylation is described in Gaudet et al.6 All reagents were purchased from Sigma unless otherwise stated. Briefly, type-I collagen (Elastin Products Company, C857) was modified by reacting the free amines of lysine residues with methacrylate groups to create collagen methacrylamide (CMA). The carboxyl group of methacrylic acid was activated with 1-ethyl-3-(3-(dimethylamino)propyl) carbodiimide (EDC) and N-hydroxysuccinimide (NHS) in MES buffer. This mixture was added to collagen at 3.75 mg/mL in 0.02 N acetic acid to form CMA. CMA was dialyzed, lyophilized, and resuspended in 0.02 N acetic acid.
Rheology
Collagen or CMA was mixed in 1 mL batches containing 20 μL of HEPES, 136 μL of 0.15 N NaOH, 100 μL of 10× PBS, 67 μL of PBS (Fisher Scientific), and 677 μL of type-I collagen or CMA (3.75 mg/mL) to form a 2.5 mg/mL suspension.6 To assess the influence of temperature on the mechanical properties of the hydrogel, a sample of 200 μL was loaded into a 600 μm gap between a 20 mm top parallel plate and the bottom parallel plate of a Kinexus Ultra rotational rheometer (Malvern Instruments) at room temperature. The temperature was increased to 37 °C at a rate of 10 °C/min with a Peltier-controlled stage and then was held at 37 °C for 20 min to allow the sample to self-assemble. The temperature was then decreased to 4 °C and increased again to 37 °C at a rate of 2 °C/min. During these temperature changes, the sample was continuously oscillated at 1 rad/s, 0.5% strain while measuring the resultant torque to acquire the temperature-dependent storage and loss moduli of the collagen hydrogels in shear.
To assess the extent of thermal reversibility, a 200 μL sample was prepared as previously described from either collagen or CMA and loaded onto the rheometer. The temperature was increased to 34 °C from 4 °C at a rate of 10 °C/min, held at 34 °C for 5 min, decreased to 4 °C at a rate of 10 °C/min, held at 4 °C for 5 min, and repeated for a total of 10 cycles while the sample was oscillated and resultant storage modulus was measured as described above. The temperature was raised to 34 °C in these experiments as other work has cited some gel denaturation can occur at 37 °C.20
Scanning Electron Microscopy
Multiple collagen and CMA gels were prepared as above and incubated at 37 °C on 12 mm glass coverslips for 2 h. Of these, one CMA sample was disassembled by cooling the gel for 30 min at 4 °C and reassembled by reincubating at 37 °C. Gels were then dehydrated in a series of aqueous acetone solutions (25, 50, 75, and 95%) for 15 min each and then placed in 100% acetone overnight. Samples were critical-point dried (CPD 020, Balzers Union Limited, Balzers, Liechtenstein), sputter coated with gold/palladium (SCD 004, Balzers Union Limited, Balzers, Liechtenstein), and imaged via SEM (Amray 1830I, Amray Inc., Bedford, MA).
Transmission Electron Microscopy
Collagen and CMA suspensions were made as described above in microfuge tubes and placed in the incubator at 37 °C to self-assemble for 30 min. One sample of CMA was placed at 4 °C to disassemble for 30 min and then placed back in the incubator to allow for reassembly for 30 min. A 10 μL sample of the supernatant of the gel was placed on a Petri dish, and an extra-thick carbon 300 mesh copper grid (Electron Microscopy Sciences) was placed face-down on top of each droplet for 5 min. Filter paper was used to remove the sample from the TEM grid. To stain, a 10 μL droplet of 1% phosphotungstic acid (Electron Microscopy Sciences) was placed on a Petri dish, and the TEM grid was placed face-down on top of the droplet for 5 min. Filter paper was used to remove the staining agent, and the samples were dried overnight prior to imaging. TEM studies were carried out using a JEM-100CX TEM microscope (JEOL).
Light Scattering
Light scattering (LS) measurements were performed on a Zetasizer Nano ZS (Malvern Instruments, U.K.) with a 3 mW He–Ne laser at λ = 633 nm, collecting backscattered light at θ = 173°. The sample temperature during measurements was controlled to within ±0.1 °C by a built-in Peltier element. Scattering intensities and autocorrelation functions were determined from the average of either three or five correlation functions, with a typical acquisition time of 60 s per correlation function. Collagen and CMA solutions were prepared either in 0.02 N acetic acid or in PBS buffer (final concentration of 0.1 mg/mL, pH 3.4 or 7 respectively). In the first set of experiments, samples were loaded into low-volume quartz batch cuvettes (ZEN2112) and equilibrated to 37 °C. Measurements were taken at 0, 5, 30, and 60 min. In the second set, samples of collagen and CMA at 2 mg/mL were diluted in PBS to a final concentration of 0.1 mg/mL, pH 7. Measurements were taken every 3 °C as the temperature was raised from 4 to 37 °C, decreased back to 4 °C, and then raised to 49 °C. The temperature was increased or decreased at a rate of 1.5 °C/min, and the temperature was equilibrated for 2 min prior to each set of measurements.
