

Abstract
G protein-coupled receptors (GPCRs), which are involved in virtually every biological process, constitute the largest family of transmembrane receptors. Many top-selling and newly approved drugs target GPCRs. In this review, we aim to recapitulate efforts and progress in combinatorial library-assisted GPCR ligand discovery, particularly focusing on one-bead-one-compound library synthesis and quantum dot-labeled cell-based assays, which both effectively enhance the rapid identification of GPCR ligands with higher affinity and specificity.
G protein-coupled receptors as drug targets
G protein-coupled receptors (GPCRs) are characterized by an extracellular N-terminus and an intracellular C-terminus connected by seven transmembrane α-helical segments (TM-1 to TM-7). GPCRs are therefore also known as seven-transmembrane domain receptors (7 TM receptors) or heptahelical receptors. The transmembrane domains are composed of three intracellular (IL-1, IL-2 and IL-3) and three extracellular loops (EL-1, EL-2 and EL-3) [1,2,3] (Figure 1). GPCRs in the human genome are generally organized into five families based on their sequence and similar structure [4]: rhodopsin (family A), secretin (family B), glutamate (family C), adhesion (family D) and frizzled/taste 2 (family E). The rhodopsin family, which contains four main groups (α, β, γ and δ) with 13 subbranches, is the largest family. Although the exact size of the human genome GPCR superfamily is uncertain, approximately 800 different human genes have been predicted based on genome-sequence analysis, 701 of which in the rhodopsin family [5].
Figure 1.

The structure and function of G protein-coupled receptors with extracellular signaling molecules (ligands) targeting varied binding sites.
GPCRs represent one of the most important classes of proteins due to their critical role in cell signaling. Extracellular signaling molecules (ligands) can be recognized at varied binding sites (Figure 1). These ligands activate inside signal transduction pathways, and ultimately lead to the activation or inactivation of a particular signaling pathway, hence a specific cellular response. The ligands are varied in type (including light-sensitive compounds, odors, pheromones, hormones, growth factors and neurotransmitters) and size (from small molecules to peptides to large proteins) [6]. The signaling pathways involved mediate almost every important physiological process in humans, such as the sense of sight and smell, behavioral and mood regulation, immune system activity and inflammation, as well as autonomic nervous system transmission [7]. The elucidation of the structure and function of GPCRs and GPCR ligands is a result of collaborated efforts, including the birth of cryoelectron microscopy [8] and the discovery of the rhodopsin structure [9]. For instance, Alfred Gilman and Martin Rod-bell (Nobel Prize in Physiology or Medicine 1994), Brian Kobilka and Robert Lefkowitz (Nobel Prize in Chemistry 2012) are recognized for their discovery of G protein structures and the role of these proteins in signal transduction in cells [10,11].
Numerous diseases and disorders, such as allergies, anxiety, asthma, congestive heart failure, glaucoma, hypertension, migraine, nocturnal heartburn, Parkinson’s, psychosis, schizophrenia and ulcers [12,13], have all been linked to mutations and polymorphisms in GPCRs. This makes GPCRs essential and potential drug targets for the pharmaceutical industry [14]. Currently, approximately 30–50% of all registered drugs act on GPCRs [15,16]. Furthermore, naturally occurring small molecules, such as adenosine, adrenaline, dopamine, prostaglandins, somatostatin, as well as drug-like small molecules, such as caffeine, morphine, heroin and histamine, all target GPCRs. Of the 90 new molecular entities approved by the US FDA in the past 3 years (2010–2012), 17 drugs target GPCRs, indicating that therapeutics for GPCRs are still a main focus for new drugs [17]. The chemical structures of some recently approved GPCR-targeting drugs are shown (Figure 2), including one approved in 2013 [18].
Figure 2. Selected newly approved drugs targeting G protein-coupled receptors.

The chemical structure, drug name, trade name, year of launch and G protein-coupled receptor target are given.
Approximately 60 GPCRs out of the total 800 have been targeted by existing drugs; for instance, the most commonly targeted receptors are histamine H1, α1A adrenergic, muscarinic M1, dopamine D2, muscarinic M2, 5-HT2A, α2A adrenergic and muscarinic M3. However, the vast majority of GPCRs have not yet been explored. Furthermore, one of the major challenges in drug development for GPCRs is the limited availability of structural data on GPCRs. Early studies only revealed the structure of visual pigment rhodopsin [9]. It was not until the period 2007–2011 that medium-to high-resolution crystal structures of several new GPCRs were revealed, including the β1 and β2 adrenergic receptors [19,20], adenosine A2A receptor [21], chemokine CXCR4 receptor [22] and dopamine D3 receptor [23]. Therefore, most of the registered drugs that act on GPCRs are derived from ligand-based drug-design strategies, and since only a small number of GPCRs have been targeted by current pharmaceuticals, huge efforts are now being made to exploit the remaining receptors, including approximately 120 members for which no existing ligands have ever been identified (known as orphan receptors) [24].
This review discusses the efforts and progress in combinatorial library development, and the identification of GPCR ligands via one-bead-one-compound (OBOC) library high-throughput screening. In addition, various strategies used in quantum dot-labeled cell-based screening methods for the rapid identification of GPCR ligands with higher affinity and specificity are presented.
Combinatorial libraries
Combinatorial chemistry, first reported in the early 1980s, is regarded as one of the most important recent advances in medicinal chemistry [25]. The essence of combinatorial chemistry is that a large range of analogues can be synthesized using the same reaction conditions and the same reaction vessels. In this way, a very large number of compounds with high molecular diversity can be synthesized by a simple methodology at a far lower cost than using traditional synthetic chemistry [26]. Combinatorial chemistry libraries are usually constructed with subunits with different R group positions. For each R group position there are a variety of building blocks that can be incorporated to generate complexity. Combinatorial library methods were first applied to peptides and oligonucleotides [27,28]. Since then, the field has been expanded to include peptidomimetics, synthetic oligomers, small molecules and oligosaccharides [29].
Peptide libraries
Peptides are particularly well suited for combinatorial synthesis. First, there is a considerable collection of amino amides that act as the subunits, including both natural amino acids and other commercially available unnatural amino acids used as alternative building blocks to extend the diversity of the peptide library. Second, the synthesis of a peptide library can be achieved effectively by virtue of the solid-phase amide bond-forming chemistry using Fmoc-protected subunits (Figure 3). Usually, the library is synthesized on solid phase, mostly on resin beads [30]. With technological advancements, the whole synthetic procedure can be performed using fully automated instruments [31].
Figure 3. Comparison of peptide and peptoid.
