

Abstract
We explored both structure–activity relationships among substituted oxyoxalamides used as the primary pharmacophore of inhibitors of the human sEH and as a secondary pharmacophore to improve water solubility of inhibitors. When the oxyoxalamide function was modified with a variety of alkyls or substituted alkyls, compound 6 with a 2-adamantyl group and a benzyl group was found to be a potent sEH inhibitor, suggesting that the substituted oxyoxalamide function is a promising primary pharmacophore for the human sEH, and compound 6 can be a novel lead structure for the development of further improved oxyoxalamide or other related derivatives. In addition, introduction of substituted oxyoxalamide to inhibitors with an amide or urea primary pharmacophore produced significant improvements in inhibition potency and water solubility. In particular, the N,N,O-trimethyloxyoxalamide group in amide or urea inhibitors (26 and 31) was most effective among those tested for both inhibition and solubility. The results indicate that substituted oxyoxalamide function incorporated into amide or urea inhibitors is a useful secondary pharmacophore, and the resulting structures will be an important basis for the development of bioavailable sEH inhibitors.
Keywords: Substituted oxyoxalamides, Human soluble epoxide hydrolase, Inhibitors
1. Introduction
Arachidonic acid, a ω-6 polyunsaturated fatty acid, plays important roles in cellular signaling as a lipid second messenger and is also a precursor in the production of oxidative metabolites known as eicosanoids by enzymes such as cyclooxygenase, lipoxygenase, and cytochrome P450. Prostanoids1,2 and leukotrienes3 are major metabolic products of arachidonic acid by cyclooxygenase and lipoxygenase, respectively. These two pathways are largely inflammatory and induce inflammation, pain, and asthma,1–3 making the both enzymes current pharmaceutical targets for relief from the symptoms. The third branch of arachidonic acid cascade involves oxidation by cytochrome P450 to produce several inflammatory hydroxylated metabolites and the corresponding lipid epoxides formed at the olefinic centers and known as epoxyeicosatrienoic acids (EETs). EETs have been reported as a new class of lipid mediators with important biological functions.4 The endogenous epoxy lipids, EETs, influence blood pressure by modulating cardiac output, vascular resistance, and urinary composition.5–11 In addition, vascular inflammation and pain are modulated by the action of EETs.9,10 However, the metabolism of the epoxy functionality of the EETs to the vicinal diols by soluble epoxide hydrolase (sEH) dramatically diminishes the biological activities.6 Many reports have shown that the treatment of potent human sEH inhibitors increases EET levels and reduces blood pressure and inflammatory responses in in vitro and in vivo experimental models,5–11 suggesting that human sEH is a promising pharmacological target for the treatment of cardiovascular and other diseases.
A number of urea compounds with a variety of substituents are highly potent inhibitors of the human sEH.12–21 The best optimization of urea derivatives affords specific inhibition potency for the target enzyme in a range of less than 1 nM. Structure–activity relationship studies indicate that a carbonyl group and a single proton donating NH group of urea function are essential for making it an effective primary pharmacophore to inhibit the enzyme activity. Functionalities such as amides and carbamates with both a carbonyl group and an NH group are, therefore, known to produce potent inhibition as a primary pharmacophore, while ester or carbonate functions without a proton donating NH group yield no inhibition for the target enzyme.12,22–24 Many of these compounds are difficult to formulate because they are high melting liphophilic solids. These formulation problems can be solved by reducing the melting point and crystal stability, increasing water solubility, and increasing potency. On the other hand, when a variety of functionalities including amides, esters, ketones, and ethers are incorporated as a secondary pharmacophore remote from the catalytic site in potent urea inhibitors, dramatic changes in inhibition potency are not observed, rather significant improvement in physical properties is often obtained,12 implying that primary inhibition of the human sEH depends on the structure of primary pharmacophores and secondary pharmacophores are useful for improving physical properties and potency. In the present study, we first investigated replacement of the primary pharmacophore with a series of substituted oxyoxalamides and then used oxyoxalamides as a second series to replace the secondary pharmacophore using the classical amide and urea primary pharmacophores. In both series, potent compounds were found with improved water solubility.