Circular Dichroism
Circular dichroism (CD) spectroscopy measurements were taken using an Aviv model 400 spectrometer (Aviv Biomedical Inc., Lakewood, NJ). CMA and type-I collagen samples were prepared at 2 mg/mL in 0.02 N acetic acid or PBS to a final concentration of 0.1 mg/mL, pH 3.4 or 7, respectively, and then loaded into optically matched 0.1 cm pathlength quartz cuvettes (model 110-OS; Hellma USA). The sample ellipticity was measured in three separate experiments (1 nm intervals, 10 s averaging). First, the ellipticity of 0.1 mg/mL samples of collagen and CMA either in 0.02 N acetic acid or in PBS was measured from 200 to 260 nm at 4 °C. Next, with the same samples, the ellipticity at 222 nm (the wavelength that nominally indicates triple-helical content) of samples was measured as the temperature was increased from 4 to 60 °C at a rate of 0.33 °C/step with a 2 min equilibration time.21 Lastly, the ellipticity of 0.1 mg/mL samples of collagen and CMA in PBS, pH 7 was measured from 200 to 260 nm as the temperature was stabilized at 4 °C, raised to induce fibrillogenesis, decreased to allow for gel disassembly, re-raised to allow for reassembly, and finally increased to 50 °C to a point of gel denaturation. In detail, samples were held at 4 °C for 5 min. The temperature was increased to 37 °C at a rate of 10 °C/min and held at 37 °C for 10 min to allow for self-assembly. The temperature was decreased to 4 °C at a rate of 2 °C/min, and the sample was incubated at 4 °C for 10 min to allow for disassembly. Again, the temperature was increased to 37 °C at the same rate and incubation time described previously for reassembly. Lastly, for gel denaturation, the temperature was increased to 50 °C at a rate of 2 °C/min and held at 50 °C for 10 min. All ellipticity measurements were corrected for the buffer baseline.
Results
CMA Is Rapidly and Repeatedly Thermoreversible
As previously shown in Gaudet et al, collagen and CMA self-assembled into hydrogels between 2 and 3 min after raising the temperature from room temperature to 37 °C.6 The storage modulus (G′) of collagen (∼450 Pa) was approximately 40% greater than that of CMA (∼250 Pa) (Figure 1). As the temperature decreased, an initial increase in the storage modulus of natural collagen was observed until ∼33 to 34 °C, followed by a reduction. In contrast, the storage modulus of CMA decreased steadily with temperature. Whereas collagen remained a gel with a positive storage modulus throughout the temperature sweep, CMA lost the ability to store energy (G′ ≈ 0 Pa) as the temperature approached 4 °C. As the temperature was increased to 37 °C, CMA reassembled into a hydrogel with a slightly higher storage modulus (G′ ≈ 280 Pa) than after the initial assembly.
Figure 1.

Self-assembly and “cold-melt” real-time rheological data of the storage moduli (G′) of collagen (solid line) and CMA (dashed line) while the temperature (thick solid line) was increased to 37 °C decreased to 4 °C and increased to 37 °C at a rate of 2 °C/min. Self-assembly of collagen and CMA gels is observed as an increase in storage modulus around t = 0–3 min. Cold melt of CMA gels is observed as a decrease in storage modulus following t = 20 min.
To demonstrate the extent of CMA thermoreversibility, the storage modulus was monitored as the temperature was rapidly increased and decreased multiple times (Figure 2B). The gelation of CMA rapidly occurred each time the temperature reached 34 °C. Additionally, with each cycle, as the temperature decreased to 4 °C, there was a concomitant decrease in the storage modulus. In contrast, collagen did not demonstrate this same behavior, although the storage modulus changed slightly with temperature (Figure 2A). The thermoreversibility of CMA gelation is robust to multiple cycles of heating and cooling.
Figure 2.

Self-assembly and “cold-melt” rheological data of (A) collagen (G′ −) and (B) CMA (G′ −) with respect to temperature (---). The temperature was cycled between 4 and 34 °C at a rate of 10 °C/min with a dwell time of 5 min for a total of 10 cycles. CMA continues to show rapid disassembly and reassembly as demonstrated by a decrease and increase in the storage modulus, respectively, even after 10 temperature cycles. Collagen exhibits some change in storage modulus with respect to temperature; however, it remains a hydrogel throughout the temperature sweeps.