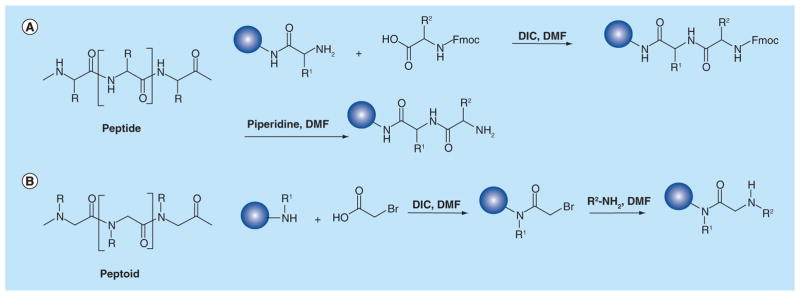
(A) Structure of peptide, and solid-phase Fmoc-method for peptide synthesis. (B) Structure of peptoid, and two-step submonomer solid-phase synthesis of peptoid.
Alternatively, peptide libraries can also be prepared biologically, for example, using a phage-display approach. First reported by Smith in 1985, the key to peptide phage display technology is to express peptides on the surface of bacteriophage as fusions with capsid proteins [32]. This can be achieved by incorporating a peptide encoding gene into a capsid structural protein encoding gene. A phage-displayed peptide library containing billions of peptides presented on phage particles can then be screened simultaneously for the desired activity [33]. Over the past two decades, phage-display technology has been influential in many scientific fields including drug discovery and drug-target validation.
Peptoid libraries
While peptide libraries are rich sources of combinatorial molecules, certain undesirable properties, such as sensitivity to proteases, make native peptides less than ideal for certain applications. To address this issue, there has been much focus on the design, synthesis and the application of a variety of other biopolymer mimetics. Peptoids, first reported by Simon et al. [34,35], are the most well-known examples of nonpeptide compounds. Peptoids are oligomers of N-substituted glycine (NSG) units, which are ideal for combinatorial approaches to drug discovery. Large libraries can be easily synthesized from readily available primary amines using the two-step submonomer solid-phase synthesis method developed by Zuckermann et al. [36] (Figure 3). Peptoids possess distinctive advantages including:
Enhanced stability toward proteolysis;
Resistance to denaturation induced by solvent, temperature or chemicals since secondary structures in peptoids do not involve hydrogen bonding;
Better cell penetration;
Thus, the use of peptoid libraries in drug discovery is rapidly gaining popularity. Drugs over the counter derived from peptoids are still undergoing optimization, although this a fast-developing and promising field.
Small-molecule libraries
The application of combinatorial libraries is not only limited to peptides and peptidomimetics, but also successfully extended to nonpeptide-like small molecules [39,40]. These structurally and chemically diverse small molecules, exhibiting characteristics not present in peptides, are important as drug candidates. Small-molecule combinatorial libraries can be classified using several different criteria, such as the design of the library, novelty of structures, structural features of building blocks and chemical strategy. Lam et al. [29] divided small-molecule libraries into four categories: acyclic libraries assembled in linear fashion, libraries built using a preformed scaffold, libraries including a heterocyclization step and structurally heterogeneous libraries. Due to the favorable pharmacokinetic properties of many small organic molecules (<600 molecular weight), the design, synthesis and evaluation of libraries of these compounds has significantly expanded the application of combinatorial chemistry in the development of therapeutic agents [41]. There are advantages and disadvantages of each type of combinatorial library. The library chosen depends largely on the nature of the target, the assay system and the resources available. In some cases, a combination of approaches may be appropriate.
GPCR ligands from OBOC combinatorial libraries
OBOC combinatorial libraries
There are many distinct methods for combinatorial library synthesis. Among them, the solid-phase OBOC method is a powerful and widely used approach. The OBOC method was first introduced in 1991 by Lam et al. [27] for the synthesis of peptide libraries; since then, this approach has been successfully applied to many other combinatorial libraries, such as the peptoid library [42,43]. As indicated by its name, in an OBOC library, one single bead (the polymeric solid support) contains only one type of compound although there may be up to 1013 copies of the same compound on a single bead depending on its size and capacity [44].
A ‘split-and-pool’ synthetic protocol, which was first developed by Houghten et al. [45], is a key method for ensuring that each bead displays only one compound. Between synthetic steps, the solid supports are combined, mixed and redistributed. The resulting stochastic distribution of solid supports provides approximately equal numbers of synthetic intermediates in each of the next reactions [46] (Figure 4). Using this methodology, a large OBOC library (106–108 members) can be synthesized rapidly, with each bead expressing only one compound and each compound exhibiting equal distribution in the library. More importantly, OBOC libraries significantly facilitate the subsequent screening assay, making it quick and straightforward, in both solid phase and solution phase.
Figure 4. ‘Split-and-pool’ synthetic protocol for one-bead-one-compound library.

The four different shapes of triangle, star, square and inverted triangle represent four different monomers used in library synthesis.
High-affinity GPCR ligands identified from OBOC libraries
Combinatorial libraries serve as powerful platforms for the quick identification of new GPCR ligands. The examples discussed in this section provide highlights of some of the key efforts for screening against a wide variety of GPCRs (Figure 5).
Figure 5. Hit compounds as G protein-coupled receptors ligands identified from one-bead-one-compound combinatorial libraries.

MSH: Melanotropin.
Zuckermann et al. [47] used competitive radioligand-binding assays to screen a 5000 synthetic peptoid library for GPCRs ligands. They discovered two hit peptoid trimers, CHIR 2279 (1) and CHIR 4531 (2), which bind to the α1-adrenergic receptor and μ-opiate receptors with Ki values of 5 and 6 nM, respectively.
Jayawickreme et al. [48] identified several novel α-melanotropin (α-MSH) receptor antagonists from a peptide library consisting of 31,360 structurally different candidates. A functional bioassay was conducted by monitoring the pigment translocation induced by peptides. The dose–response curves of the hit peptides were further measured using microtiter plate assays. Finally, 153N-6 (3) was identified as the most potent MSH receptor antagonist with an IC50 value of 11 ± 7 nM to decrease receptor activation. This finding is important since α-MSH stimulates melanogenesis, and also plays a role in feeding behavior, energy homeostasis and sexual activity.
Appell et al. [49] created a 56,000-member combinatorial library to identify compounds that inhibit the binding of labeled ligands to two related GPCRs. OBOC library beads arrayed at 20 beads per well were first photolyzed to cleave 50% of the compounds, which after drying were redissolved at a concentration of 0.5–1 μM. Beads in positive wells were then collected and redistributed at one bead per well to rephotolyzed thoroughly for secondary screening. Finally, 86 unique structures were identified as active against one receptor and 24 were active against the other using a Wallac Mcrobeta scintillation counter on fluorometer.
Heizmann et al. [50] tested a peptoid library containing 328,509 compounds and successfully identified new ligands for both α-MSH receptor and GRP-preferring bombesin receptor. The KD values calculated from competition binding data were 1.58 μM (4) and 3.4 μM (5), respectively.
