

Abstract
Proteins are synthesized by the ribosome and generally must fold to become functionally active. Although it is commonly assumed that the ribosome affects the folding process, this idea has been extremely difficult to demonstrate. We have developed an experimental system to investigate the folding of single ribosome-bound stalled nascent polypeptides with optical tweezers. In T4 lysozyme, synthesized in a reconstituted in vitro translation system, the ribosome slows the formation of stable tertiary interactions and the attainment of the native state relative to the free protein. Incomplete T4 lysozyme polypeptides misfold and aggregate when free in solution, but they remain folding-competent near the ribosomal surface. Altogether, our results suggest that the ribosome not only decodes the genetic information and synthesizes polypeptides, but also promotes efficient de novo attainment of the native state.
Proteins can spontaneously fold into their native structures under appropriate conditions (1). In vitro, some small proteins and single domains attain their native structures within microseconds (2), whereas topologically complex and larger proteins may require many seconds to fold (3) and often populate folding intermediates along the way (4, 5). In vivo, however, folding is not necessarily limited to full-length proteins or domains. Proteins can begin to fold before they are fully synthesized and while still bound to the ribosome (6–12). Moreover, during protein synthesis on the ribosome (13), elongation rates are regulated by factors including tRNA abundance (14), codon order (15), and mRNA secondary structure (16). Decreasing these rates locally (17, 18) or globally (19) can affect the folding efficiency of newly synthesized proteins. The complete synthesis of even small proteins (100 amino acids or less) requires at least several seconds at a maximum rate of ~20 amino acids per second in Escherichia coli (20), giving nascent-chain segments sufficient time to conformationally equilibrate in the environment of the ribosome, perhaps adopting structures that are distinctly different from the native protein fold (7, 21). However, the observation of folding transitions in ribosome-bound nascent proteins has not been possible, and a detailed analysis has been limited to computational approaches (22, 23).
We have developed an experimental system to directly probe the folding of single ribosome-bound nascent chains (24, 25) by subjecting them to force using optical tweezers (Fig. 1 and figs. S1 to S3). The force is applied between the nascent chain and the large ribosomal subunit. Because force acts locally (26, 27), we can selectively perturb the stability of ribosome-bound nascent polypeptides without disrupting the structural integrity of the ribosome. We studied a cysteine-free version of T4 lysozyme (28), a monomeric cytosolic protein composed of two globular regions, or subdomains (Fig. 1, C and D). T4 lysozyme folding has been studied in ensemble (29–31) and single-molecule (27) experiments. The native fold requires interactions between the N- and C-terminal sequences whose synthesis is separated in time during vectorial translation by the ribosome.
Fig. 1.
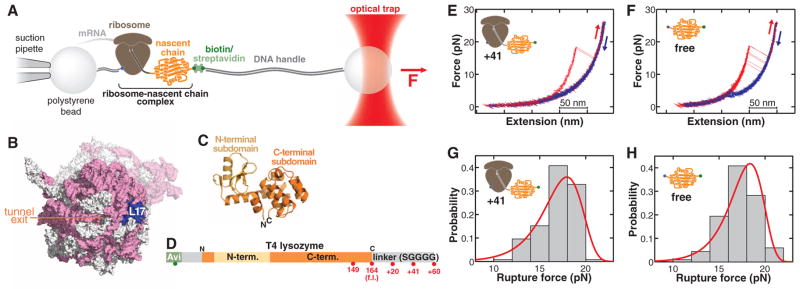
(A) Schematic of the molecular assembly for optical-tweezers experiments. Force can be applied to the nascent polypeptide by moving the optical trap relative to the pipette. (B) Surface representation of the ribosome [Protein Data Bank identification numbers (PDB IDs): 2aw4 and 2avy] showing the opening of the ribosomal exit tunnel in the large subunit and the location of ribosomal protein L17, serving as the attachment site in the optical-tweezers experiments. Ribosomal RNA, pink; ribosomal proteins, white; L17, blue. The small subunit is shown in a semitransparent rendering. (C) Cartoon diagram of T4 lysozyme (PDB ID: 4lzm). The N-terminal subdomain (light orange) is composed of residues 13 to 59; the C-terminal subdomain (dark orange) comprises residues 1 to 12 and 60 to 164. (D) Primary structure diagram of the protein construct translated for optical-tweezers experiments (32). Red spheres indicate stalling positions along the sequence. f.l., full length. (E and F) Force-extension traces of T4 lysozyme unfolding (red) and refolding (blue) on the ribosome (41–amino acid linker) (E) and free in solution (F). The protein unfolds in one cooperative transition near 17 pN. (G and H) Rupture-force histograms (gray bars) for unfolding of the ribosome-bound (G) and free (H) protein. Red lines, rupture force distribution reconstructed from the force-dependent lifetimes (fig. S5).
