

Abstract
One of the primary components of the East Indian sandalwood oil (EISO) is α-santalol, a molecule that has been investigated for its potential use as a chemopreventive agent in skin cancer. Although there is some evidence that α-santalol could be an effective chemopreventive agent, to date, purified EISO has not been extensively investigated even though it is widely used in cultures around the world for its health benefits as well as for its fragrance and as a cosmetic. In the current study, we show for the first time that EISO-treatment of HaCaT keratinocytes results in a blockade of cell cycle progression as well as a concentration-dependent inhibition of UV-induced AP-1 activity, two major cellular effects known to drive skin carcinogenesis. Unlike many chemopreventive agents, these effects were not mediated through an inhibition of signaling upstream of AP-1, as EISO treatment did not inhibit UV-induced Akt, or MAPK activity. Low concentrations of EISO were found to induce HaCaT cell death, although not through apoptosis as annexin V and PARP cleavage were not found to increase with EISO treatment. However, plasma membrane integrity was severely compromised in EISO-treated cells, which may have led to cleavage of LC3 and the induction of autophagy. These effects were more pronounced in cells stimulated to proliferate with bovine pituitary extract and EGF prior to receiving EISO. Together, these effects suggest that EISO may exert beneficial effects upon skin, reducing the likelihood of promotion of pre-cancerous cells to actinic keratosis (AK) and skin cancer.
Keywords: sandalwood oil, ultraviolet light, cell death, autophagy
Introduction
Ultraviolet light induces tumor promoting events in keratinocyte cells that, if allowed to proceed, will ultimately lead to the development of pre-cancerous conditions such as actinic keratosis (AK) and skin tumors, including papillomas and squamous cell carcinoma (SCC). In humans, these promotion events result from constant and unavoidable exposure to the sun over many decades, a fact which highlights the need to identify new chemopreventive strategies that can be applied over a lifetime. One strategy includes topical formulations of novel natural products that are non-toxic to normal cells, but which prevent the outgrowth of tumors through a mechanism that targets UV-induced promotion events.
Sandalwood oils are essential oils commonly used as fragrances for body oils and incense as well as in medicines and cosmetics. Sandalwood oils have many well-known health benefits due to their antiinflammatory and anti-septic properties, among others [1]. The principal commercial oils are steam distilled from the wood of two species of sandalwood trees: East Indian sandalwood (Santalum album) and West Australian sandalwood (Santalum spicatum). The compositions of the oils from the two types of trees are remarkably different with the fragrance and quality of East Indian sandalwood oil (EISO) considered superior to Western Australian sandalwood oil (WASO). One clear difference between the two oils is the content of α-santalol, which can vary from ~20% of the total oil content in WASO to more than 50% in EISO.
α-Santalol is one of the primary components of sandalwood oil and has been recently investigated for its chemopreventive properties. In fact, purified α-santalol, as well as sandalwood oil, has previously been demonstrated to prevent skin tumor development in mice [2-7]. Cell-based studies have found that α-santalol activates proapoptotic caspases, induces G2/M cell cycle arrest and blocks inflammation, which may be responsible for the prevention of tumor development after UV exposure [8, 9]. Although these studies identified some of the chemopreventive properties of α-santalol, little is known about the essential oil from which it was extracted and its potential value in preventing UV-induced skin cancer.
In the current study, we investigated the effects of EISO on cultured HaCaT keratinocytes. HaCaT cells were established from adult sun-damaged skin and are well characterized as representing an initiated human keratinocyte cell line expressing mutant dysfunctional p53 and a defective NF-kB signaling pathway, both of which are commonly found in UV-initiated keratinocytes in human skin [10-12]. We investigated the use of EISO on cells irradiated with UVB light. Although UVB light comprises only 1-10% of solar UV light, UVB acts as a complete carcinogen capable of activating signaling pathways in HaCaT cells known to stimulate cell proliferation and survival, including p38, JNK, ERK and PI3-K upstream of activator protein-1 (AP-1) transcription factor activation. UVB-induced AP-1 activity has been linked to cellular proliferation and survival, and in mouse skin AP-1 activation has been demonstrated to be a major cause of skin cancer [13]. In addition, we investigated the effects of EISO on cell cycle progression and cell membrane integrity. We determined that the effects of EISO are more pronounced in proliferating cells than in quiescent cells. Loss of cell membrane integrity and expression of a prominent marker of autophagy were both clearly more prominent in cells stimulated to grow than in serum-starved quiescent cells. These findings suggest that EISO may be valuable as a topical nondestructive chemopreventive agent through selective targeting of proliferative cancer and pre-cancerous cells.
Materials and Methods
Materials
EISO was provided by Santalis Pharmaceuticals, Inc. (San Antonio, TX). Annexin V antibodies, bovine pituitary extract (BPE) and epidermal growth factor (EGF) were obtained from Life Technologies/Invitrogen (Grand Island, NY). Propidium Iodide was purchased from Sigma-Aldrich (St. Louis, MO). Antibodies used for Western blot analysis of signaling proteins (phospho-ERK, phospho-p38, phospho-JNK, phospho-Akt) were all purchased from Cell Signaling Technology (Danvers, MA). All other reagents were purchased from Sigma-Aldrich.
Cells
The human keratinocyte cell line, HaCaT, was established from cells obtained from adult sun damaged skin and have been described previously[10-12]. HaCaT cells contain UV-signature mutations and express mutant dysfunctional p53 and a defective NF-kB signaling pathway, which are common findings in UV-initiated keratinocytes in human skin. However, these cells maintain normal signaling in response to UV irradiation compared to normal human keratinocytes. HCL-14 and FL-30 cells are HaCaT-derived cell lines that stably express a luciferase reporter gene driven by a portion of the human collagenase I promoter containing a single activator protein-1 (AP-1) binding site, or a full length human c-fos promoter, respectively [14, 15]. The cells were cultured in Dulbecco's modified Eagle's Medium (DMEM) with 10% fetal bovine serum (FBS) and 100 units/ml penicillin/streptomycin at 37°C and in 5% CO2. In experiments using growth arrested cells, the cells were cultured to 80–90% confluence and maintained in serum-free DMEM (SFM) for 24 h prior to UVB exposure. In experiments using proliferating cells, 25 μg/ml (0.25% v/v) BPE and 0.2 ng/ml EGF was added back to cells after serum starvation for 3 hr prior to subsequent treatment.