Collagen and CMA Fibril Structures Are Similar
Cold-melted and reassembled CMA imaged via SEM had similar fibril formation compared to collagen and CMA gels (Figure 3), indicating that cold-melting and reassembling of the hydrogel did not significantly affect fibril formation. Additionally, collagen, CMA, and cold-melted and reassembled CMA imaged via TEM demonstrated canonical D-periodic banding, which is a hallmark of ordered, nativelike collagen assembly (Figure 4).
Figure 3.

SEM images of (A) self-assembled collagen, (B) self-assembled CMA, and (C) cold-melted and reassembled CMA gels at 37 °C imaged at 5000×. The scale bar length is 10 μm. Each gel is composed of a network of fibers.
Figure 4.

TEM images of fibers of (A) self-assembled collagen, (B) self-assembled CMA, and (C) cold-melted and reassembled CMA gels at 37 °C imaged at 80 000×. The scale bar length is 0.2 μm. All samples contain fibers that demonstrate approximate D-banding.
LS Demonstrates Higher-Order Structure Formation in Collagen and CMA Samples
Light scattering allows us to characterize the particle size in solution on the orders of tens of nanometers to micrometers. However, given that the hydrodynamic properties of collagen fibers deviate significantly from the idealized behavior of spherical particles, we used the derived count rate to classify smaller and larger species based on their scattering intensity. Samples of both materials in acetic acid displayed constant and similar scattering intensities throughout the 60 min duration of the experiment, which was consistent with the size distribution of soluble units being the same (Figure 5). This result demonstrates that the methacrylation process does not change the particle size of collagen compared to CMA in its soluble form. In PBS, collagen and CMA began to self-assemble immediately after samples were placed at 37 °C, as indicated by the immediate increase in light scattering and large standard deviation. Scattering intensities of collagen samples were initially larger than those of CMA (Figure 5). After 5 min, both samples had fully self-assembled, as shown by consistent scattering intensities (5, 30, and 60 min time points). In combination with the rheological characterization, this result suggests that the rapid assembly of both collagen and CMA occurs on the same time scale.
Figure 5.

Light scattering of self-assembled collagen and CMA in acetic acid (AcA) and in PBS (open circle, filled circle, open triangle, and filled triangle, respectively) at 37 °C at 0, 5, 30, and 60 min. The scattering intensities were similar for collagen and CMA in each of the diluents. The scattering intensity was unchanged in acetic acid, where the low pH prevents self-assembly, but increased quickly for samples in PBS to form relatively stable structures in size by 5 min after the temperature was increased. Error bars ± standard deviation.
CMA Higher-Order Structure Reversibly Disassembles and Reassembles with Decreasing and Increasing Temperature
The size distribution of higher-order structures of collagen and CMA was also characterized during cold denaturation and reassembly, similar to the rheology experiment. Measurements were taken every 3 °C as the temperature was raised from 4 to 37 °C, decreased to 4 °C, reraised to 37 °C, and then further increased to 49 °C. Collagen and CMA in PBS had similar scattering intensities from 4 °C until the temperature for self-assembly was reached (Figure 6). Collagen self-assembled prior to CMA, shown again by an increase in the rate of light scattering and therefore an increase in size. As temperature was decreased back to 4 °C, the CMA scattering intensity first increased but then decreased, whereas the collagen scattering intensity remained relatively constant (Figure 6). This is indicative of a decrease in the fibril size of CMA, whereas the collagen fibril size was maintained. As the temperature was increased to 37 °C, the CMA scattering intensity increased, suggestive of CMA reassembly and an increase in fibril size.
Figure 6.

Light scattering measurements of collagen and CMA in PBS (solid line and dashed line, respectively) were taken every 3 °C as the temperature increased from 4 to 37 °C, decreased back to 4 °C, and increased to 49 °C. Upon self-assembly, collagen maintained a relatively constant scattering intensity. However, the scattering intensity for CMA decreased upon cooling and then increased again upon reheating. Error bars ± standard deviation.
Collagen and CMA Triple-Helix Signal Decreases Coincident with Self-Assembly
Using circular dichroism (CD) spectroscopy, which is a measure of protein secondary structure, we observe multiple transitions in the mean residue ellipticity (MRE) at 222 nm upon heating collagen from 4 °C. The number of transitions and the temperatures at which they occur depend both on the system, CMA versus collagen, and the solution pH. Under acidic conditions, type-I collagen will form a folded triple helix that is unable to assemble further into higher-order structures.22 Consistent with this, both collagen and CMA had a positive MRE band indicative of a triple-helical supercoiled polyproline-II secondary structure at 4 °C in acetic acid (Figure 7A). Both proteins denatured at 42 °C, indicating that lysine methacrylation does not directly impact the stability of the triple helix itself (Figure 7B).