Oxytocin is a mammalian neurohypophysial hormone that plays an important role in the neuroanatomy of intimacy, specifically in sexual reproduction. Evans et al. [51] prepared a library of 1296 1,4-benzodiazepines for the discovery of selective oxytocin antagonist using scintillation proximity binding assay. The most potent compound GW405212 (6) was discovered with an IC50 value as low as 5 nM.
The melanocortin (MC) receptors are GPCRs with five different isoforms, MC1R–MC5R. Particularly, MC4R is known to be an excellent drug target for the treatment of obesity. Kruijtzer et al. [52] synthesized peptoid–peptide hybrids and investigated the library on cells expressing different MC receptor subtypes. Iuga et al. [53] also aimed at the identification of novel melanocortin receptor agonists. By using a hexapeptide library they discovered several related novel peptides, with the most potent (7) displaying an EC50 value of 0.1 ± 0.03 μM to stimulate pigment dispersion. The structures of 1–7 are shown in Figure 5.
Progress of OBOC library screening methods
All combinatorial library methods involve three main steps: preparation of the library, screening of the library components and determination of the chemical structures of active compounds [29]. These steps are closely linked, and each of them is indispensable for the final successful identification of GPCR ligands. Numerous efforts have been made to improve the efficacy and decrease the cost of these procedures; encouraging advances have been achieved [54,55]. In this section, we focus on recent progress in the use of screening methods for the identification of high-affinity GPCR ligands based on OBOC libraries.
A reliable high-throughput assay is essential for successfully screening a combinatorial library. Both solid-phase and solution-phase assays have been developed for OBOC libraries. For solid-phase assays, ligands are still attached to the beads; while cleavage of ligands from the beads is required before screening during solution-phase assays. Considerable advances in screening technology have been made, which significantly enhance the efficiency of rapidly identifying GPCR ligands.
The most straightforward way to screen high-affinity GPCR ligands is to detect the binding behavior of target GPCRs to ligand libraries. For molecular targets that are intrinsically colored or fluorescent, a bead library can be screened directly [56]. However, in some cases, indirect screening is required in which a reporter group is attached to the target. The reporter group can be an enzyme, a radioactive isotopes, a color dye or a fluorescent probe [29]. Generally, the color intensity or fluorescence of the target-attached bead after incubating with the receptors is proportional to the binding affinity of the ligand.
Traditional screening methods
Lam et al. [27] reported the enzyme-linked colorimetric assay in 1991. In this method, the peptide library is incubated with a receptor–enzyme complex (e.g., receptor-alkaline phosphatase conjugate). After washing, the beads are then mixed with the chromogenic substrates, which are a combination of nitroblue tetrazolium (NBT) and 5-bromo-4-chloro-3-indolyl phosphate. After the hydrolysis of phosphate by alkaline phosphatase, NBT is reduced to formazan turning the positive beads purple. However, NBT can also be reduced by any trace amount of reducing agent in the beads [57]. A dual-color detection scheme has been developed later in order to eliminate the false positive beads [58].
Turck described a method to screen OBOC libraries against radiolabeled receptor targets [59]. The peptide resin beads were incubated with radiolabeled receptor molecule and, subsequently, immobilized in a thin layer of agarose. Resin beads that carry acceptor-specific sequences were identified by autoradiography and subjected to automated gas-phase sequencing.
While there are many other available ways to label a protein for a library screening, fluorescent tags have many advantages, including high sensitivity [60]. Fluorescence intensity can be measured using a fluorescence reader, or imaged by a fluorescence microscope. Furthermore, fluorescence-based screening methods using a fluorescence-activated cell sorter have also been developed [61,62,63].
The proper selection of bead material and synthetic protocol as well as the fluorescent marker is critical to this technique [64]. TentaGel™ resin beads (Rapp Polymere Ltd; Tuebingen, Germany), which are produced by cografting PEG units onto a low-crosslinked polystyrene [65], is now the most widely used solid support for on bead library screen considering its uniform size. However, the TentaGel beads exhibit a high-level, broad-wavelength intrinsic fluorescence, particularly in the green region of the spectrum [66]. This autofluorescence rendered the use of many organic fluoresceins for screening experiments impractical and, therefore, considerate efforts have been made to overcome this limitation.
Since the intensity of the bead fluorescence drops off significantly in the red region of the spectrum, Alluri et al. [66] evaluated Texas Red florescent dye labeled protein as potential targets in the screening process. After incubating the labeled protein with the bead library for 1 h and then washing the beads thoroughly, the beads were identified visually under a fluorescence microscope. It was observed that positive hit beads were clearly brighter than the surrounding beads. Although the reduced background is tolerable, it is still noticeable, requiring careful and painstaking visual analysis of beads under a microscope field, which is a tedious process.
Quantum dot-labeled screening methods
Olivos et al. [67] successfully solved the above problem by using the unique fluorescent properties of quantum dots (QDs). QDs are semiconductor nanocrystals that show unique optical properties, including size-tunable light emission, simultaneous excitation of multiple fluorescence colors, high signal brightness, long-term photostability and multiplex capabilities [68,69,70]. In contrast to traditional fluorescent organic dyes, one of the critical features of QDs is that they absorb energy in a broad and continuous region of the spectrum while have narrow and symmetrical emission peaks [71]. Streptavidin (SA)-coated QDs that emit in the red region of the spectrum (λmax = 608 nm) and can detect biotinylated proteins captured on beads are commercially available.
Olivos et al. [67] made a comparison of the performance of QDs to Texas Red in a protein binding assay with peptides displayed on TentaGel beads. Beads displaying ubiquitin-binding peptides, negative controls or positive controls were incubated with biotin-labeled ubiquitin. After washing, the beads were exposed to either Texas Red-labeled SA or SA-coated QDs (Figure 6). It was observed that in Texas Red-labeled group, the hits were brighter than background, but with low levels of contrast. On the other hand, in SA-coated QDs, after employing the excitation filter used for 4′,6-diamidino-2-phenylindole dye, hit beads exhibited a bright red fluorescence while negative control beads were bright green. These results indicate that QDs provide a striking color difference between positive and negative beads without being obscured by the autofluorescence of the beads. In the following genuine library-screening experiment, the authors further demonstrated that QD-based detection allowed reliable selection of true hits in the midst of many other beads with high efficacy. This red-emitting QDs labeling method was later successfully applied by Lim et al. [72] to identify an inhibitor of the proteasome 19S regulatory particle from a 32,768-member peptoid OBOC library (compound 8, Table 1).
Figure 6. On-bead screening assay using streptavidin-coated quantum dot.

Beads displaying ligands that are able to bind to a receptor protein are labeled with quantum dot through specific interaction between biotin and streptavidin.
Table 1.