EISO treatment
HaCaT cells were treated with EISO diluted to a 1000× stock concentration in DMSO. Control cells were treated in the same manner with DMSO alone. Cells were treated for 1 hr prior to irradiation with ultraviolet B light. HaCaT cells were washed once in PBS and irradiated with a dose of 250 J/m2 UVB using a bank of two SF20 UVB lamps (National Biological Corp., Beachwood, OH) providing a peak emission of 313 nm. Control cells were treated in the same manner and mock irradiated. Following irradiation, HaCaT cells were again washed with PBS and returned to DMEM. During the post-irradiation incubation, cells were again treated with the indicated dose of EISO.
MTS assay
An MTS assay to assess cell viability after EISO treatment was performed using the CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega). HaCaT cells were plated in 96 well plates and cultured and treated with EISO as indicated in triplicate. At the end of the treatment period, DMEM containing EISO was removed and replaced with 100 μ1 fresh SFM and 20 μ1 MTS substrate and incubated at 37°C for 30 minutes for color development. Absorbance was read at 490 nm and triplicate values averaged. Results representative of n = 3.
Apoptotic analysis
HaCaT keratinocytes were serum-starved for 24 hr before pretreatment with EISO (0.0005% or 0.001% in DMSO) for 1 hr prior UVB irradiation. Cells were then washed with PBS and irradiated with 250 J/m2 of UVB. Then the cells were incubated with EISO (0.0005% or 0.001%) in serum-free DMEM media for an additional 6.5 hours. Floating cells were collected and pooled with adherent cells removed with trypsin. Cells were spun briefly and cell pellets incubated with Alexa 488-conjugated anti-annexin V (AnnV) antibodies and propidium iodide for 30 minutes at room temperature in the dark. Labeled cells were counted by flow cytometry, collecting a total of 10,000 data points per treatment condition using a FACSCanto II device (BD Biosciences).
Cell cycle analysis
HaCaT cells were starved in SFM for 24 hr. Cell culture medium was then changed to SFM or to SFM supplemented with 25 μg/ml bovine pituitary extract and 0.2 ng/ml EGF to stimulate cellular proliferation. After 3 hr incubation, HaCaT cell were treated with EISO for 24 hr. At the end of the treatment period, adherent cells were detached with trypsin and combined with floating cells, pelleted by centrifugation and incubated in 70% EtOH overnight at -20°C. The following day, cells were resuspended in PBS and incubated with 40 μg/ml propidium iodide (PI) and 0.5 mg/ml RNase A for 15 minutes. Single-color flow cytometric-based cell cycle analysis was performed using a BD FACScan (BD Biosciences) equipped with an air-cooled 15-mW argon ion laser tuned to 488 nm. Propidium iodide was detected in the FL2 detector through a 585/42 bandpass filter. List mode data files were acquired and analyzed using CellQuest PRO software (BD Biosciences). A total of 10,000 events were recorded and analyzed. Analysis of DNA content yielded individual populations of cells in G1 or G2/M phases, with S phase cells having intermediate DNA content. Data are expressed as % cells in each cell cycle phase. Three independent experiments were normalized so that the DMSO control equals 1 and the results were pooled for statistical analysis.
Western blotting
Serum-starved HaCaT cells were pretreated with BPE/EGF and/or EISO as described above, irradiated with 250 J/m2 UVB, and incubated in SFM supplemented with the same pretreatment compounds. HaCaT cells were lysed in RIPA buffer containing protease and phosphatase inhibitors as described previously [16]. Membranes were blocked in Tris buffered saline containing 0.1% Tween-20 (TBST) and either 5% nonfat dry milk or 5% BSA (for phospho-specific antibodies). Primary antibodies in 5% BSA were incubated overnight at 4°C. After washing in TBST, membranes were incubated with HRP-conjugated secondary antibodies and then washed extensively in TBST. Antigen-antibody complexes were detected using Amersham ECL Detection Reagent (GE Healthcare, Buckinghamshire, UK). Results representative of n = 3.
Luciferase reporter assays
HCL-14 and FL-30 cells were treated and UVB-irradiated in triplicate for each independent experiment. Twelve hours after exposure to UVB, cells were lysed and a total of 20 μg protein per sample replicate were assayed for luciferase activity according to the manufacturer's instructions for the Luciferase Assay System (Promega, Madison, WI) using a TD 20/20 luminometer (Turner Designs, Sunnyvale, CA). The experiment triplicates were averaged and the means from each independent experiment were subsequently averaged and analyzed by Student's t test for statistical significance. Results representative of n = 3.
Fluorescein diacetate hydrolysis
Cultured HaCaT cells were treated with BPE/EGF and/or EISO as described above and then adherent cells were detached by trypsinization and pooled with floating cells. Cells were pelleted, resuspended in PBS, and incubated with 2 μg/ml non-fluorescent fluorescein diacetate (FDA) for 15 min in the dark prior to analysis of FDA hydrolysis to fluorescein and retention of the fluorescent signal by flow cytometry. The emission fluorescence of cellular fluorescein was detected and recorded through a 530/30 bandpass filter in the FL1 detector. List mode data files were acquired and analyzed using CellQuest PRO software. Results representative of n = 2.
Immunocytochemistry
HaCaT cells plated on 8-well chamber slides were treated with EISO as indicated for 24 hr. At the end of the treatment period, cells were fixed in 2% paraformaldehyde in PBS for 30 min, permeabilized with 1% Triton X-100/PBS for 30 min, blocked in 2% goat serum for 1 hr, and incubated with rabbit anti-LC3 A/B primary antibodies (Cell Signaling Technologies, Danvers, MA) for 1 hr. Cells were then washed and incubated with Alexa Fluor 488 conjugated anti-rabbit secondary antibodies. Excess antibody was washed five times, and cells were mounted in Vectashield mounting media containing DAPI, a nuclear stain (Vector Laboratories, Burlingame, CA).