Using a reconstituted in vitro translation system supplemented with E. coli ribosomes (32), we first translated the protein with an unstructured C-terminal extension of 41 amino acids such that the entire T4 lysozyme sequence emerges from the narrow ribosomal exit tunnel (33). This experimental design allows us to study the folding dynamics of the full-length protein on the ribosome. When we stretched the molecule by continuously increasing the tension applied across the nascent chain (“force ramp”), we observed single rips in the resulting force-extension traces, representing cooperative unfolding events (Fig. 1E). Puromycin-release experiments confirmed that these signals originated from ribosome-bound nascent proteins (fig. S4).
The mechanical unfolding pathways of free and ribosome-bound full-length T4 lysozyme are very similar: The latter unfolds at a mean force (FUnf) of 17.0 ± 2.0 pN (N = 125 unfolding events) at a pulling speed of 100 nm/s (Fig. 1G). Using the wormlike chain (WLC) model (34), we calculated a contour length increase upon unfolding (ΔLC) of 59.9 ± 2.1 nm, consistent with the value expected for full-length T4 lysozyme (164 amino acids × 0.36 nm per amino acid − 0.9 nm corresponding to the folded end-to-end distance = 58.1 nm). Experiments with the free protein in the absence of the ribosome (Fig. 1, F and H) revealed similar unfolding characteristics (FUnf = 17.2 ± 1.8 pN, ΔLC = 60.1 ± 0.9 nm, N = 453), confirming that the entire protein is able to emerge from the ribosomal tunnel by means of the 41–amino acid linker (33). Analysis of the unfolding force distributions (35) of free and ribosome-bound polypeptides (Fig. 1, G and H) also reveals similar distances to the transition state (Δ‡xfree = 2.3 ± 0.5 nm, Δx‡ribosome-bound = 2.0 ± 0.2 nm) and native-state lifetimes (fig. S5). Thus, the ribosome-bound protein folds to the same native structure as the free protein and unfolds through the same pathway in the pulling experiments.
Whereas the unfolding transitions of free and ribosome-bound T4 lysozyme are indistinguishable in our experiments, the refolding exhibits marked differences: During repeated cycles of pulling and relaxation, the free protein virtually always refolds, but the ribosome-bound protein refolds only 28% of the time and lacks a well-defined refolding transition in the force-ramp measurements (fig. S6). We carried out “force-clamp” experiments (fig. S7) to better resolve the folding transition. From measurements of the refolding time, τrefold (Fig. 2A), we estimated apparent refolding rates at 3.6 pN (Fig. 2B). The free protein folds rapidly with an apparent rate (kapp) of 5.4 s−1 [95% confidence interval (CI): 4.4 s−1, 6.5 s−1]. Remarkably, the folding rate of the ribosome-bound protein (with a 41–amino acid C-terminal linker) is 0.012 s−1 (95% CI: 0.004 s−1, 0.024 s−1), more than two orders of magnitude slower than the free protein.
Fig. 2.

(A) Example extension-versus-time trace of T4 lysozyme refolding at constant force (5 pN). Before transitioning to the native state (N) at ~0.5 s, the protein visits a folding intermediate (I). Enlargement of the first second of the trace (red dashed box) reveals that the intermediate is visited immediately before folding to the native state (arrowhead), demonstrating that the intermediate is on-pathway. U, unfolded state. (B) Apparent refolding rates for ribosome-bound T4 lysozyme with 41–amino acid (+41) and 60–amino acid (+60) linkers and for the free protein (free). Error bars: 95% CIs. (C) Example trace of ribosome-bound protein (60–amino acid linker) re-folding at 5 pN. (D) Apparent refolding rates of free protein (blue circles) and +60 linkers (red triangles) at 150 mM (filled symbols) and 500 mM (open symbols) KCl. Increasing the salt concentration mitigates the effect of the ribosome on the refolding rate. (E) Population of the folding intermediate before refolding. The unfolded and intermediate states are equally populated (P = 0.5) at a force of ~3.6 pN, both in the free and the ribosome-bound protein.