Application of the quantum dot-labeled screening method to identify hit ligands from one-bead-one-compound combinatorial libraries.
| Structure of ligands | Target | Activity | Screening methods | Ref. |
|---|---|---|---|---|
|
8 |
Proteasome 19S RP | Inhibit the protein unfolding activity of 19S RP with IC50 value of 3 μM | On-bead red-emitting QDs labeling method |
[72] |
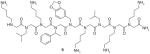 9 |
VEGFR2 | Bind to VEGFR2 with KD value of approximately 2 μM | On-bead two-color QDs labeling method |
[74] |
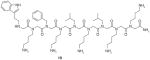 10 |
VEGFR2 | Bind to VEGFR2 with KD value of approximately 2 μM | ||
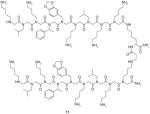 11 |
VEGFR2 | Bind to VEGFR2 with KD value of approximately 30 nM. Inhibit VEGFR2 activation with IC50 value of approximately 1 μM | ||
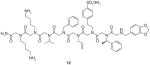 12 |
OxR1 | Antagonist of OxR1 with IC50 of approximately 139 μM | On-bead two-color QDs labeling method accelerated with magnetic beads | [80] |
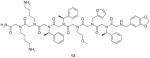 13 |
OxR1 | Antagonist of OxR1 with IC50 of approximately 205 μM | ||
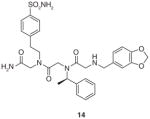 14 |
OxR1 | Antagonist of OxR1 with IC50 of approximately 51 μM |
OxR1: Orexin receptor 1; QD: Quantum dot; RP: Regulatory particle; VEGFR2: VEGF receptor 2.
One critical problem during library screening against a target protein is how to eliminate nonspecific or false-positive hits. Practical solutions include the use of a high salt- and detergent-containing buffer, lowering the concentration of target protein (100–500 nM), or employing a 1000- to 10,000-fold excess of cleared Escherichia coli lysate as competitor proteins. Using competitor proteins is particularly effective in screening against relatively hydrophobic, ‘sticky’ targets, which often exhibit good affinity but are rarely specific [73].
In 2008, Udugamasooriya et al. [74,75] developed a novel on-bead two-color (OBTC) QDs-labeled screening technology, which eliminated the requirement for introducing such protein competitors, and made it easy to identify ligands for the specific receptor of interest. Using this approach, a maximum amount of 100,000 OBOC beads (TentaGel beads) can be used, which are equilibrated with cell culture media before introduction of the cells. At the same time, cells expressing receptors of interest are labeled with a red QD (Qtraker® 655), while primary cells lacking corresponding receptors are labeled with a green QD (Qtraker 565). Observation under the fluorescence microscope is required to confirm the success of cell staining. After mixing at a 1:1 ratio, the two groups of cells are then incubated with library beads in polypropylene tubes, which are shaken gently. After equilibration for the optimized time period, the beads are washed and transferred to tissue culture dishes for observation under a fluorescence microscope. With proper filter sets, all the colors (blue from beads, red and green from cells) can be observed at the same time. Beads that bind both red and green QDs-labeled cells imply that the ligands on them act as nonspecific ligands. Therefore, only the beads that exclusively bind to red cells are considered specific ligands and are later manually picked up using pipette under microscope. Cells and other debris are stripped off from the positive beads using 1% SDS solution, and the structures of the ligands on beads are elucidated using mass spectrum.
Several factors need to be taken into account and to be optimized before the OBTC screening (e.g., the design of cell pair, the density of cells and beads and equilibration time) [75]. Common cell types ideal for this kind of screening include HEK-293, Chinese hamster ovary and HeLa. In some cases, the stable transfection method is required to overexpress the specific receptor of interest on cell surface. The density and the equilibration time depend on the binding affinity of the cells to compounds, and these factors are usually optimized using a small-scale pilot study.
The above screening assay can be very efficient. Usually, the identification of hit ligands from a 100,000 OBOC library can be completed in a single day, although the optimization of screening conditions ahead may take a few days or weeks. Meanwhile, this method is very low cost since only a fluorescence microscope is required for the screening without the need for other specialized, expensive equipment [76]. More importantly, cell-based screen allows the receptors of interest to be displayed in a relatively natural cellular environment. This particularly makes sense for GPCR ligands, and other integral membrane receptors screenings, because the standard screening methods developed for soluble proteins could not be easily employed for membrane receptors [77,78,79].
Udugamasooriya et al. [74] successfully applied the above two-color, cell-based screening method to identify specific peptoids binding to VEGF receptor 2 (VEGFR2). The two hit ligands are shown in Table 1. Both 9 and 10 bound to VEGFR2 with KD values of about 2 μM. Homodimeric derivatives of 9 and 10 were further synthesized, and 11 was finally discovered to bind VEGFR2 with KD of 30 nM. An in vitro function assay demonstrated that 11 was an antagonist of VEGF-dependent receptor activation (Table 1).
Based on the above OBTC screening protocol, Qi et al. [80] further incorporated a magnetic isolation procedure to screen a 3 million peptoid library for orexin receptor 1 (OxR1) ligands. OxR1 is a GPCR involved in a number of interesting metabolic events and a potential drug target for insomnia, diabetes and drug addiction. In this experiment, Chinese hamster ovary cells labeled with red or green QDs were mixed and exposed to the OBOC library. After thorough washing, the beads were exposed to anti-OxR1 polyclonal antibody from rabbit and, after incubation and further washing, iron oxide particles Dynabeads® M-280 coated with sheep anti-rabbit IgG secondary antibody were added. The suspension was then placed in a conical tube; after gentle shaking, the tube was placed in a holder that positioned a powerful magnet at the side of the tube (Figure 7A). TentaGel beads that display peptoids able to bind to OxR1 were held there, while others settled to the bottom of the tube. After removal of all of the unmagnetized beads with a pipette, those retained by the magnet (only a small fraction of the original library) were further examined under fluorescent microscope (Figure 7B). This methodology remarkably increases the efficiency by shortening the most tedious step of previously published bead screening techniques, and allows millions of different compounds to be screened rapidly.
Figure 7. A two-color, cell-based magnetic screen to identify orexin receptor 1-binding ligands.

(A) Representation of the screening procedure. (B) Fluorescence microscopic image of cells labeled with quantum dots and beads. The bead observed to bind only red quantum dot-labeled cells represents one of the positive hits out of 3 million beads. OxR1 peptoid antagonists (12 & 13) and the further developed peptoid trimer (14) are shown in Table 1.
CHO: Chinese hamster ovary; OxR1: Orexin receptor 1.
Reproduced with permission from [80] © The Royal Society of Chemistry (2010).
Using this method, Qi et al. [80] successfully identified two hit compounds (12 & 13, Table 1) as OxR1 antagonists. Furthermore, guided by a structure–activity relationship study, a peptoid trimer (14, Table 1) that kept the same pharmacophore of two N-terminal residues in 12 and 13 was synthesized and proved to be an even more effective OxR1 antagonists than 12 and 13. These results strongly demonstrate the feasibility and reliability of identifying GPCRs ligands using both the combinatorial chemistry and traditional medicinal chemistry methods. Taken together, this approach provides a platform for the use of peptoid library and two-color QD-labeled cell-based screening for rapidly identifying GPCR ligands. One can imagine that this method would be applicable to other types of OBOC libraries as long as the library sequences can be encoded and identified. Furthermore, constructing expression vectors for GPCRs is affordable. This screening technology can be readily applied to isolate ligands with high affinity and specificity against many GPCRs in a high-throughput fashion with low cost.