Results
EISO is cytotoxic in cultured HaCaT keratinocytes at low concentrations
A major active component of sandalwood oil, α-santalol, has been reported to induce apoptosis in skin and cancer cell lines [3, 4, 16-22]. However, little is known about the cellular response to pure sandalwood oil. Understanding this response is crucial if essential oils are to be used safely in cosmetic formulations. For initial testing, an MTS assay was performed to assess the general cellular effect of EISO on cell proliferation and/or viability in cultured HaCaT cells (Figure 1A). MTS conversion to a soluble formazan product occurs in the presence of live cells and the extent of conversion is dependent on cell number. HaCaT cells were serum starved for 24 hr and then treated with EISO. Cells treated with EISO up to 0.0005% were not adversely affected, but instead showed an increase or no effect in proliferation and/or viability compared to DMSO-treated controls. HaCaTs began to show reduced conversion of the MTS substrate at concentrations above 0.0005% EISO. Percent of MTS conversion was 53% of the DMSO control at 0.001% EISO and approximately 3% at concentrations of 0.002% and higher, suggesting a very narrow therapeutic index in cultured HaCaT cells. Cell density also impacted the effectiveness of EISO treatment (Figure 1B). Increased cell densities reduced the effectiveness of EISO treatment at both 0.001% and 0.002% final EISO concentration. This factor was taken into account in all subsequent experiments.
Figure 1. MTS assay to determine functional dose range of EISO in HaCaT cells.
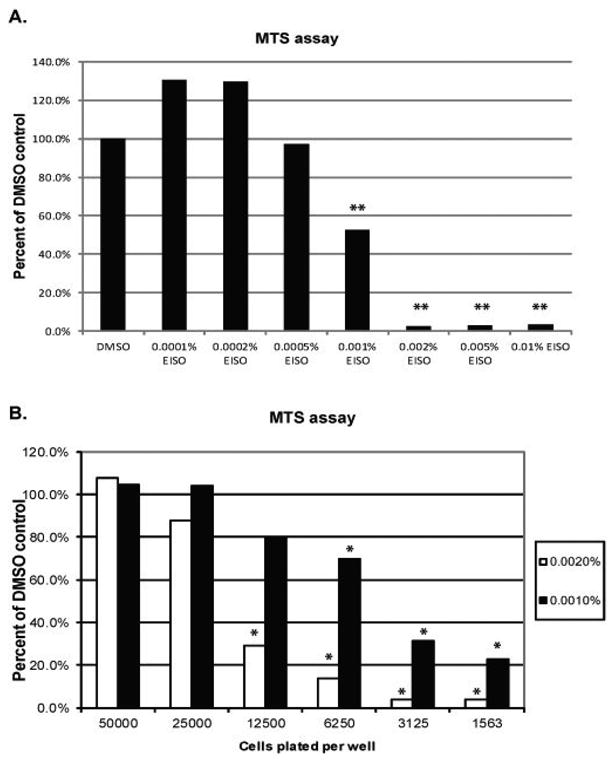
(A) HaCaT keratinocytes grown in a 96-well plate were serum-starved for 24 hr and then treated with a range of EISO from 0.0001-0.01% diluted in DMSO in triplicate for a period of 24 hr. (B) HaCaT cells were plated at varying cell densities so that the initial number of cells per well ranged from 1,563 to 50,000 cells using serial 1:1 dilutions of cells into DMEM. Again, cells were serum starved for 24 hr before treatment with 0.001% or 0.002% EISO in triplicate for an additional 24 hr. In all experiments, cell growth medium was spiked with MTS substrate for a period of 15 minutes and then an OD was obtained using a wavelength of 490 nm. Triplicate values were averaged and means expressed as a percentage of the reading for DMSO-treated control cells. In (A), triplicate wells were treated with DMSO, and in (B) a triplicate cell control was run for each density plated.
EISO induces cell cycle arrest
To determine whether the result of reduced MTS conversion was due to inhibition of proliferation or induction of cell death, cell cycle analysis and assessment of apoptosis by annexin V staining were both performed on serum-starved EISO-treated HaCaT cells. We found that EISO did not induce apoptotic death of HaCaT cells, (Figure 2A). EISO treatment (0.0005% and 0.001%) resulted in no significant increase in annexin V staining nor was there an increase in propidium iodide staining. Moreover, there was no significant difference observed between EISO treated cells compared to control cells after UVB-induced apoptosis. Cell cycle analysis revealed that in serum-starved cells, EISO induced S-phase arrest in a concentration-dependent manner, suggesting that EISO was stimulating quiescent G0/G1 phase cells to enter S phase while simultaneously preventing them from progressing completely through S phase (Figure 2B). There was also a mild dose-dependent rise in cells in the G2/M phase. To better understand this effect of EISO, a second experiment was performed in which cells were first stimulated to proliferate prior to EISO treatment. Serum-starved HaCaT cells were treated with a cocktail of bovine pituitary extract (BPE) and epidermal growth factor (EGF) for 3 hr before treatment with EISO. Cell cycle analysis revealed a trend toward G2/M block observed with the highest dose of 0.002% EISO, but experimental variability precluded statistical significance for these findings (data not shown). These data indicate an anti-proliferative, yet not apoptotic effect of EISO on cultured HaCaT cells.
Figure 2. Apoptosis/cell death by annexin V/PI staining and cell cycle analysis in EISO-treated HaCaT cells.
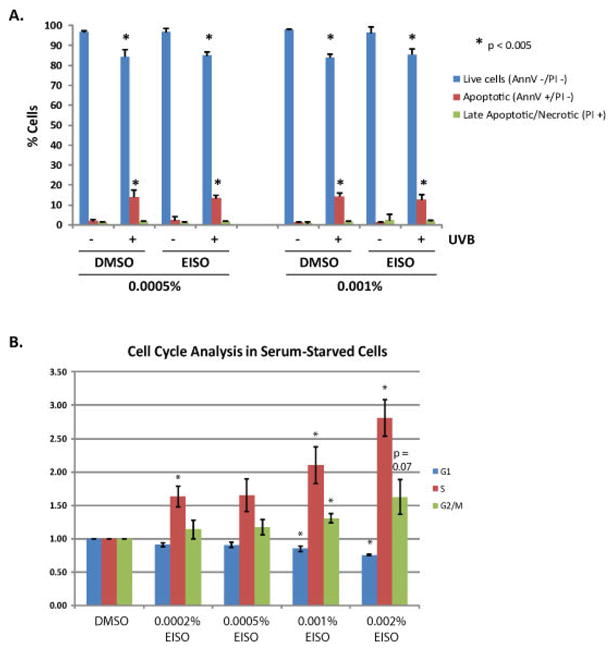
(A)) HaCaT keratinocytes were serum-starved for 24 hr and then treated with EISO (0.0005% or 0.001% in DMSO) for 1 hr prior to UVB irradiation, after which the treatments were added back to the cells. After a 6.5 hr rest the cells were incubated with Alexa 488-conjugated anti-annexin V (AnnV) antibodies and propidium iodide. Labeled cells were counted by flow cytometry, collecting a total of 10,000 data points per treatment condition. Data are expressed as percent of cells AnnV-/PI- (Live cells), AnnV+/PI- (Apoptotic cells) or PI+ regardless of AnnV status (Late apoptotic/necrotic). Data represent means±SD of three independent samples for each treatment condition. Asterisks indicate significant differences compared to control/mock cells (P value < 0.05). (B-C) Cell cycle analysis was performed on serum-starved cells treated with EISO for 24 hr or on cells first challenged with bovine pituitary extract and epidermal growth factor for 3 hr to initiate cell cycle entry before EISO treatment. Floating and adherent cells were collected and pooled. After spinning, cell pellets were fixed overnight in 70% EtOH and treated with RNase and PI for 15 minutes. Flow cytometry was used to collect 10,000 data points and data is expressed as percent of cells in G1, S, or G2/M phase based on relative cellular DNA content per cell.