Figure 7.

Circular dichroism spectroscopy wavelength scan and temperature melt of collagen and CMA samples in acetic acid (AcA) or PBS (open circle, filled circle, open triangle, and filled triangle, respectively). (A) All samples have a positive ellipticity peak wavelength at 222 nm. (B) The ellipticity peak at 222 nm was monitored as the temperature was increased from 4 to 60 °C. For both collagen and CMA in PBS, the positive peak at 222 nm is lost at a temperature consistent with the onset of self-assembly and is replaced by a strong negative peak until the proteins denature.
In contrast, an additional strong negative MRE band at 222 nm was observed for both CMA and collagen when heated in PBS (Figure 7B). This new transition coincided with a temperature range (30–50 °C) that overlaps with temperatures for self-assembly and denaturation observed with LS and rheology, suggesting that this negative band is diagnostic of higher-order structure.
If thermoreversibility is occurring at the level of protein structure, we would expect the positive and negative MRE bands corresponding to the folded triple helix and higher-order assembly to be repeatedly observed upon cycles of heating and cooling. Wavelength scans were performed on collagen and CMA samples in PBS buffer in a temperature sweep experiment to evaluate the secondary structure at specific temperatures for self-assembly, cold-melting, and reassembly. Heating collagen and CMA from 4 °C (Figure 8A) to 37 °C (Figure 8B) resulted in significant shifts in the CD spectrum from positive bands at 222 nm to strong negative bands at ∼206 nm, which produced a negative ellipticity at 222 nm. After returning the samples to 4 °C to allow for cold denaturation, the collagen spectrum was unchanged, indicating the preservation of the structures formed upon heating, whereas CMA regained the characteristic triple-helical peak at 222 nm (Figure 8C). Another round of heating drove the loss of the positive MRE band for CMA at 222 nm, whereas the collagen spectrum remained unchanged (Figure 8D). Both samples fully denatured and lost all secondary structure upon a temperature increase to 50 °C (Figure 8E).
Figure 8.

Circular dichrosim wavelength scan from 200 to 260 nm of collagen (circle) and CMA (triangle) samples in PBS during cold-melt and reassembly. The temperature was equilibrated to 4 °C, raised to 37 °C, decreased to 4 °C, increased to 37 °C, and then raised to 50 °C while wavelength scans were conducted. (A) In PBS, both collagen and CMA have positive ellipticity peaks at 222 nm initially at 4 °C. (B) At 37 °C, both peaks disappear. (C) After returning to 4 °C, CMA regains its positive ellipticity at 222 nm while collagen does not. (D) When the temperature is returned to 37 °C, CMA loses the peak at 222 nm and forms an identical structure to (B). Collagen remains the same. (E) At 50 °C, collagen and CMA denature and lose secondary structure.
Discussion
We have demonstrated and characterized the thermoreversible self-assembly of CMA caused by methacrylation of the lysine residues on type-I collagen. Previous work in our laboratory has used an EDC cross-linking reaction to add bioactive peptide fragments to type-I collagen gels,23−25 but we have not observed the same phenomenon seen in the methacrylated collagen gels. Type-I collagen and methacrylated collagen gels showed minimal differences in initial characterization–the sample ellipticity, fiber diameter, and count using SEM and self-assembly properties were similar.6 However, in characterizing and developing CMA for other applications, we found that the methacrylated collagen gels spontaneously disassembled when the temperature was decreased to below 10 °C. This thermoreversible property was demonstrated in rheological measurements of the storage modulus, LS measurements of fibril size via scattering intensity, and CD measurements of the triple-helix secondary structure. Cold-melted and reassembled CMA gels formed fibrous networks and demonstrated appropriate D-banding similar to that of collagen or CMA hydrogels, indicating that the characteristic collagen structure is preserved. CMA exhibited a decrease in light scattering, indicative of a decrease in fibril size, a drop to zero shear modulus, and a return to the hallmark triple-helical CD signal following cooling, and reassembled to a hydrogel that exhibited a higher storage modulus and a decrease of the triple-helix signal at physiological temperature (Figure 9B). As the scattering intensity at 4 °C did not decrease to its initial value, the CMA fibrils that formed may not have completely disassembled, which could potentially lead to larger or more fibrils and a stronger gel following reassembly, as was observed in the cold-melt LS and rheology. Conversely, type-I collagen self-assembly was shown to be irreversible on the same time scale using these same measurements. Although the triple-helix signal decreases in both collagen and CMA samples, the collagen storage modulus remains positive, indicating that a gel-like state is maintained, and light scattering intensities remain relatively constant throughout the temperature sweep (Figure 9A).
Figure 9.