Future perspective
The application of QDs to the library screening assay has significantly enhanced the efficacy and accuracy for the identification of GPCR ligands with strong affinity and high specificity. Based on the principles of the above-mentioned one- and two-color QD methods, it is conceivable that multicolor QD-labeled cell-based screenings is a promising trend in this field. In fact, multicolor QDs have already proved their potential in many other fields, such as biological imaging, molecular diagnosis and multiplexed optical coding [68,81,82]. Using multicolor QD approach, cells expressing different GPCRs of interest can be labeled with QDs of different colors. All the QD-labeled cells can be mixed in equal ratios and exposed to the combinatorial library. When examining the beads, a single color-bound bead correlates to a highly selective ligand for a specific receptor on the cells. Beads attached with multicolored cells correlate with nonselectively binding ligands (Figure 8).
Figure 8. On-bead multicolor screen with quantum dot labeling for the identification of ligands for G protein-coupled receptors.
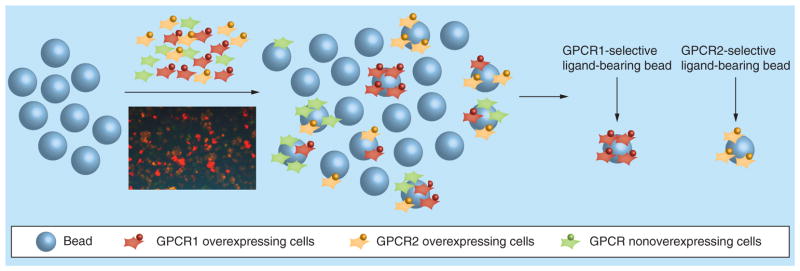
Cells overexpressing GPCR1, GPCR2, or nonoverexpressing cells are labeled with Qtracker® 655 (red), Qtracker 585 (yellow), or Qtracker 565 (green) quantum dots. After incubation with quantum dots-labeled cells, beads with only red cells are selectively chosen to identify the GPCR1 specific ligand. The beads with only orange cells are selectively chosen to identify the GPCR2 specific ligand as potential hits.
GPCR: G protein-coupled receptor.
One of the key advantages of this method is that it permits quick and straightforward high-throughput screening for different GPCR ligands at one time using one library. This is especially desirable for the screening of receptors originated from homologous genes, such as OxR1 and OxR2, providing powerful tools for the identification of selective GPCR ligands as well as dual- or even multi-target ligands.
It is also noteworthy that the application of QD-labeled screening is not only limited to binding assays, but can also be extended to function assays. For example, with the application of the QD tagging strategy, Garske et al. [83] developed a novel and high-throughput method for determining deacetylase SIRT1 substrate specificity by using an OBOC acetyl peptide library. Beads were first biotinylated and labeled with SA-coated QDs. The brightest beads after fluorescent sorting therefore represented the ones which were most preferentially deacetylated by SIRT1. Using the same method, it might be possible to conduct functional screening assays of combinatorial libraries against GPCRs. The above-mentioned screening methods can sometimes be combined to perform more stringent screening thus providing more information for the GPCRs drug discovery.
In conclusion, GPCRs-based drug discovery continues to be a major area of pharmaceutical research given the fundamental role of GPCRs in signaling transduction and the large size of GPCRs superfamily in the human genome. Considerable opportunities still exist for GPCR drug discovery since only a small number of GPCRs have been targeted by current pharmaceuticals. Combinatorial libraries provide an efficient route for achieving great molecular diversity at a low cost; in addition, they are powerful tools for ligand-based GPCRs drug design and development. Through the comprehensive application of solid-phase synthetic chemistry, combinatorial chemistry, in particular multicolor QD-labeled cell-based screening assay methods, the rapid identification of high-affinity GPCR ligands from combinatorial libraries can be made available, thereby providing promising opportunities for future drug discovery.
Key Terms.
One-bead-one-compound library
Important type of combinatorial library in which one single bead only displays one kind of compound, although there may be millions of the same compound on a single bead.
Peptoids
Poly-N-substituted glycines, a class of peptidomimetics whose side chains are appended to the nitrogen atom of the peptide backbone, rather than to the α-carbons (as they are in amino acids).
Split-and-pool-TentaGel
Methodology for solid-phase library (especially one-bead-one-compound library) synthesis. Between synthetic steps, the solid supports are combined, mixed and redistributed.
™ resins: Grafted copolymer beads that consist of a low-crosslinked polystyrene matrix and PEG. TentaGel beads are currently the most widely used solid support for one-bead-one-compound combinatorial library synthesis.
Quantum dots
Nanocrystals made of semiconductor materials that are small enough to display quantum mechanical properties.
Executive summary.
G protein-coupled receptors as drug targets
G protein-coupled receptors (GPCRs) are the largest and the most important classes of proteins due to their critical role in cell signaling.
While they are considered essential drug targets only a small number of GPCRs have been targeted by current drugs. Great efforts are now being made to exploit the remaining receptors.
Combinatorial libraries
A huge number of compounds with high molecular diversity can be synthesized quickly using combinatorial chemistry.
Combinatorial libraries can be applied to peptides, peptidomimetics (e.g., peptoids), and nonpeptide-like small molecules. Libraries are usually synthesized using solid-phase chemistry.
One-bead-one-compound libraries, usually synthesized by a split-and-pool method, provide a straightforward platform for library screening.
Progress of library screening methods for the identification of GPCR ligands
The autofluorescence of TentaGel™ resin obscures the screening assays. Labeling proteins with Texas Red florescent dye partially reduces the background of TentaGel resin, but the results are still far from satisfying. This problem is successfully solved using the unique fluorescent properties of quantum dots (QDs).
The screening assays are further improved using a two-color QD-labeled, cell-based screening approach. Receptor-expressing cells and receptor non-expressing cells are labeled with two different QDs. Ligands with high affinity and specificity can be identified rapidly without employing protein competitors.
Future perspective
It is conceivable that cell-based screening will be further improved using multicolor QDs, allowing different GPCRs to be screened simultaneously.