The 41–amino acid linker is long enough to allow the entire protein to emerge from the exit tunnel but restricts the folding environment to the immediate vicinity of the ribosomal polypeptide exit site. Increasing the linker length by 19 amino acids provides ~2.1 nm at 3.6 pN (calculated based on a WLC model) of additional separation from the ribosomal surface and increases the folding rate: This construct, harboring a 60–amino acid linker, folds with kapp = 0.24 s−1 (95% CI: 0.21 s−1, 0.28 s−1) (Fig. 2B), significantly faster than the rate observed with the shorter linker, but still more slowly than the free protein (Fig. 2B, also compare Fig. 2C with 2A). We used this construct to characterize the folding of the ribosome-bound protein in more detail.
Given the high negative charge density of the ribosomal RNA (rRNA) moiety, the observed deceleration in folding is likely mediated by electrostatic interactions of the ribosomal surface with charged residues in the nascent chain (36). Increasing the potassium chloride concentration from 150 to 500 mM, which results in more effective screening of electrostatic interactions (32), increased the folding rate of the ribosome-bound protein, whereas the folding rates of the free protein were less sensitive to salt (Fig. 2D). Thus, the effect of the ribosome on the folding of the nascent polypeptide is mediated, at least in part, by electrostatic interactions.
Close inspection of the force clamp traces revealed that, before folding to the native state (N), the protein transiently and reversibly samples an intermediate (I) from the unfolded state (U) (Fig. 2, A and C), exhibiting bistability, or “hopping.” In almost all traces examined, folding to the native state then proceeds from the intermediate state (Fig. 2A, arrowhead), indicating that the latter is an on-pathway state and, therefore, obligatory for productive attainment of the native state. The I-to-N transition is practically irreversible at the refolding force; thus, the folding pathway can be written as
From the extension changes in the force-clamp traces, we estimate that between 96 and 108 residues participate in the formation of the folding intermediate. These dimensions are consistent with a folding intermediate that is formed largely by the C-terminal T4 lysozyme subdomain (32) and might be related to a previously described “hidden intermediate” (37).
The probability of occupying the state I (relative to U) during hopping (fig. S8) is similar in the presence and absence of the ribosome (Fig. 2E). From the force at which U and I are equally populated (F1/2 ≈ 3.6 pN) and the observed change in extension between U and I at that force (ΔxU-I ≈ 10 nm), we estimate that I is stabilized by a Gibbs free energy of unfolding [ΔG0unfolding (intermediate)] of = 3.0 kcal/mol (5.1kBT, where kB is the Boltzmann constant and T is temperature) relative to U (38) for both the free and the ribosome-bound protein. This value is small compared with the stability of the folded protein [ΔG0unfolding (native) = 14.1 kcal/mol (30)], indicating that the intermediate is lacking many of the interactions that stabilize the natively folded protein. It is also smaller than the reported stability of the “hidden” intermediate, perhaps because the N-terminal A helix is not part of the intermediate observed in our experimental geometry (32).
To explore the ribosomal effect within the folding pathway, we used a Bayesian Hidden Markov model (BHMM) approach to conduct a kinetic analysis of the force-clamp data (Fig. 3A) (32). The values for the rates of the U-to-I and I-to-U transitions (kU-I and kI-U, respectively) are essentially the same for the free and the ribosome-bound protein (60–amino acid linker). In contrast, kI-N of the ribosome-bound protein is smaller by at least one order of magnitude compared with that of the free protein (Fig. 3A). The BHMM analysis also yields estimates of the extensions of each species (fig. S9). Interestingly, the unfolded protein, both free and ribosome-bound, is more compact than predicted from a WLC model (34) at forces below 4 pN (Fig. 3B). This deviation from the model is consistent with a compaction of the unfolded protein, perhaps reflecting transient local secondary structure formation at those low forces before cooperative folding transitions. For the free protein, the unfolded-state extensions gradually increase to the values predicted by the WLC model when the force is raised to 5 pN. Notably, the unfolded state remains more compact in the ribosome-bound protein (over the limited force range accessible in these measurements; Fig. 3B). Thus, the ribosomal interactions appear to have a dual effect: deceleration of native tertiary-structure formation and stabilization of a compacted state before folding (Fig. 3C), presumably mediated at least in part by electrostatic interactions.
Fig. 3.