AP-1 activity is inhibited by EISO, but not through blocking of UV-induced MAPK or PI3-K activation
Activation of AP-1 is critically important in UVB-induced skin carcinogenesis [13]. Treatment of HaCaT cells with EISO up to a concentration of 0.0005% resulted in a concentration-dependent reduction in AP-1 activity measured through the use of a luciferase reporter cell line, HCL-14 cells (Figure 3A). Concentrations of 0.001% EISO and above resulted in significant detachment of HCL-14 cells from the culture dish, causing AP-1 activity to be unmeasurable. PI3-K and p38 MAPK are known to be activated by UVB and coordinate upstream signaling that results in c-fos gene expression leading directly to increased AP-1 activity in HaCaT cells [23]. HaCaT cells were treated with 0.0005% EISO to determine the effect of the compound on signaling upstream of AP-1. Figure 3B shows that EISO treatment slightly potentiated UVB-induced phosphorylation of p38, ERK and Akt, instead of causing the expected response of inhibition of activity. EISO greatly potentiated phosphorylation of JNK by UVB. Therefore, the signaling through PI3-K, p38, ERK and JNK were intact in the presence of EISO, suggesting that AP-1 inhibition was through an alternate mechanism. To confirm this finding, we measured UVB-induced c-fos promoter activity in FL-30 cells and observed no significant decrease in c-fos-induced luciferase expression, consistent with the lack of inhibition of upstream signaling (Figure 3C). These findings suggest that the effects of EISO are not mediated though inhibition of UVB-induced signaling.
Figure 3. EISO inhibited UV-induced AP-1 activity without affecting signaling pathways upstream of AP-1 known to be induced by UV.
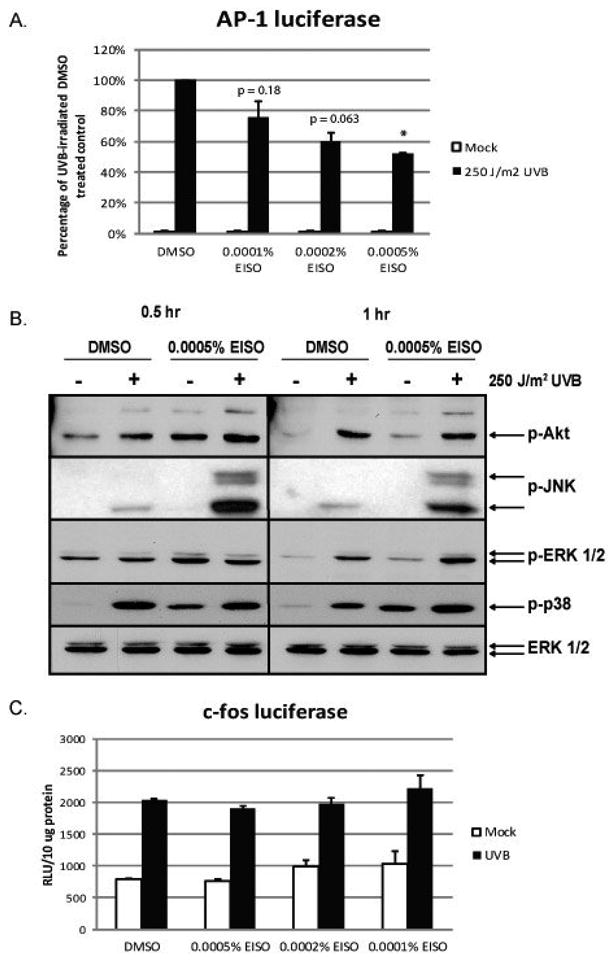
(A) HCL-14 cells, a HaCaT cell line stably expressing a portion of the human collagenase I gene (containing an AP-1 binding site) driving a firefly luciferase reporter gene, were treated with EISO for 1 hr prior to UV irradiation. After exposure to UV, media containing the same pretreatment (DMSO or EISO at the indicated concentration) was added back to the cells for 12 hr before cell lysates collected. Luciferase activity was measured and normalized to total protein content. Data are expressed as percent of the UV-induced luciferase activity in DMSO (vehicle)-treated control cells. (B) HaCaT cells were pretreated with EISO for 1 hr, irradiated with UVB and then returned to media containing pretreatments for 0.5 or 1 hr. Cell lysates were subjected to Western analysis using antibodies against phospho-Akt, phospho-JNK, phospho-ERK 1/2, and phospho-p38. Total ERK 1/2 was also analyzed to show that equal protein was loaded on the gel across all treatments. (C) FL-30 cells, a HaCaT cell line that stably expresses a full-length human c-fos promoter driving a firefly luciferase reporter gene, were pretreated with EISO for 1 hr before irradiation with UV and then returned to media containing pretreatments. Cell lysates were collected 6 hr post UV-irradiation and analyzed for luciferase activity, which was normalized to total protein.