Footnotes
For reprint orders, please contact reprints@future-science.com
Financial & competing interests disclosure
The authors would like to acknowledge financial support from start-up support from University of Florida to XQ and in part by the NIH/NCATS Clinical and Translational Science Award to the University of Florida UL1 TR00064 to XQ. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as:
• of interest
•• of considerable interest
- 1.Kroeze WK, Sheffler DJ, Roth BL. G-protein-coupled receptors at a glance. J Cell Sci. 2003;116(24):4867–4869. doi: 10.1242/jcs.00902. [DOI] [PubMed] [Google Scholar]
- 2.Gruber CW, Muttenthaler M, Freissmuth M. Ligand-based peptide design and combinatorial peptide libraries to target G protein-coupled receptors. Curr Pharm Des. 2010;16(28):3071–3088. doi: 10.2174/138161210793292474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosenbaum DM, Rasmussen SGF, Kobilka BK. The structure and function of G-protein-coupled receptors. Nature. 2009;459(7245):356–363. doi: 10.1038/nature08144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fredriksson R, Lagerstrom MC, Lundin LG, Schioth HB. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol. 2003;63(6):1256–1272. doi: 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- 5.Bjarnadottir TK, Gloriam DE, Hellstrand SH, Kristiansson H, Fredriksson R, Schioth HB. Comprehensive repertoire and phylogenetic analysis of the G protein-coupled receptors in human and mouse. Genomics. 2006;88(3):263–273. doi: 10.1016/j.ygeno.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 6.Marinissen MJ, Gutkind JS. G-protein-coupled receptors and signaling networks: emerging paradigms. Trends Pharmacol Sci. 2001;22(7):368–376. doi: 10.1016/s0165-6147(00)01678-3. [DOI] [PubMed] [Google Scholar]
- 7•.Katritch V, Cherezov V, Stevens RC. Structure-function of the G protein-coupled receptor superfamily. Annu Rev Pharmacol Toxicol. 2013;53:531–556. doi: 10.1146/annurev-pharmtox-032112-135923. Useful review on G protein-coupled receptor (GPCR) structure and function. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frank J, Zhu J, Penczek P, et al. A model of protein-synthesis based on cryoelectron microscopy of the E-coli ribosome. Nature. 1995;376(6539):441–444. doi: 10.1038/376441a0. [DOI] [PubMed] [Google Scholar]
- 9.Palczewski K, Kumasaka T, Hori T, et al. Crystal structure of rhodopsin: a G protein-coupled receptor. Science. 2000;289(5480):739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 10.Clark RB. Profile of Brian K Kobilka and Robert J Lefkowitz, 2012 Nobel laureates in chemistry. Proc Natl Acad Sci USA. 2013;110(14):5274–5275. doi: 10.1073/pnas.1221820110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rasmussen SG, Devree BT, Zou Y, et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature. 2011;477(7366):549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nat Rev Mol Cell Biol. 2002;3(9):639–650. doi: 10.1038/nrm908. [DOI] [PubMed] [Google Scholar]
- 13.Heng BC, Aubel D, Fussenegger M. An overview of the diverse roles of G-protein coupled receptors (GPCRs) in the pathophysiology of various human diseases. Biotechnol Adv. 2013;31(8):1676–1694. doi: 10.1016/j.biotechadv.2013.08.017. [DOI] [PubMed] [Google Scholar]
- 14.George SR, O’Dowd BF, Lee SR. G-protein-coupled receptor oligomerization and its potential for drug discovery. Nat Rev Drug Discov. 2002;1(10):808–820. doi: 10.1038/nrd913. [DOI] [PubMed] [Google Scholar]
- 15.Tyndall JDA, Pfeiffer B, Abbenante G, Fairlie DP. Over one hundred peptide-activated G protein-coupled receptors recognize ligands with turn structure. Chem Rev. 2005;105(3):793–826. doi: 10.1021/cr040689g. [DOI] [PubMed] [Google Scholar]
- 16.Jimonet P, Jager R. Strategies for designing GPCR-focused libraries and screening sets. Curr Opin Drug Discov Dev. 2004;7(3):325–333. [PubMed] [Google Scholar]
- 17.Garland SL. Are GPCRs still a source of new targets? J Biomol Screen. 2013;18(9):947–966. doi: 10.1177/1087057113498418. [DOI] [PubMed] [Google Scholar]
- 18.Thompson CA. Macitentan approved by FDA to delay progression of PAH. Am J Health Syst Pharm. 2013;70(23):2054–2054. doi: 10.2146/news130076. [DOI] [PubMed] [Google Scholar]
- 19.Warne T, Serrano-Vega MJ, Baker JG, et al. Structure of a beta(1)-adrenergic G-protein-coupled receptor. Nature. 2008;454(7203):486–492. doi: 10.1038/nature07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rasmussen SGF, Choi HJ, Rosenbaum DM, et al. Crystal structure of the human β(2) adrenergic G-protein-coupled receptor. Nature. 2007;450(7168):383–388. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- 21.Jaakola VP, Griffith MT, Hanson MA, et al. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322(5905):1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu BL, Chien EYT, Mol CD, et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science. 2010;330(6007):1066–1071. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chien EYT, Liu W, Zhao QA, et al. Structure of the human dopamine D3 Receptor in complex with a D2/D3 selective antagonist. Science. 2010;330(6007):1091–1095. doi: 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lappano R, Maggiolini M. G protein-coupled receptors: Novel targets for drug discovery in cancer. Nat Rev Drug Discov. 2011;10(1):47–60. doi: 10.1038/nrd3320. [DOI] [PubMed] [Google Scholar]
- 25.Lazo JS, Wipf P. Combinatorial chemistry and contemporary pharmacology. J Pharmacol Exp Ther. 2000;293(3):705–709. doi: 10.1002/chin.200043259. [DOI] [PubMed] [Google Scholar]
- 26.Kodadek T. The rise, fall and reinvention of combinatorial chemistry. Chem Commun. 2011;47(35):9757–9763. doi: 10.1039/c1cc12102b. [DOI] [PubMed] [Google Scholar]
- 27.Lam KS, Salmon SE, Hersh EM, Hruby VJ, Kazmierski WM, Knapp RJ. A new type of synthetic peptide library for identifying ligand-binding activity. Nature. 1991;354(6348):82–84. doi: 10.1038/354082a0. [DOI] [PubMed] [Google Scholar]
- 28.Scott JK, Smith GP. Searching for peptide ligands with an epitope library. Science. 1990;249(4967):386–390. doi: 10.1126/science.1696028. [DOI] [PubMed] [Google Scholar]