(A) Kinetic rates along the T4 lysozyme folding pathway. The rate of the final, irreversible step (kI-N) along the folding pathway is significantly slower for the ribosome-bound protein (60–amino acid linker). (B) Distance changes upon unfolding (N → U, open symbols) and refolding (U → N, filled symbols) at various forces. At low forces, the distance is shorter than expected from a WLC model (gray line), indicating a partial compaction of the polypeptide that does not resist forces above 4 pN. The compact structure is stabilized in the ribosome-bound protein. Error bars: 95% CIs. (C) Schematic folding energy landscapes of free and ribosome-bound T4 lysozyme. The height of the barrier from I to N is affected by the ribosome, resulting in a decrease in kI-N.
How might the ribosome contribute to efficient de novo protein folding? So far, we have described folding of the entire protein. To study folding before completion of synthesis, we translated the N-terminal 149 amino acids of T4 lysozyme, so that ~110 to 120 residues of the full T4 lysozyme sequence (164 residues) exited the ribosome (~70% of the full protein), whereas 30 to 40 amino acids remained within the exit tunnel. Upon stretching the ribosome-bound polypeptide, the force-extension curves did not reveal any cooperative folding or unfolding transitions (fig. S10). Even when we extended the T4 lysozyme sequence by a 20–amino acid linker (so that ~144 to 154 out of the 164 amino acids of T4 lysozyme are outside the tunnel), we did not detect folding (Fig. 4A), presumably because the C-terminal residues, which interact with the N-terminal A helix in the native structure, are still sequestered within the ribosomal exit tunnel.
Fig. 4.

(A) Force-extension curves of a T4 lysozyme (164 amino acids) with a 20–amino acid C-terminal extension bound to the ribosome (~150 amino acids outside the ribosomal tunnel), exhibiting no defined unfolding transitions. (B) A free protein fragment of 149 amino acids misfolds into a heterogeneous ensemble of structures that unfold over a wide rage of forces. (C) Comparison of unfolding events recorded for the free protein and the 149–amino acid fragment. The full-length protein unfolds within a narrow, stochastic range of forces and extension changes, whereas the unfolding transitions of the isolated fragment are highly heterogeneous.
Attempts to express soluble fragments in E. coli were unsuccessful (fig. S11). Thus, we used puromycin-modified DNA oligonucleotides (39) to release the in vitro translated 149–amino acid fragment from the ribosome (32). In contrast to the lack of transitions observed on the ribosome, the released fragment unfolded at a range of forces and contour length changes (Fig. 4B), indicating that it adopts a highly heterogeneous ensemble of structures, some of which exhibit considerable (mechanical) stability. Given the homogeneous unfolding of the complete protein, it is unlikely that all of these structures represent productive, on-pathway species. Rather, the diverse behavior of this fragment is probably due to misfolding, aggregation, entanglement of several polypeptides immobilized in close vicinity, or interactions of the misfolded protein with the bead surface, none of which are observed for the ribosome-bound nascent protein. Thus, the ribosome appears to prevent misfolding of the incomplete protein through a kinetic mechanism, effectively acting as a molecular chaperone for nascent polypeptides. This chaperone activity is probably mediated by the surface surrounding the polypeptide exit tunnel and is distinct from the previously described “protein folding activity of the ribosome” mediated by the 26S rRNA (40). Mechanisms that keep proteins in a folding-competent conformation may be particularly important if C-terminal residues, which are synthesized last, are required for productive folding, as in T4 lysozyme and other proteins (21, 41–43).
In the cell, molecular chaperones interact with nascent polypeptides during their synthesis (44). The in vitro experiments described here suggest that the ribosome contributes to efficient de novo folding in several ways. Polypeptide compaction, an early event during protein folding (45), is promoted by the ribosome and may, in conjunction with the spatial arrangement within polysomes (46), limit aberrant interactions among nascent chains. Rather than acting as an inert “wall,” which would be expected to increase folding rates entropically (47, 48), the ribosome slows folding of T4 lysozyme, presumably by attracting positively charged residues and repelling negatively charged ones within the same nascent polypeptide chain. Thus, it can bias the conformational search of the protein and its folding rate. This mechanism may complement the mode of action of other molecular chaperones that bind their substrates through hydrophobic interactions and may be particularly pronounced for T4 lysozyme and other basic proteins that represent a large group in most proteomes (49). However, such a mechanism should be operative even for proteins harboring a net negative charge and may apply at least to cytosolic proteins, which are held in close proximity to the ribosomal surface during synthesis. Our findings may represent a paradigm for how the ribosome can, in principle, affect nascent-chain folding. The system is easily adaptable for investigating proteins other than T4 lysozyme and should be amenable to observing nascent-chain elongation in real time. These future experiments will shed more light onto how protein folding is tuned to synthesis.