EISO treatment compromises plasma membrane integrity
Serum-starved HaCaT cells were treated with EISO and/or BPE/EGF and loaded with cell-permeable, non-fluorescent fluorescein diacetate (FDA). Conversion of FDA to fluorescein and retention of the signal was measured over a period of 12 hrs in 3 hr increments. We discovered that in cells stimulated with BPE/EGF to induce proliferation, sensitivity to EISO treatment was much higher than in quiescent serum-starved cells, resulting in a more rapid loss of fluorescein signal (Figure 4 – compare 4C R7 population to that of 4D R7). Cells were also labeled with propidium iodide to confirm that the plasma and nuclear membranes were compromised during the EISO treatment. Through flow cytometric analysis, we identified a population of cells with intact DNA which confirmed that early in the treatment period (6 hr), cell membranes were being compromised by EISO but biochemical markers of cell death were not apparent.
Figure 4. Proliferating HaCaT cells were more sensitive to EISO-induced plasma membrane damage than quiescent cells.
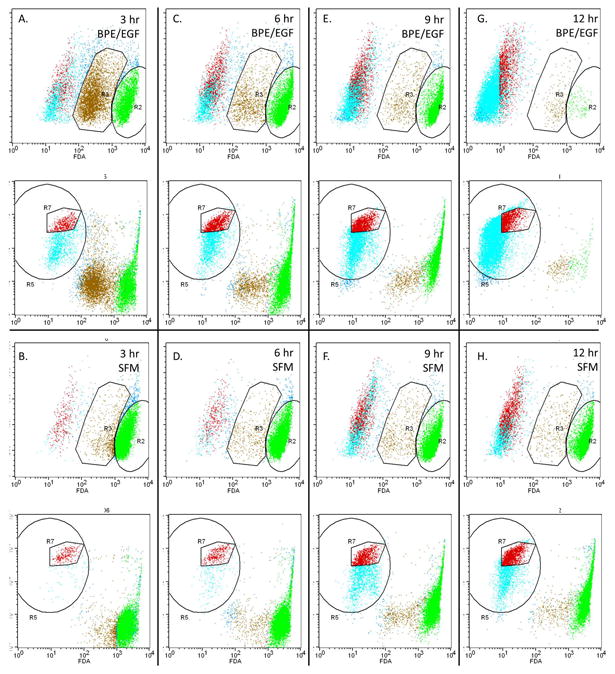
HaCaT cells were starved in serum-free DMEM for 24 hr. Fresh media supplemented with bovine pituitary extract and epidermal growth factor (A, C, E, G) or not supplemented (B, D, F, H) was added to the cells for 3 hr prior to treatment with EISO. At the indicated times, cells were detached by trypsinization and pooled with floating cells. Cells were pelleted, resuspended in PBS and incubated with fluorescein diacetate and propidium iodide for 15 minutes before analysis by flow cytometry. Using control cells loaded with individual dyes, regions of cells were identified as live cells with uncompromised membranes (R2), live cells with compromised membranes (R3), or severely compromised cells. Serum-starved EISO-treated cells (6 hr; panel D) displayed 2 distinct PI-stained populations of actively cycling cells in G1 and G2 phases (R7) that was used to confirm the population of cells with compromised membranes (evidenced by reduced fluorescein retention) but that were still alive.
LC3 expression and cleavage increases as a result of EISO treatment
We were interested in determining whether the blockade of cell proliferation, AP-1 activation, and compromised membrane integrity involved the induction of autophagy of EISO-treated HaCaT cells. To assess this, we treated serum-starved and BPE/EGF-stimulated cells as indicated and harvested lysates to measure the expression and cleavage of LC3 by Western blotting and immunocytochemistry (ICC), since LC3 is a key biomarker in the autophagic process [24]. ICC was performed on serum-starved cells only since EISO treatment of BPE/EGF resulted in a significant loss of cells due to detachment. ICC revealed that expression of LC3 was increased by very low doses of EISO (0.0002%) and was predominantly nuclear and perinuclear (Figure 5). Western blot analysis of both serum-starved and BPE/EGF stimulated cells revealed that 0.001% EISO induced accumulation of LC3 II in both populations, although this was less pronounced in quiescent cells (Figure 6). Irradiation with UVB also induced LC3 II, and this induction was again less dramatic in serum-starved cells. The effect of EISO and UV irradiation on LC3 processing was additive, resulting in the LC3 II band being largest in BPE/EGF+EISO+UVB treated HaCaT cells. Poly ADP ribose polymerase (PARP) cleavage, a marker of apoptosis, was induced as expected in control cells treated with UVB both in serum starved and BPE/EGF-stimulated conditions. Exposure to 0.001% EISO + DMSO resulted in minor PARP cleavage in both treatment conditions. However, this cleavage was not notably increased when EISO-treated cells were exposed to UVB, although LC3 II clavage was highest in these samples (lanes 4 and 8 in Figure 6).
Figure 5. Induction of autophagy in HaCaT cells treated with low doses of EISO.
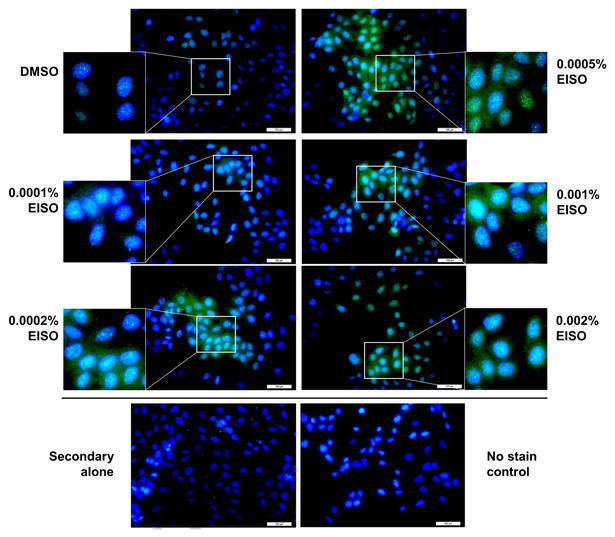
HaCaT cells plated on glass chamber slides were serum starved and treated with EISO for 24 hr. HaCaT cells were fixed and stained using antibodies against LC3 to demonstrate the induction of autophagy. Nuclear and perinuclear LC3 staining was observed in EISO-treated HaCaT cells, even at low doses.
Figure 6. UV-induced LC3 processing and expression increased and PARP cleavage decreased in EISO-treated proliferating and quiescent HaCaT cells.