Approximate correlations among (A) collagen and (B) the CMA storage modulus (G′, Pa) (solid line), derived count rate (DCR, cps) (dot-dashed line), and mean residue ellipticity (MRE, 103 deg cm2 dmol–1 res–1) at 222 nm (small dashed line) derived from rheology, DLS, and temperature-melt CD spectroscopy experiments, respectively (Figures 1, 6, and 7). Discrete MRE measurements at 222 nm obtained from wavelength scans at each temperature are also presented (filled circle) (Figure 8). Initially, at 4 °C, collagen and CMA are triple-helical and unassembled. At 37 °C, collagen and CMA exhibit a negative MRE and begin to form higher-order structures indicated by an increase in DCR and G′. Returning to 4 °C, CMA loses the ability to store energy, exhibiting a storage modulus of ∼0 Pa, although DCR has not reached its initial value, while the storage modulus of collagen slightly decreases, although the DCR remains relatively constant. The MRE of CMA at 4 °C has returned to a positive value, while the MRE of collagen stays negative. Finally, at 37 °C, CMA reassembles as seen by increases in G′ and DCR and a return to a negative MRE value, while collagen G′, DCR, and MRE remain constant.
Additionally, rheology experiments demonstrate a correlation between the rate of change of the storage modulus of CMA and the cooling/heating rates compared to collagen (Figures 1 and 2). Initial assembly of both collagen and CMA occurs quickly (Figure 1, 2 to 3 min). In both experiments, the rate of change of the storage modulus of CMA follows either rate, 2 or 10 °C/min, upon disassembly and reassembly (Figures 1 and 2B). The lack of hysteresis upon multiple cycles is demonstrated by full sol–gel transitions occurring at the specified rate in the cooling/heating schedule in each experiment. In contrast, collagen does not exhibit complete disassembly/reassembly and maintains a hydrogel state with a positive storage modulus. However, slow disassembly and reassembly seen with a slow cooling/heating rate (Figure 1) may lead to the accumulating hysteresis seen in rapid disassembly/reassembly experiments (Figure 2A).
Furthermore, thermoreversible behavior spanned multiple concentrations, where low concentrations are required when using CD spectroscopy and LS to prevent scattering, and high concentrations are required to discern changes in the storage modulus using rheology. Interestingly, despite these differences, there was good agreement of the dynamic, temperature-dependent changes in the data across testing modalities, specifically, a lag in initial self-assembly of CMA relative to collagen (Figure 1) and a correlation of the loss (and regain) of the triple-helical signal in CD with assembly (and disassembly) from the other modalities.
CD spectroscopy results of self-assembled collagen and CMA exhibited trends that to our knowledge have not been shown before in the literature. Often, shorter fragments of collagen have been characterized using these methods under acidic conditions at physiological temperature. Similar to results with our collagen and CMA under acidic conditions, previous work with full-length rat tail tendon collagen has shown that collagen and a glycidyl methacrylated collagen display thermal denaturing at temperatures of around 40 and 35 °C under acidic conditions, respectively.26 However, CD spectroscopy of our self-assembled collagen and CMA under physiological conditions lacked the positive ellipticity peak for triple-helical structure. We hypothesize that collagen and CMA remain triple-helical in structure but have exhibited a change in the CD spectroscopy signal. On the basis of the TEM results, both self-assembled collagen and CMA exhibit D-periodic banding in the fibrils, indicating triple-helical structure. This is consistent with previous studies in rat tail collagen, showing triple-helical structure in aggregate fibrils using X-ray diffraction.27,28 Therefore, as this change in CD signal is observed in both collagen and CMA, it may be due to differences in intermolecular interactions as a result of higher-order assembly, which can perturb the circular dichroism of the protein backbone. Such perturbations can be caused by aromatic amino acids, which have been shown to be involved in direct contacts between triple-helical units in the higher-order assembly.29−31 This novel observation will be studied in the future to understand the molecular determinants of the negative MRE value at 222 nm.