2. Results and discussion
2.1. Chemistry
Substituted oxyoxalamide derivatives (3–15) and N-(benzyloxy)-2-(adamant-2-ylamino)acetamide (16) in Tables 1 and 2 were synthesized as outlined in Scheme 1. Ethyl (chlorocarbonyl)formate was reacted with an alkyl- or a cycloalkyl-amine (Scheme 1A) or adamant-2-ylamine (Scheme 1B) in dichloromethane, followed by hydrolysis with 1 N NaOH in ethanol to provide the corresponding (carbamoyl)formic acid in approximately 80–95% yield. The formic acid was then coupled with benzyloxyamine (Scheme 1A) or with a substituted oxyamine (Scheme 1B) using 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide (EDCI) and 4-dimethylaminopyridine (DMAP) in dichloromethane to yield compounds 3–12, 14, and 15 in 50–85% yield.23 As seen in Scheme 1B, substituted oxyamines for compounds 10–12 were prepared by the reaction of a corresponding bromide with N-hydroxyphthalimide in N,N-dimethylformamide (DMF), which was followed by the addition of hydrazine in trichloromethane (40–50%). N-Methylation of compound 6 with iodomethane (CH3I) in the presence of potassium carbonate (K2CO3) in DMF afforded compound 13 in 55% yield (Scheme 1C). Substitution of methyl 2-bromoacetate with adamant-2-ylamine in dichloromethane and hydrolysis of the ester with 1 N NaOH in methanol gave the corresponding adamantylaminoacetic acid in approximately 85% yield. Then, coupling of this acid with benzyloxyamine using EDCI/DMAP in dichloromethane produced compound 16 in 70% yield (Scheme 1D).
Table 1.
Inhibition of human sEH by substituted oxyoxalamide derivatives
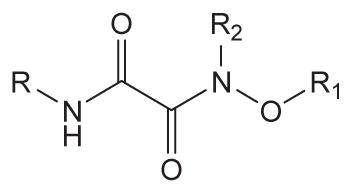
| ||||
|---|---|---|---|---|
| No. | R | R1 | R2 | Human sEH IC50 (nM)a |
| 3 |
|
|
H | >10,000 |
| 4 |
|
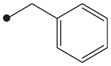
|
H | >10,000 |
| 5 |
|
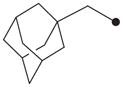
|
H | 838 |
| 6 |
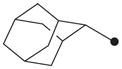
|
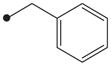
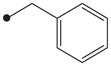
|
H | 50 |
| 7 |
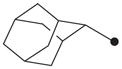
|
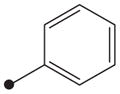
|
H | >10,000 |
| 8 |
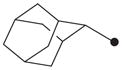
|
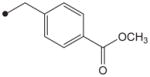
|
H | 1500 |
| 9 |
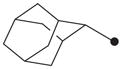
|
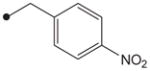
|
H | >10,000 |
| 10 |
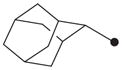
|
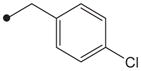
|
H | 2300 |
| 11 |
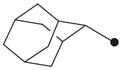
|
|
H | 4400 |
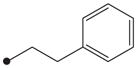
| 12 |
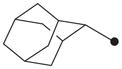
|
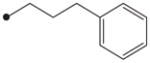
|
H | 550 |
| 13 |
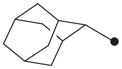
|
|
CH3 | 3200 |
| 14 |
|
CH3 | CH3 | >10,000 |
| 15 |
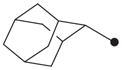
|
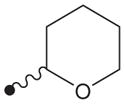
|
H | >10,000 |
Test compounds prepared in DMSO were reacted with human sEH (1 nM) for 10 min in 25 mM Bis–Tris/HCl buffer (202 μL; pH 7.0) at 30 °C. The fuorescent substrate (CMNPC; [S] = 5 μM) was then introduced to the incubation mixture. Inhibition potency against the human sEH was determined by measuring the appearance of the 6-methoxy-2-naphthaldehyde with an excitation wavelength of 330 nm and an emission wavelength of 465 nm for 10 min on a fluorometer. Results are averages of three separate measurements. See the Supplementary data for the detailed procedures.