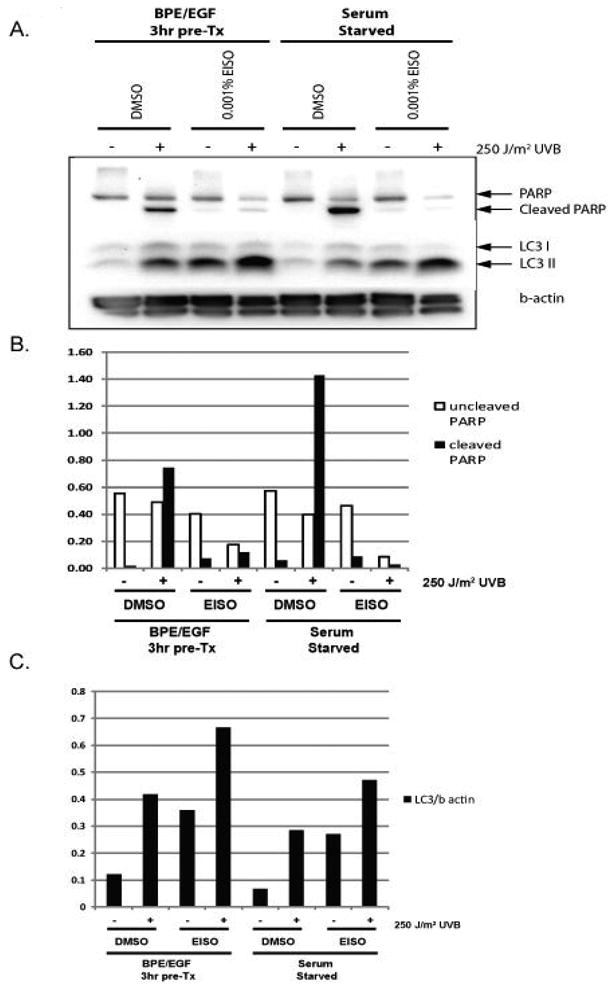
Serum-starved and BPE/EGF-treated HaCaT cells were treated with EISO for 16 hr. Adherent and floating cells were pooled and lysed in RIPA buffer for analysis of cellular proteins by Western blotting. PARP cleavage was induced by UV-irradiation, as expected, but EISO treatment blocked UV-induced apoptosis in both serum-starved and BPE/EGF treated cells. LC3 expression and processing were increased by UV in both BPE/EGF treated cells and serum-starved cells. EISO treatment increased LC3 processing in both treatment groups, and augmented the UVB response. Proliferating cells produced the most LC3 II after UVB treatment compared to controls.
Discussion
In the current study, our goal was to evaluate EISO as a novel chemopreventive agent, particularly because it is already in widespread use as a cosmetic, a fragrance and traditional medicine but also since the properties of one of the primary components of the oil, α-santalol, has been investigated as a preventive agent against skin cancer. It is interesting that although sandalwood oil has been utilized by cultures across the world for its health benefits for centuries, very few studies on the chemopreventive properties of the oil have been published [2, 5]. We propose that there are several potential mechanisms by which EISO may act as a chemopreventive agent, and in the current study we investigated the effect of EISO on apoptosis and cell death, cell proliferation, and inhibition of key signaling pathways that mediate the cellular response to UV. Recently, a study using purified α-santalol in vivo indicated that UVB-induced apoptosis, inflammation, proliferation and cell cycle control were all being affected by treatment with this compound, the net effect being significant reduction in UV-induced tumorigenesis in SKH-1 mice [6]. The EISO used in this study contains 45-50% α-santalol. We were interested in utilizing the extract instead of purified α-santalol because many cosmetics and natural remedies use the full extract, suggesting that the presence of other components may affect functionality. To our knowledge, this is the first time that purified EISO has been evaluated as an agent suitable for use as a chemopreventive substance against skin carcinogenesis.
We determined that treatment of cultured HaCaT keratinocytes with EISO alone does not induce apoptotic cellular responses, contrary to what has been previously reported for treatment with purified α-santalol [19]. However, EISO did induce growth arrest in an interesting manner that was dependent on the proliferative state of the cells. In quiescent (serum and hormone-starved) cells primarily in the G1/G0 phase, EISO-treated cells entered into S-phase but then primarily failed to progress into the G2 or M phase except at the highest EISO doses 24 hr post treatment. In proliferating HaCaT cells (serum-starved cells stimulated with BPE and EGF for 3 hr prior to treatment), EISO treatment resulted in a trend toward cell cycle blockade in the G2/M phase, although sample variability precluded finding significance in this experiment (data not shown). G2/M phase blockage has previously been reported in skin cells and in prostate cancer cells treated with α-santalol [21]. One possible explanation for this noted difference of the effect of EISO in quiescent versus proliferating cells is that the S-phase checkpoint through which the quiescent cells failed to progress was already passed by the proliferating cells. This suggests that there are at least two points in the cell cycle at which cell proliferation is inhibited by treatment with EISO. Since skin cells are largely quiescent in vivo, this finding supports the hypothesis that EISO has chemopreventive properties against the development of skin cancer.
We next investigated signaling responses commonly activated in keratinocytes by UV light to identify a possible mechanism by which EISO was inhibiting cell growth and proliferation. Information on the effects of sandalwood oil or α-santalol in this context is unavailable, as previous studies have either not investigated the effect or any findings from such studies have not been reported. To our surprise, unlike many other agents being investigated for chemopreventive activities, EISO had no inhibitory effect on the UV-stimulated PI3-K/Akt signaling pathway or on MAPK signaling pathways, instead slightly stimulating activation of these pathways even in control conditions. Interestingly, we discovered that UV-induced AP-1 signaling was significantly inhibited by EISO treatment and that the inhibition occurred in a dose-dependent manner. However, consistent with our finding that signaling pathways upstream of AP-1 activity were not affected by EISO treatment, c-Fos promoter activity was not inhibited by EISO. These findings argue that EISO may also elicit chemopreventive action by direct inhibition of AP-1 activity, a major known causative factor in UV-induced skin cancer [13]. There is precedence for direct inhibition of UV-stimulated AP-1 by other natural products in the literature [25].