The characterizations presented in this article were motivated by the discovery of thermoreversible CMA self-assembly. Previous work has shown that collagen fibrillogenesis is partially dependent on the telopeptide region. The amino acid sequences of bovine type-I collagen show that the C-terminal of the telopeptide region contains a substantial number of lysines, the amino acid to which the methacrylic acid is conjugated. The α1 chain contains 56 lysines while the α2 chain contains 49 lysines, of which each chain contains 17 lysines on the C-terminal telopeptide (UniProtKB/Swiss-Prot, sequences P02453.3 and P02465.2). On the basis of previous characterization of CMA, approximately 20% of the collagen triple helix is methacrylated.6 The dense population of lysines in the telopeptide region may account for delays in CMA self-assembly compared to collagen, as seen in the Rheology section. The triple helix is intact in the methacrylated collagen, but as shown in the CD spectra, the “assembly” of CMA is delayed compared to that of collagen, and denaturation occurred more rapidly, indicating that the CMA fibrils are metastable compared to collagen fibrils. We have previously used similar EDC cross-linking methods to couple bioactive peptides to collagen but did not observe thermoreversible behavior, suggesting that the thermoreversibility is specific to the molecule added and perhaps methacrylation uniquely. We hypothesize that the differences in intermolecular associations during the self-assembly of collagen and CMA account for the thermoreversibility seen in the methacrylated collagen. As described in Kar et al., higher-order assembly at physiological temperatures is predicted to occur through two steps: (1) partial unfolding of the native triple-helical state to a “loosened” triple-helical state, followed by (2) the aggregation of loosened triple-helical molecules.32 Of these steps, it is unlikely that there are differences in the native triple-helical state of collagen and CMA based on the CD and LS results under acidic conditions. A mechanism that explains differences in aggregation may account for the thermoreversibility of CMA. The telopeptide region is particularly important in catalyzing self-assembly and is involved in the formation of covalent cross-links between adjacent triple helices.33,34 If a significant proportion of the lysines in this region are methacrylated, then a perturbation of interactions between the telopeptide and triple helix may account for the formation of a less stable aggregate of CMA compared to collagen. This aggregate form may be unstable at cooler temperatures, accounting for the thermoreversibility of CMA compared to that of type-I collagen. Future studies will characterize where methacrylates are conjugated and, specifically, if methacrylated lysine residues in the telopeptide region of type-I collagen are the source of the protein’s thermoreversibility.
Thermoreversible biomaterials have a variety of applications in 3D cell culture, drug delivery, and tissue engineering. On the basis of the intrinsic properties of the biomaterial, constructs may have vastly different temperature-dependent properties, which direct the potential applications for that material. CMA self-assembly and cold-denaturing temperatures match those of other previously established thermoreversible biomaterials, as it loses bulk mechanical properties at temperatures of less than 10 °C and forms a soft hydrogel (∼250 Pa) at physiological temperature. Current thermoreversible biomaterials are primarily synthetic hydrogels and have been utilized for tissue engineering applications as scaffolds for corneal wound repair, constructs for cell encapsulation, and drug delivery.35−39 To our knowledge, CMA is the first thermoreversible, collagen-based hydrogel that has the ability to reversibly self-assemble into a fibrillar network at physiological temperature (above 20 °C) and is biodegradable through natural enzymes, biofunctional through cross-linking methods, and cytocompatible.4,5 Future studies will aim to utilize these unique properties of CMA for soft tissue engineering applications.
Conclusions
In this study, we have examined the temperature-dependent, reversible self-assembly of CMA that does not occur with type-I collagen. Collagen and CMA both demonstrate triple-helical structure in soluble form but also lose evidence of triple-helical content during self-assembly under physiological conditions. When cooled, CMA gels disassemble from a fibrillar gel to a solution. Commensurate with this disassembly, light scattering decreases, indicating a decrease in fibril size, and the triple-helix signal is recovered. Following a temperature increase to 37 °C, CMA gels reassemble, the rate of light scattering increases, and the triple-helix secondary structure signal changes. Conversely, collagen gels maintain secondary and fibrillar structure throughout the temperature sweep, demonstrating irreversible self-assembly, at least over the time scale examined in these studies. Overall, the cause for disassembly and reassembly of CMA gels is a result of the collagen methacrylation. CMA tertiary structure may be disrupted in the telopeptide region due to the presence of the methacrylates, allowing for disassembly at cool temperatures. CMA may serve as a model collagen-like protein for a comparison of flexibility, stability, and self-assembly to collagen or for cold-denaturation studies. Furthermore, CMA gels have new properties as a tissue-engineered scaffold; in addition to photolabile modifications of gel mechanical properties, CMA can be utilized as a thermoreversible self-assembled hydrogel. These hydrogels can be utilized in tissue engineering, cell encapsulation, and microenvironment design and as a drug-delivery system.
Acknowledgments
We thank Dr. I. John Khan for his expertise and guidance in this work. This project was supported in part by National Science Foundation grants ARRA-CBET0846328 and DMR-0907273, National Institutes of Health Office of the Director DP2-OD-006478-1, and fellowships from the New Jersey Commission on Spinal Cord Research (08-2937-SCR-E-0), the National Science Foundation (NSF DGE 0801620, IGERT on the Integrated Science and Engineering of Stem Cells), and Rutgers-UMDNJ Biotechnology Training Program (NIH grant number 5T32GM008339-20).