- 29••.Lam KS, Lebl M, Krchnak V. The ‘one-bead-onecompound’ combinatorial library method. Chem Rev. 1997;97(2):411–448. doi: 10.1021/cr9600114. Review on one-bead-one-compound (OBOC) combinatorial library method. [DOI] [PubMed] [Google Scholar]
- 30.Chen CL, Strop P, Lebl M, Lam KS. [12] one bead-one compound combinatorial peptide library: different types of screening. Methods Enzymol. 1996;267:211–219. doi: 10.1016/s0076-6879(96)67014-1. [DOI] [PubMed] [Google Scholar]
- 31.Bartak Z, Bolf J, Kalousek J, et al. Design and construction of the automatic peptide library synthesizer. Methods. 1994;6(4):432–437. [Google Scholar]
- 32.Smith GP. Filamentous fusion phage – novel expression vectors that display cloned antigens on the virion surface. Science. 1985;228(4705):1315–1317. doi: 10.1126/science.4001944. [DOI] [PubMed] [Google Scholar]
- 33.Molek P, Strukelj B, Bratkovic T. Peptide phage display as a tool for drug discovery: targeting membrane receptors. Molecules. 2011;16(1):857–887. doi: 10.3390/molecules16010857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Simon RJ, Kania RS, Zuckermann RN, et al. Peptoids – a modular approach to drug discovery. Proc Natl Acad Sci USA. 1992;89(20):9367–9371. doi: 10.1073/pnas.89.20.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zuckermann RN. Peptoid origins. Biopolymers. 2011;96(5):545–555. doi: 10.1002/bip.21573. [DOI] [PubMed] [Google Scholar]
- 36•.Zuckermann RN, Kerr JM, Kent SBH, Moos WH. Efficient method for the preparation of peptoids [oligo(N-substituted glycines)] by submonomer solid-phase synthesis. J Am Chem Soc. 1992;114(26):10646–10647. First report of the submonomer solid-phase synthesis of peptoids. [Google Scholar]
- 37.Miller SM, Simon RJ, Ng S, Zuckermann RN, Kerr JM, Moos WH. Proteolytic studies of homologous peptide and N-substituted glycine peptoid oligomers. Bioorg Med Chem Lett. 1994;4(22):2657–2662. [Google Scholar]
- 38.Kwon Y-U, Kodadek T. Quantitative evaluation of the relative cell permeability of peptoids and peptides. J Am Chem Soc. 2007;129(6):1508–1509. doi: 10.1021/ja0668623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thompson LA, Ellman JA. Synthesis and applications of small molecule libraries. Chem Rev. 1996;96(1):555–600. doi: 10.1021/cr9402081. [DOI] [PubMed] [Google Scholar]
- 40.Liu DR, Schultz PG. Generating new molecular function: a lesson from nature. Angew Chem Int Ed. 1999;38(1–2):36–54. [Google Scholar]
- 41.Ellman JA. Design, synthesis, and evaluation of small-molecule libraries. Acc Chem Res. 1996;29(3):132–143. [Google Scholar]
- 42.Figliozzi GM, Goldsmith R, Ng SC, Banville SC, Zuckermann RN. [25] synthesis of N-substituted glycine peptoid libraries. Methods Enzymol. 1996;267:437–447. doi: 10.1016/s0076-6879(96)67027-x. [DOI] [PubMed] [Google Scholar]
- 43.Reddy MM, Bachhawat-Sikder K, Kodadek T. Transformation of low-affinity lead compounds into high-affinity protein capture agents. Chem Biol. 2004;11(8):1127–1137. doi: 10.1016/j.chembiol.2004.05.013. [DOI] [PubMed] [Google Scholar]
- 44.Lam KS, Lehman AL, Song A, et al. [17] synthesis and screening of “one-bead one-compound” combinatorial peptide libraries. Methods Enzymol. 2003;369:298–322. doi: 10.1016/S0076-6879(03)69017-8. [DOI] [PubMed] [Google Scholar]
- 45.Houghten RA, Pinilla C, Blondelle SE, Appel JR, Dooley CT, Cuervo JH. Generation and use of synthetic peptide combinatorial libraries for basic research and drug discovery. Nature. 1991;354(6348):84–86. doi: 10.1038/354084a0. [DOI] [PubMed] [Google Scholar]
- 46.Tan DS. Diversity-oriented synthesis: exploring the intersections between chemistry and biology. Nat Chem Biol. 2005;1(2):74–84. doi: 10.1038/nchembio0705-74. [DOI] [PubMed] [Google Scholar]
- 47••.Zuckermann RN, Martin EJ, Spellmeyer DC, et al. Discovery of nanomolar ligands for 7-transmembrane G-protein-coupled receptors from a diverse N-(substituted)glycine peptoid library. J Med Chem. 1994;37(17):2678–2685. doi: 10.1021/jm00043a007. First report of the discovery of GPCR ligands from peptoid library. [DOI] [PubMed] [Google Scholar]
- 48.Jayawickreme CK, Quillan JM, Graminski GF, Lerner MR. Discovery and structure-function analysis of alpha-melanocyte-stimulating hormone antagonists. J Biol Chem. 1994;269(47):29846–29854. [PubMed] [Google Scholar]
- 49.Appell KC, Chung TDY, Ohlmeyer MJH, Sigal NH, Baldwin JJ, Chelsky D. Biological screening of a large combinatorial library. J Biomol Screen. 1996;1(1):27–31. [Google Scholar]
- 50.Heizmann G, Hildebrand P, Tanner H, et al. A combinatorial peptoid library for the identification of novel MSH and GRP/bombesin receptor ligands. J Recept Signal Transduct Res. 1999;19(1–4):449–466. doi: 10.3109/10799899909036664. [DOI] [PubMed] [Google Scholar]
- 51.Evans B, Pipe A, Clark L, Banks M. Identification of a potent and selective oxytocin antagonist, from screening a fully encoded differential release combinatorial chemical library. Bioorg Med Chem Lett. 2001;11(10):1297–1300. doi: 10.1016/s0960-894x(01)00201-3. [DOI] [PubMed] [Google Scholar]
- 52.Kruijtzer JaW, Nijenhuis WaJ, Wanders N, Gispen WH, Liskamp RMJ, Adan RaH. Peptoid-peptide hybrids as potent novel melanocortin receptor ligands. J Med Chem. 2005;48(13):4224–4230. doi: 10.1021/jm0490033. [DOI] [PubMed] [Google Scholar]
- 53.Iuga AO, Reddy VB, Lerner EA. Identification of novel hexapeptide agonists at the xenopus laevis melanophore melanocortin receptor. Peptides. 2005;26(11):2124–2128. doi: 10.1016/j.peptides.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 54.Gallop MA, Barrett RW, Dower WJ, Fodor SPA, Gordon EM. Applications of combinatorial technologies to drug discovery. 1 Background and peptide combinatorial libraries. J Med Chem. 1994;37(9):1233–1251. doi: 10.1021/jm00035a001. [DOI] [PubMed] [Google Scholar]
- 55.Gordon EM, Barrett RW, Dower WJ, Fodor SPA, Gallop MA. Applications of combinatorial technologies to drug discovery. 2 Combinatorial organic-synthesis, library screening strategies, and future-directions. J Med Chem. 1994;37(10):1385–1401. doi: 10.1021/jm00036a001. [DOI] [PubMed] [Google Scholar]