We were interested in assessing the effects of EISO chemistry on HaCaT cells, specifically on plasma membrane integrity. The hydrophobic properties of the oil could potentially compromise plasma membrane integrity and cause cell death through a mechanism that is independent of the induction of apoptotic signaling or inhibition of cell survival mechanisms, as was seen with Annexin V staining, PARP cleavage and Akt activation. We found that cellular retention of a cell permeable, hydrolyzable fluorescein molecule, fluorescein diacetate, was compromised by treatment of cells with EISO in a dose-dependent manner, although the dose response occurred in a very narrow treatment window arguing that there is a maximum tolerance threshold for EISO in HaCaT cells. We monitored the effect of EISO on plasma membrane integrity over a period of 12 hr in both quiescent and in hormone-stimulated proliferating HaCaT cells and found that proliferating cells were more sensitive to EISO treatment than quiescent cells, although plasma membrane integrity was compromised in both. The reason for this differential effect is unknown, but it could be postulated that the membrane is more fluid in cells undergoing cell division, which may prime the cell membranes for more effective disruption by EISO treatment. Regardless of the cause of this observation, this finding highlights the fact that proliferative cells within an organ containing predominantly quiescent cells, like those found in a neoplasm of the skin, may be more responsive to treatment with EISO. In addition to this finding, we observed an increase in the apparent expression and processing of LC3, indicating an autophagic response to EISO treatment, and the significance of this response is unknown. Autophagy is a complex mechanism with a number of potential cellular effects, including the removal of organelles damaged by cytotoxic treatment as a way for tumor cells to escape chemotherapeutic treatment and as an alternative method to initiate cell death when apoptotic mechanisms are inactive or inhibited. However, it is now appreciated that cell death occurs both through, and in spite of, autophagy [26-29]. Our findings regarding LC3 induction and processing in response to EISO treatment along with a severe impairment of plasma membrane integrity and the lack of an apoptotic response suggests that EISO treatment induces organelle damage that, if sufficiently severe, ultimately leads to cell death.
α-Santalol, and its isomer, β-santalol, are the primary components of EISO, and although some of the cellular effects of treatment with the α-isomer have been investigated, it is likely that there are multiple targets for these two compounds. Additionally, there are numerous other ingredients in EISO that likely induce cellular responses, and it is therefore to be expected that treatment with the essential oil mixture will have multiple specific cellular effects.
Conclusion
In summary, we determined that EISO treatment of HaCaT cells resulted in:
a concentration-dependent inhibition of AP-1 activity through a mechanism independent of upstream signaling pathway inhibition,
an inhibition of cellular proliferation, as evidenced by a blockade of cell cycle progression,
a severely compromised plasma membrane resulting in an impaired ability to retain an intracellular fluorophore, and
induction of LC3 processing in the absence of any evidence of apoptosis, suggesting an initiation of autophagy.
Due to the complex nature of EISO, it is unclear as to whether these cellular effects are dependent on one another or if they occur independently. However, each of these effects suggests the need for continued study of EISO for use in protective and beneficial skin treatments. For example, the inhibition of AP-1 signaling and cell cycle progression may have occurred as a direct result of EISO treatment or since AP-1 activity drives cellular proliferation. However, it is also conceivable that cell proliferation was inhibited due to the direct effects of EISO on cellular membranes as a mechanism by which the cells conserve energy and first repair the membrane damage before resuming normal proliferation. Regardless of the precise mechanism, the blockade of cellular growth and the induction of cell death, to a greater extent in cells stimulated to divide, both support the hypothesis that EISO acts as a chemopreventive agent in skin cells. These studies are the first of their kind to investigate potential chemopreventive properties of purified East Indian sandalwood oil and support the hypothesis that EISO would be an effective treatment for pre-cancerous conditions such as AK and the prevention of skin cancer.
Acknowledgments
This work was supported by: NIH grants: Arizona Cancer Center Support Grant CA23074, Chemoprevention of Skin Cancer Program, Project Grant CA27502, Southwest Environmental Health Sciences Center Support Grant ES06694.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Priest DB. A potent natural antibacterial and anti-inflammatory derived from Australian sandalwood for cosmetic personal use. Cosmetic News. 2004;27:107–109. [Google Scholar]
- 2.Dwivedi C, Abu-Ghazaleh A. Chemopreventive effects of sandalwood oil on skin papillomas in mice. Eur J Cancer Prev. 1997;6:399–401. doi: 10.1097/00008469-199708000-00013. [DOI] [PubMed] [Google Scholar]
- 3.Dwivedi C, Guan X, Harmsen WL, Voss AL, Goetz-Parten DE, Koopman EM, Johnson KM, Valluri HB, Matthees DP. Chemopreventive effects of alpha-santalol on skin tumor development in CD-1 and SENCAR mice. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2003;12:151–156. [PubMed] [Google Scholar]
- 4.Dwivedi C, Valluri HB, Guan X, Agarwal R. Chemopreventive effects of alpha-santalol on ultraviolet B radiation-induced skin tumor development in SKH-1 hairless mice. Carcinogenesis. 2006;27:1917–1922. doi: 10.1093/carcin/bgl058. [DOI] [PubMed] [Google Scholar]
- 5.Dwivedi C, Zhang Y. Sandalwood oil prevent skin tumour development in CD1 mice. Eur J Cancer Prev. 1999;8:449–455. doi: 10.1097/00008469-199910000-00011. [DOI] [PubMed] [Google Scholar]
- 6.Santha S, Dwivedi C. alpha-Santalol, a skin cancer chemopreventive agent with potential to target various pathways involved in photocarcinogenesis. Photochemistry and photobiology. 2013;89:919–926. doi: 10.1111/php.12070. [DOI] [PubMed] [Google Scholar]