Supplementary Material
Acknowledgments
We thank S. Marqusee for the T4 lysozyme source plasmid and for helpful discussions, O. Dudko for help with the analysis of force-ramp data, and J. R. Moffitt and members of the Bustamante and Tinoco laboratories for helpful discussions on the manuscript. C.M.K. acknowledges support from the NIH K99 Award 5K99GM086516; C.M.K. and J.D.C. acknowledge funding from the QB3 Institute, Berkeley (Distinguished Postdoctoral Fellowship); D.H.G. acknowledges the NSF’s Graduate Research Fellowship; I.T. acknowledges support from NIH grant 5R01GM10840 and the Human Frontiers of Science Program; and C.B. acknowledges support from NIH grant 5R01GM32543.
Footnotes
References and Notes
- 1.Anfinsen CB. Science. 1973;181:223. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 2.Kubelka J, Chiu TK, Davies DR, Eaton WA, Hofrichter J. J Mol Biol. 2006;359:546. doi: 10.1016/j.jmb.2006.03.034. [DOI] [PubMed] [Google Scholar]
- 3.Plaxco KW, Simons KT, Baker D. J Mol Biol. 1998;277:985. doi: 10.1006/jmbi.1998.1645. [DOI] [PubMed] [Google Scholar]
- 4.Brockwell DJ, Radford SE. Curr Opin Struct Biol. 2007;17:30. doi: 10.1016/j.sbi.2007.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Udgaonkar JB. Annu Rev Biophys. 2008;37:489. doi: 10.1146/annurev.biophys.37.032807.125920. [DOI] [PubMed] [Google Scholar]
- 6.Ellis JP, Bakke CK, Kirchdoerfer RN, Jungbauer LM, Cavagnero S. ACS Chem Biol. 2008;3:555. doi: 10.1021/cb800059u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Evans MS, Sander IM, Clark PL. J Mol Biol. 2008;383:683. doi: 10.1016/j.jmb.2008.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hsu ST, et al. Proc Natl Acad Sci USA. 2007;104:16516. doi: 10.1073/pnas.0704664104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khushoo A, Yang Z, Johnson AE, Skach WR. Mol Cell. 2011;41:682. doi: 10.1016/j.molcel.2011.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu J, Deutsch C. Nat Struct Mol Biol. 2005;12:1123. doi: 10.1038/nsmb1021. [DOI] [PubMed] [Google Scholar]
- 11.Woolhead CA, McCormick PJ, Johnson AE. Cell. 2004;116:725. doi: 10.1016/s0092-8674(04)00169-2. [DOI] [PubMed] [Google Scholar]
- 12.Bhushan S, et al. Nat Struct Mol Biol. 2010;17:313. doi: 10.1038/nsmb.1756. [DOI] [PubMed] [Google Scholar]
- 13.Green R, Noller HF. Annu Rev Biochem. 1997;66:679. doi: 10.1146/annurev.biochem.66.1.679. [DOI] [PubMed] [Google Scholar]
- 14.Varenne S, Buc J, Lloubes R, Lazdunski C. J Mol Biol. 1984;180:549. doi: 10.1016/0022-2836(84)90027-5. [DOI] [PubMed] [Google Scholar]
- 15.Cannarozzi G, et al. Cell. 2010;141:355. doi: 10.1016/j.cell.2010.02.036. [DOI] [PubMed] [Google Scholar]
- 16.Qu X, et al. Nature. 2011;475:118. doi: 10.1038/nature10126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kimchi-Sarfaty C, et al. Science. 2007;315:525. doi: 10.1126/science.1135308. [DOI] [PubMed] [Google Scholar]
- 18.Zhang G, Hubalewska M, Ignatova Z. Nat Struct Mol Biol. 2009;16:274. doi: 10.1038/nsmb.1554. [DOI] [PubMed] [Google Scholar]
- 19.Siller E, DeZwaan DC, Anderson JF, Freeman BC, Barral JM. J Mol Biol. 2010;396:1310. doi: 10.1016/j.jmb.2009.12.042. [DOI] [PubMed] [Google Scholar]
- 20.Liang ST, Xu YC, Dennis P, Bremer H. J Bacteriol. 2000;182:3037. doi: 10.1128/jb.182.11.3037-3044.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chow CC, et al. Biochemistry. 2003;42:7090. doi: 10.1021/bi0273056. [DOI] [PubMed] [Google Scholar]