- 56.Lam KS, Zhao ZG, Wade S, Krchnak V, Lebl M. Identification of small peptides that interact specifically with a small organic dye. Drug Dev Res. 1994;33(2):157–160. [Google Scholar]
- 57.Hornik V, Hadas E. Self-encoded, highly condensed solid phase-supported peptide library for identification of ligand-specific peptides. React Funct Polym. 1994;22(3):213–220. [Google Scholar]
- 58.Lam KS, Wade S, Abdullatif F, Lebl M. Application of a dual-color detection scheme in the screening of a random combinatorial peptide library. J Immunol Methods. 1995;180(2):219–223. doi: 10.1016/0022-1759(94)00316-o. [DOI] [PubMed] [Google Scholar]
- 59.Turck CW. Radioactive screening of synthetic peptide libraries. Methods. 1994;6(4):396–400. [Google Scholar]
- 60.Crivat G, Taraska JW. Imaging proteins inside cells with fluorescent tags. Trends Biotechnol. 2012;30(1):8–16. doi: 10.1016/j.tibtech.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen JK, Lane WS, Brauer AW, Tanaka A, Schreiber SL. Biased combinatorial libraries – novel ligands for the SH3 domain of phosphatidylinositol 3-kinase. J Am Chem Soc. 1993;115(26):12591–12592. [Google Scholar]
- 62.Doran TM, Kodadek T. A liquid array platform for the multiplexed analysis of synthetic molecule–protein interactions. ACS Chem Biol. 2013;9(2):339–346. doi: 10.1021/cb400806r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Needels MC, Jones DG, Tate EH, et al. Generation and screening of an oligonucleotide-encoded synthetic peptide library. Proc Natl Acad Sci USA. 1993;90(22):10700–10704. doi: 10.1073/pnas.90.22.10700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lebl M, Krchnak V, Sepetov NF, et al. One-bead one-structure combinatorial libraries. Biopolymers. 1995;37(3):177–198. doi: 10.1002/bip.360370303. [DOI] [PubMed] [Google Scholar]
- 65.Quarrell R, Claridge TDW, Weaver GW, Lowe G. Structure and properties of TentaGel resin beads: implications for combinatorial library chemistry. Mol Diversity. 1996;1(4):223–232. doi: 10.1007/BF01715526. [DOI] [PubMed] [Google Scholar]
- 66.Alluri PG, Reddy MM, Bachhawat-Sikder K, Olivos HJ, Kodadek T. Isolation of protein ligands from large peptoid libraries. J Am Chem Soc. 2003;125(46):13995–14004. doi: 10.1021/ja036417x. [DOI] [PubMed] [Google Scholar]
- 67••.Olivos HI, Bachhawat-Sikder K, Kodadek T. Quantum dots as a visual aid for screening bead-bound combinatorial libraries. Chembiochem. 2003;4(11):1242–1245. doi: 10.1002/cbic.200300712. First indication of using quantum dots for on bead screening of combinatorial libraries. [DOI] [PubMed] [Google Scholar]
- 68.Jin S, Hu YX, Gu ZJ, Liu L, Wu HC. Application of quantum dots in biological imaging. J Nanomater. 2011;2011:1–13. [Google Scholar]
- 69.Michalet X, Pinaud FF, Bentolila LA, et al. Quantum dots for live cells, in vivo imaging, and diagnostics. Science. 2005;307(5709):538–544. doi: 10.1126/science.1104274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Medintz IL, Uyeda HT, Goldman ER, Mattoussi H. Quantum dot bioconjugates for imaging, labelling and sensing. Nat Mater. 2005;4(6):435–446. doi: 10.1038/nmat1390. [DOI] [PubMed] [Google Scholar]
- 71.Watson A, Wu XY, Bruchez M. Lighting up cells with quantum dots. Biotechniques. 2003;34(2):296–303. doi: 10.2144/03342bi01. [DOI] [PubMed] [Google Scholar]
- 72.Lim HS, Archer CT, Kodadek T. Identification of a peptoid inhibitor of the proteasome 19S regulatory particle. J Am Chem Soc. 2007;129(25):7750–7751. doi: 10.1021/ja072027p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kodadek T, Bachhawat-Sikder K. Optimized protocols for the isolation of specific protein-binding peptides or peptoids from combinatorial libraries displayed on beads. Mol Biosyst. 2006;2(1):25–35. doi: 10.1039/b514349g. [DOI] [PubMed] [Google Scholar]
- 74.Udugamasooriya DG, Dineen SP, Brekken RA, Kodadek T. A peptoid ‘antibody surrogate’ that antagonizes VEGF receptor 2 activity. J Am Chem Soc. 2008;130(17):5744–5752. doi: 10.1021/ja711193x. [DOI] [PubMed] [Google Scholar]
- 75••.Udugamasooriya DG, Kodadek T. On-bead two-color (OBTC) cell screen for direct identification of highly selective cell surface receptor ligands. Curr Protoc Chem Biol. 2012;4:35–48. doi: 10.1002/9780470559277.ch110199. Detail protocols concerning on-bead two-color cell based screening of OBOC libraries. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kodadek T. Rethinking screening. Nat Chem Biol. 2010;6(3):162–165. doi: 10.1038/nchembio.303. [DOI] [PubMed] [Google Scholar]
- 77.Osmond RI, Crouch MF, Dupriez VJ. An emerging role for kinase screening in GPCR drug discovery. Curr Opin Mol Ther. 2010;12(3):305–315. [PubMed] [Google Scholar]
- 78.Black CB, Duensing TD, Trinkle LS, Dunlay RT. Cell-based screening using high-throughput flow cytometry. Assay Drug Dev Technol. 2011;9(1):13–20. doi: 10.1089/adt.2010.0308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Maynard JA, Lindquist NC, Sutherland JN, et al. Surface plasmon resonance for high-throughput ligand screening of membrane-bound proteins. Biotechnol J. 2009;4(11):1542–1558. doi: 10.1002/biot.200900195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80••.Qi X, Astle J, Kodadek T. Rapid identification of orexin receptor binding ligands using cell-based screening accelerated with magnetic beads. Mol Biosyst. 2010;6(1):102–107. doi: 10.1039/b915611a. Example showing rapid identifying of GPCR ligands from OBOC library using quantum dots labeled screening method. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Han MY, Gao XH, Su JZ, Nie S. Quantum-dot-tagged microbeads for multiplexed optical coding of biomolecules. Nat Biotechnol. 2001;19(7):631–635. doi: 10.1038/90228. [DOI] [PubMed] [Google Scholar]
- 82.Smith AM, Dave S, Nie SM, True L, Gao XH. Multicolor quantum dots for molecular diagnostics of cancer. Expert Rev Mol Diagn. 2006;6(2):231–244. doi: 10.1586/14737159.6.2.231. [DOI] [PubMed] [Google Scholar]
- 83.Garske AL, Denu JM. SIRT1 top 40 hits: Use of one-bead, one-compound acetyl-peptide libraries and quantum dots to probe deacetylase specificity. Biochemistry. 2006;45(1):94–101. doi: 10.1021/bi052015l. [DOI] [PMC free article] [PubMed] [Google Scholar]