- 7.Chilampalli C, Zhang X, Kaushik RS, Young A, Zeman D, Hildreth MB, Fahmy H, Dwivedi C. Chemopreventive effects of combination of honokiol and magnolol with alpha-santalol on skin cancer developments. Drug discoveries & therapeutics. 2013;7:109–115. [PubMed] [Google Scholar]
- 8.Santha S, Bommareddy A, Rule B, Guillermo R, Kaushik RS, Young A, Dwivedi C. Antineoplastic effects of alpha-santalol on estrogen receptor-positive and estrogen receptor-negative breast cancer cells through cell cycle arrest at G2/M phase and induction of apoptosis. PloS one. 2013;8:e56982. doi: 10.1371/journal.pone.0056982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sharma M, Levenson C, Bell RH, Anderson SA, Hudson JB, Collins CC, Cox ME. Suppression of Lipopolysaccharide-stimulated Cytokine/Chemokine Production in Skin Cells by Sandalwood Oils and Purified alpha-santalol and beta-santalol. Phytotherapy research: PTR. 2013 doi: 10.1002/ptr.5080. [DOI] [PubMed] [Google Scholar]
- 10.Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. The Journal of cell biology. 1988;106:761–771. doi: 10.1083/jcb.106.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ehrhart JC, Gosselet FP, Culerrier RM, Sarasin A. UVB-induced mutations in human key gatekeeper genes governing signalling pathways and consequences for skin tumourigenesis. Photochem Photobiol Sci. 2003;2:825–834. doi: 10.1039/b302281a. [DOI] [PubMed] [Google Scholar]
- 12.Lehman TA, Modali R, Boukamp P, Stanek J, Bennett WP, Welsh JA, Metcalf RA, Stampfer MR, Fusenig N, Rogan EM, et al. p53 mutations in human immortalized epithelial cell lines. Carcinogenesis. 1993;14:833–839. doi: 10.1093/carcin/14.5.833. [DOI] [PubMed] [Google Scholar]
- 13.Cooper SJ, MacGowan J, Ranger-Moore J, Young MR, Colburn NH, Bowden GT. Expression of dominant negative c-jun inhibits ultraviolet B-induced squamous cell carcinoma number and size in an SKH-1 hairless mouse model. Molecular cancer research: MCR. 2003;1:848–854. [PubMed] [Google Scholar]
- 14.Gonzales M, Bowden GT. Ultraviolet B (UVB) induction of the c-fos promoter is mediated by phospho-cAMP response element binding protein (CREB) binding to CRE and c-fos activator protein 1 site (FAP1) cis elements. Gene. 2002;293:169–179. doi: 10.1016/s0378-1119(02)00723-0. [DOI] [PubMed] [Google Scholar]
- 15.Chen W, Borchers AH, Dong Z, Powell MB, Bowden GT. UVB irradiation-induced activator protein-1 activation correlates with increased c-fos gene expression in a human keratinocyte cell line. The Journal of biological chemistry. 1998;273:32176–32181. doi: 10.1074/jbc.273.48.32176. [DOI] [PubMed] [Google Scholar]
- 16.Olson ER, Melton T, Dong Z, Bowden GT. Stabilization of quercetin paradoxically reduces its proapoptotic effect on UVB-irradiated human keratinocytes. Cancer Prevention Research. 2008;1:362–368. doi: 10.1158/1940-6207.CAPR-08-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arasada BL, Bommareddy A, Zhang X, Bremmon K, Dwivedi C. Effects of alpha-santalol on proapoptotic caspases and p53 expression in UVB irradiated mouse skin. Anticancer research. 2008;28:129–132. [PubMed] [Google Scholar]
- 18.Bommareddy A, Hora J, Cornish B, Dwivedi C. Chemoprevention by alpha-santalol on UVB radiation-induced skin tumor development in mice. Anticancer research. 2007;27:2185–2188. [PubMed] [Google Scholar]
- 19.Bommareddy A, Rule B, VanWert AL, Santha S, Dwivedi C. alpha-Santalol, a derivative of sandalwood oil, induces apoptosis in human prostate cancer cells by causing caspase-3 activation. Phytomedicine. 2012;19:804–811. doi: 10.1016/j.phymed.2012.04.003. [DOI] [PubMed] [Google Scholar]
- 20.Kaur M, Agarwal C, Singh RP, Guan X, Dwivedi C, Agarwal R. Skin cancer chemopreventive agent, {alpha}-santalol, induces apoptotic death of human epidermoid carcinoma A431 cells via caspase activation together with dissipation of mitochondrial membrane potential and cytochrome c release. Carcinogenesis. 2005;26:369–380. doi: 10.1093/carcin/bgh325. [DOI] [PubMed] [Google Scholar]
- 21.Zhang X, Chen W, Guillermo R, Chandrasekher G, Kaushik RS, Young A, Fahmy H, Dwivedi C. Alpha-santalol, a chemopreventive agent against skin cancer, causes G2/M cell cycle arrest in both p53-mutated human epidermoid carcinoma A431 cells and p53 wild-type human melanoma UACC-62 cells. BMC Res Notes. 2010;3:220. doi: 10.1186/1756-0500-3-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang X, Dwivedi C. Skin cancer chemoprevention by alpha-santalol. Front Biosci (Schol Ed) 2011;3:777–787. doi: 10.2741/s186. [DOI] [PubMed] [Google Scholar]
- 23.Bowden GT. Prevention of non-melanoma skin cancer by targeting ultraviolet-B-light signalling. Nat Rev Cancer. 2004;4:23–35. doi: 10.1038/nrc1253. [DOI] [PubMed] [Google Scholar]
- 24.Tanida I, Ueno T, Kominami E. LC3 conjugation system in mammalian autophagy. The international journal of biochemistry & cell biology. 2004;36:2503–2518. doi: 10.1016/j.biocel.2004.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dickinson SE, Melton TF, Olson ER, Zhang J, Saboda K, Bowden GT. Inhibition of activator protein-1 by sulforaphane involves interaction with cysteine in the cFos DNA-binding domain: implications for chemoprevention of UVB-induced skin cancer. Cancer research. 2009;69:7103–7110. doi: 10.1158/0008-5472.CAN-09-0770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Steeves MA, Dorsey FC, Cleveland JL. Targeting the autophagy pathway for cancer chemoprevention. Curr Opin Cell Biol. 2010;22:218–225. doi: 10.1016/j.ceb.2009.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tasdemir E, Galluzzi L, Maiuri MC, Criollo A, Vitale I, Hangen E, Modjtahedi N, Kroemer G. Methods for assessing autophagy and autophagic cell death. Methods Mol Biol. 2008;445:29–76. doi: 10.1007/978-1-59745-157-4_3. [DOI] [PubMed] [Google Scholar]
- 28.Wu WK, Sakamoto KM, Milani M, Aldana-Masankgay G, Fan D, Wu K, Lee CW, Cho CH, Yu J, Sung JJ. Macroautophagy modulates cellular response to proteasome inhibitors in cancer therapy. Drug Resist Updat. 2010;13:87–92. doi: 10.1016/j.drup.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 29.Yang ZJ, Chee CE, Huang S, Sinicrope FA. The role of autophagy in cancer: therapeutic implications. Molecular cancer therapeutics. 2011;10:1533–1541. doi: 10.1158/1535-7163.MCT-11-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]