Author Contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Klouda L.; Mikos A. G. Thermoresponsive Hydrogels in Biomedical Applications. Eur. J. Pharm. Biopharm. 2008, 68, 34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billiet T.; Vandenhaute M.; Schelfhout J.; Van Vlierberghe S.; Dubruel P. A Review of Trends and Limitations in Hydrogel-Rapid Prototyping for Tissue Engineering. Biomaterials 2012, 33, 6020–41. [DOI] [PubMed] [Google Scholar]
- Jeong B.; Kim S. W.; Bae Y. H. Thermosensitive Sol–Gel Reversible Hydrogels. Adv. Drug Delivery Rev. 2002, 54, 37–51. [DOI] [PubMed] [Google Scholar]
- Zhu J.; Marchant R. E. Design Properties of Hydrogel Tissue-Engineering Scaffolds. Expert Rev. Med. Devices 2011, 8, 607–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drury J. L.; Mooney D. J. Hydrogels for Tissue Engineering: Scaffold Design Variables and Applications. Biomaterials 2003, 24, 4337–4351. [DOI] [PubMed] [Google Scholar]
- Gaudet I. D.; Shreiber D. I. Characterization of Methacrylated Type-I collagen as a Dynamic, Photoactive Hydrogel. Biointerphases 2012, 7, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricard-Blum S. The Collagen Family. Cold Spring Harbor Perspect. Biol. 2011, 3, a004978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoulders M. D.; Raines R. T. Collagen Structure and Stability. Annu. Rev. Biochem. 2009, 78, 929–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver F. H.; Freeman J. W.; Seehra G. P. Collagen Self-Assembly and the Development of Tendon Mechanical Properties. J. Biomech. 2003, 36, 1529–1553. [DOI] [PubMed] [Google Scholar]
- Lee C. H.; Singla A.; Lee Y. Biomedical Applications of Collagen. Int. J. Pharm. 2001, 221, 1–22. [DOI] [PubMed] [Google Scholar]
- Abou Neel E. A.; Bozec L.; Knowles J. C.; Syed O.; Mudera V.; Day R.; Hyun J. K. Collagen--Emerging Collagen Based Therapies Hit the Patient. Adv. Drug Delivery Rev. 2013, 65, 429–56. [DOI] [PubMed] [Google Scholar]
- Kadler K. E.; Holmes D. F.; Trotter J. A.; Chapman J. A. Collagen Fibril Formation. Biochem. J. 1996, 316, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielsen C. Mechanical Properties of Reconstituted Collagen Fibrils. Connect. Tissue Res. 1981, 9, 51–57. [DOI] [PubMed] [Google Scholar]
- Cheung H.-Y.; Lau K.-T.; Lu T.-P.; Hui D. A Critical Review on Polymer-Based Bio-Engineered Materials for Scaffold Development. Composites, Part B 2007, 38, 291–300. [Google Scholar]
- Engler A. J.; Sen S.; Sweeney H. L.; Discher D. E. Matrix Elasticity Directs Stem Cell Lineage Specification. Cell 2006, 126, 677–89. [DOI] [PubMed] [Google Scholar]
- Burdick J. A.; Vunjak-Novakovic G. Engineered Microenvironments for Controlled Stem Cell Differentiation. Tissue Eng., Part A 2009, 15, 205–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundararaghavan H. G.; Monteiro G. A.; Firestein B. L.; Shreiber D. I. Neurite Growth in 3D Collagen Gels with Gradients of Mechanical Properties. Biotechnol. Bioeng. 2009, 102, 632–43. [DOI] [PubMed] [Google Scholar]
- Schoof H.; Apel J.; Heschel I.; Rau G. Control of Pore Structure and Size in Freeze-Dried Collagen Sponges. J. Biomed Mater. Res. 2001, 58, 352–357. [DOI] [PubMed] [Google Scholar]
- Chvapil M. Collagen Sponges: Theory and Practice of Medical Applications. J. Biomed. Mater. Res. 1977, 11, 721–741. [DOI] [PubMed] [Google Scholar]
- Leikina E.; Mertts M. V.; Kuznetsova N.; Leikin S. Type I Collagen is Thermally Unstable at Body Temperature. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 1314–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persikov A. V.; Ramshaw J. A. M.; Kirkpatrick A.; Brodsky B. Electrostatic Interactions Involving Lysine Make Major Contributions to Collagen Triple-Helix Stability. Biochemistry 2005, 44, 1414–1422. [DOI] [PubMed] [Google Scholar]
- Harris J. R.; Soliakov A.; Lewis R. J. In vitro Fibrillogenesis of Collagen Type I in Varying Ionic and pH Conditions. Micron 2013, 49, 60–8. [DOI] [PubMed] [Google Scholar]