Table 2.
Inhibition of human sEH by N1-(adamant-2-yl)-N2-(benzyloxy)oxalamide (6) and N-(benzyloxy)-2-(adamant-2-ylamino)acetamide (16)
Test compounds prepared in DMSO were reacted with human sEH (1 nM) for 10 min in 25 mM Bis–Tris/HCl buffer (202 μL; pH 7.0) at 30 °C. The fluorescent substrate (CMNPC; [S] = 5 μM) was then introduced to the incubation mixture. Inhibition potency against the human sEH was determined by measuring the appearance of the 6-methoxy-2-naphthaldehyde with an excitation wavelength of 330 nm and an emission wavelength of 465 nm for 10 min on a fluorometer. Results are averages of three separate measurements. See the Supplementary data for the detailed procedures.
12-(3-Adamantan-1-yl-ureido)dodecanoic acid, which was synthesized in the reaction of 1-adamantane isocyanate with 12-aminododecanoic acid in 1,2-dichloroethanol as previously described13 and was used as a positive control for the inhibition assay in this study.
Scheme 1.

Syntheses of substituted oxyoxalamides (3–15) and N-(benzyloxy)-2-(adamant-2-ylamino)acetamide (16). (a) An alkyl- or a cycloalkyl-amine, Et3N, CH2Cl2, room temp; (b) 1 N NaOH, EtOH or MeOH, room temp; (c) benzyloxyamine (in parts A and D) or a substituted oxyamine (in part B), EDCI, DMAP, CH2Cl2, room temp; (d) (i) N-hydroxyphthalimide, Et3N, DMF, room temp, (ii) H2NNH2, 10% MeOH in CHCl3, room temp; (e) CH3I, K2CO3, DMF, room temp.
Amide-oxyoxalamide derivatives 18–26 in Table 3 were synthesized by the procedure outlined in Scheme 2. Coupling reaction of 4-aminobenzylamine with adamant-1-ylcarboxylic acid (for compounds 18–20 and 22) or with adamant-1-ylmethylcarboxylic acid (for compound 23) using EDCI/DMAP in dichloromethane afforded an amide-amine intermediate, which was used for the reaction with ethyl (chlorocarbonyl)formate in the presence of triethylamine (Et3N) in dichloromethane to make substituted carbonylformate in about 60% yield. Hydrolysis of the ester function of this intermediate with 1 N NaOH in ethanol gave the corresponding (carbamoyl)formic acid, which was further reacted with a substituted oxyamine in the presence of EDCI/DMAP coupling reagents in dichloromethane to produce compounds 18–20, 22, and 23 in a range of 50–90% yield (Scheme 2A). As depicted in Scheme 2B, coupling of adamant-1-ylmethylcarboxylic acid with mono-N-protected 4-aminoaniline using EDCI/DMAP and following N-de-protection using 4 N HCl provided the corresponding amide-amine intermediate in about 80% yield. This amide-amine was reacted with ethyl (chlorocarbonyl)formate in dichloromethane to afford the corresponding formate, which was hydrolyzed with 1 N NaOH in ethanol to give a formic acid intermediate in 85% yield. Coupling of the acid function with methoxymethylamine using EDCI/DMAP yielded compound 25 in 70% yield. In addition, compounds 21, 24, and 26 were synthesized by the alkylation of compounds 20, 23, 25, respectively, with CH3I in the presence of K2CO3 as a base in DMF in approximately 60% yield (Scheme 2C).
Table 3.