- 22.Elcock AH. PLoS Comput Biol. 2006;2:e98. doi: 10.1371/journal.pcbi.0020098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O’Brien EP, Hsu ST, Christodoulou J, Vendruscolo M, Dobson CM. J Am Chem Soc. 2010;132:16928. doi: 10.1021/ja106530y. [DOI] [PubMed] [Google Scholar]
- 24.Moffitt JR, Chemla YR, Smith SB, Bustamante C. Annu Rev Biochem. 2008;77:205. doi: 10.1146/annurev.biochem.77.043007.090225. [DOI] [PubMed] [Google Scholar]
- 25.Ashkin A, Dziedzic JM, Bjorkholm JE, Chu S. Opt Lett. 1986;11:288. doi: 10.1364/ol.11.000288. [DOI] [PubMed] [Google Scholar]
- 26.Dietz H, Rief M. Proc Natl Acad Sci USA. 2006;103:1244. doi: 10.1073/pnas.0509217103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shank EA, Cecconi C, Dill JW, Marqusee S, Bustamante C. Nature. 2010;465:637. doi: 10.1038/nature09021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matsumura M, Matthews BW. Science. 1989;243:792. doi: 10.1126/science.2916125. [DOI] [PubMed] [Google Scholar]
- 29.Cellitti J, et al. Protein Sci. 2007;16:842. doi: 10.1110/ps.062632807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Llinás M, Marqusee S. Protein Sci. 1998;7:96. doi: 10.1002/pro.5560070110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baase WA, Liu L, Tronrud DE, Matthews BW. Protein Sci. 2010;19:631. doi: 10.1002/pro.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Materials and methods and additional information are available as supporting material on Science Online.
- 33.Voss NR, Gerstein M, Steitz TA, Moore PB. J Mol Biol. 2006;360:893. doi: 10.1016/j.jmb.2006.05.023. [DOI] [PubMed] [Google Scholar]
- 34.Bustamante C, Marko JF, Siggia ED, Smith S. Science. 1994;265:1599. doi: 10.1126/science.8079175. [DOI] [PubMed] [Google Scholar]
- 35.Dudko OK, Hummer G, Szabo A. Proc Natl Acad Sci USA. 2008;105:15755. doi: 10.1073/pnas.0806085105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weinkam P, Pletneva EV, Gray HB, Winkler JR, Wolynes PG. Proc Natl Acad Sci USA. 2009;106:1796. doi: 10.1073/pnas.0813120106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cellitti J, Bernstein R, Marqusee S. Protein Sci. 2007;16:852. doi: 10.1110/ps.062632807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bustamante C, Chemla YR, Forde NR, Izhaky D. Annu Rev Biochem. 2004;73:705. doi: 10.1146/annurev.biochem.72.121801.161542. [DOI] [PubMed] [Google Scholar]
- 39.Liu R, Barrick JE, Szostak JW, Roberts RW. Methods Enzymol. 2000;318:268. doi: 10.1016/s0076-6879(00)18058-9. [DOI] [PubMed] [Google Scholar]
- 40.Voisset C, Saupe SJ, Blondel M. Biotechnol J. 2011;6:668. doi: 10.1002/biot.201100021. [DOI] [PubMed] [Google Scholar]
- 41.de Prat Gay G, et al. J Mol Biol. 1995;254:968. doi: 10.1006/jmbi.1995.0669. [DOI] [PubMed] [Google Scholar]
- 42.Neira JL, Fersht AR. J Mol Biol. 1999;285:1309. doi: 10.1006/jmbi.1998.2249. [DOI] [PubMed] [Google Scholar]
- 43.Taniuchi H. J Biol Chem. 1970;245:5459. [PubMed] [Google Scholar]
- 44.Hartl FU, Hayer-Hartl M. Science. 2002;295:1852. doi: 10.1126/science.1068408. [DOI] [PubMed] [Google Scholar]
- 45.Schuler B, Lipman EA, Eaton WA. Nature. 2002;419:743. doi: 10.1038/nature01060. [DOI] [PubMed] [Google Scholar]
- 46.Brandt F, et al. Cell. 2009;136:261. doi: 10.1016/j.cell.2008.11.016. [DOI] [PubMed] [Google Scholar]
- 47.Zhou HX, Dill KA. Biochemistry. 2001;40:11289. doi: 10.1021/bi0155504. [DOI] [PubMed] [Google Scholar]
- 48.Mittal J, Best RB. Proc Natl Acad Sci USA. 2008;105:20233. doi: 10.1073/pnas.0807742105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kiraga J, et al. BMC Genomics. 2007;8:163. doi: 10.1186/1471-2164-8-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.