- Sundararaghavan H. G.; Masand S. N.; Shreiber D. I. Microfluidic Generation of Haptotactic Gradients Through 3D Collagen Gels for Enhanced Neurite Growth. J. Neurotrauma 2011, 28, 2377–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro G. A.; Biotech M.; Fernandes A. V.; Sundararaghavan H. G.; Shreiber D. I. Positively and Negatively Modulation Cell Adhesion to Type I Collagen via Peptide Grafting. Tissue Eng., Part A 2011, 17, 1663–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masand S. N.; Perron I. J.; Schachner M.; Shreiber D. I. Neural Cell Type-Specific Responses to Glycomimetic Functionalized Collagen. Biomaterials 2012, 33, 790–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronci G.; Russell S. J.; Wood D. J. Photo-Active Collagen Systems with Controlled Triple Helix Architecture. J. Mater. Chem. B 2013, 1, 3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gathercole L. J.; Keller A. X-ray Diffraction Effects Related to Superstructure in Rat Tail Tendon Collagen. Biochim. Biophys. Acta, Protein Struct. 1978, 535, 253–271. [DOI] [PubMed] [Google Scholar]
- Brodsky B.; Hukins DW; Hulmes DJ; Miller A.; White S.; J W.-G. Low Angle X-ray Diffraction Studies on Stained Rat Tail Tendons. Biochim. Biophys. Acta 1978, 535, 25–32. [DOI] [PubMed] [Google Scholar]
- Woody R. W. Aromatic Side-Chain Contributions to the Far Ultraviolet Circular Dichroism of Peptides and Proteins. Biopolymers 1978, 17, 1451–1467. [Google Scholar]
- Kar K.; Ibrar S.; Nanda V.; Getz T. M.; Kunapuli S. P.; Brodsky B. Aromatic Interactions Promote Self-Association of Collagen Triple-Helical Peptides to Higher-Order Structures. Biochemistry 2009, 48, 7959–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cejas M. A.; Kinney W. A.; Chen C.; Vinter J. G.; Almond H. R. Jr.; Balss K. M.; Maryanoff C. A.; Schmidt U.; Breslav M.; Mahan A.; Lacy E.; Maryanoff B. E. Thrombogenic Collagen-Mimetic Peptides: Self-Assembly of Triple Helix-Based Fibrils Driven by Hydrophobic Interactions. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 8513–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar K.; Amin P.; Bryan M. A.; Persikov A. V.; Mohs A.; Wang Y. H.; Brodsky B. Self-Association of Collagen Triple Helic Peptides into Higher Order Structures. J. Biol. Chem. 2006, 281, 33283–33290. [DOI] [PubMed] [Google Scholar]
- Kuznetsova N.; Leikin S. Does the Triple Helical Domain of Type I Collagen Encode Molecular Recognition and Fiber Assembly while Telopeptides Serve as Catalytic Domains?. Effect of Proteolytic Cleavage on Fibrillogenesis and on Collagen-Collagen Interaction in Fibers. J. Biol. Chem. 1999, 274, 36083–36088. [DOI] [PubMed] [Google Scholar]
- Eyre D. R. Cross-Linking in Collagen and Elastin. Annu. Rev. Biochem. 1984, 53, 717–748. [DOI] [PubMed] [Google Scholar]
- Pratoomsoot C.; Tanioka H.; Hori K.; Kawasaki S.; Kinoshita S.; Tighe P. J.; Dua H.; Shakesheff K. M.; Rose F. R. A Thermoreversible Hydrogel as a Biosynthetic Bandage for Corneal Wound Repair. Biomaterials 2008, 29, 272–81. [DOI] [PubMed] [Google Scholar]
- Landers R.; Hubnerb U.; Schmelzeisenb R.; Mulhaupta R. Rapid Prototyping of Scaffolds Derived from Thermoreversible Hydrogels and Tailored for Applications in Tissue Engineering. Biomaterials 2002, 23, 4437–4447. [DOI] [PubMed] [Google Scholar]
- Yan H.; Saiani A.; Gough J. E.; Miller A. F. Thermoreversible Protein Hydrogel as Cell Scaffold. Biomacromolecules 2006, 7, 2776–2782. [DOI] [PubMed] [Google Scholar]
- Park J. S.; Yang H. N.; Woo D. G.; Kim H.; Na K.; Park K. H. Multi-Lineage Differentiation of hMSCs Encapsulated in Thermo-reversible Hydrogel Using a Co-culture System with Differentiated Cells. Biomaterials 2010, 31, 7275–87. [DOI] [PubMed] [Google Scholar]
- Bhattarai N.; Ramay H. R.; Gunn J.; Matsen F. A.; Zhang M. PEG-Grafted Chitosan as an Injectable Thermosensitive Hydrogel for Sustained Protein Release. J. Controlled Release 2005, 103, 609–24. [DOI] [PubMed] [Google Scholar]