Inhibition of human sEH by amide derivatives substituted with oxyoxalamide function
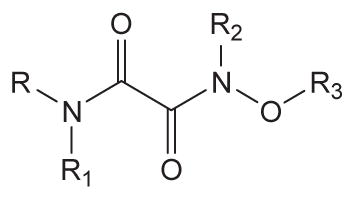
| ||||||
|---|---|---|---|---|---|---|
| No. | R | R1 | R2 | R3 | Human sEH IC50 a (nM) | Solubilityb (μM) |
| 17d |
|
>1000 | 40 | |||
| 18 |
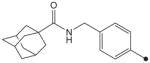
|
H | H | CH3 | 280 | NDc |
| 19 |
|
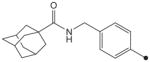
H | H |
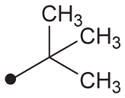
|
408 | ND |
| 20 |
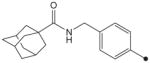
|
H | CH3 | CH3 | 190 | ND |
| 21 |
|
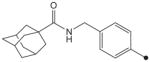
CH3 | CH3 | CH3 | 204 | ND |
| 22 |
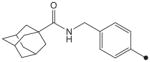
|
H | H |
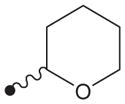
|
>1000 | ND |
| 23 |
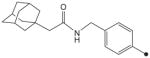
|
H | CH3 | CH3 | 69 | 156 |
| 24 |
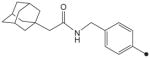
|
CH3 | CH3 | CH3 | 7.9 | 625 |
| 25 |
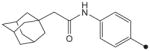
|
H | CH3 | CH3 | 35 | 78 |
| 26 |
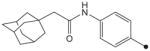
|
CH3 | CH3 | CH3 | 4.4 | 156 |
| 2e |
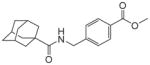
|
9.1 | 125 | |||
| IK950e |
|
14 | 625 | |||
Test compounds prepared in DMSO were reacted with human sEH (1 nM) for 10 min in 25 mM Bis–Tris/HCl buffer (202 μL; pH 7.0) at 30 °C. The fluorescent substrate (CMNPC; [S] = 5 μM) was then introduced to the incubation mixture. Inhibition potency against the human sEH was determined by measuring the appearance of the 6-methoxy-2-naphthaldehyde with an excitation wavelength of 330 nm and an emission wavelength of 465 nm for 10 min on a fluorometer. Results are averages of three separate measurements. See the Supplementary data for the detailed procedures.
Water solubility was determined by adding a variety of concentrations of a test compound prepared in DMSO to 0.1 M sodium phosphate buffer (pH 7.4) in a final ratio of 5:95 (v/v). The turbidity of the water solution was measured at 650 nm to determine solubility in water. Results are the average of triplicate determinations.
Not-determined because the inhibition results were not potent enough compared to that of other derivatives.
Amide inhibitor with no substitution by oxyoxalamide function.
Scheme 2.

Syntheses of amide derivatives with substituted oxyoxalamide. (a) adamant-1-ylcarboxylic acid (for compounds 18–20 and 22) or adamant-1-ylmethylcarboxylic acid (for compounds 23 and 25), EDCI, DMAP, CH2Cl2, room temp; (b) ethyl (chlorocarbonyl)formate, Et3N, CH2Cl2, room temp; (c) 1 N NaOH, EtOH, room temp; (d) a corresponding oxyamine (in parts A and B), EDCI, DMAP CH2Cl2, room temp; (e) 4 N HCl, CH2Cl2, room temp; (f) CH3I, K2CO3, DMF, room temp.
Urea compounds with substituted oxyoxalamide were synthesized as outlined in Scheme 3. The reaction of 4-aminobenzylamine with adamant-1-yl isocyanate in DMF gave 1-adamantyl-3-(4-aminobenzyl) urea in 100% yield.13 Compounds 28, 29, 30, and 32 were produced by the above reactions used for the syntheses of compounds 18, 19, 20, and 22 from this urea-amine intermediate in a range of 40–70% yield (Scheme 3A). N-Methylation of compound 30 using CH3I and K2CO3 as a base in DMF yielded compound 31 in 50% yield (Scheme 3B).
Scheme 3.

Syntheses of urea derivatives with substituted oxyoxalamide. (a) adamant-1-yl isocyanate, DMF, room temp; (b) ethyl (chlorocarbonyl)formate, Et3N, CH2Cl2, room temp; (c) 1 N NaOH, EtOH, room temp; (d) a corresponding oxyamine (in part A), EDCI, DMAP CH2Cl2, room temp; (e) CH3I, K2CO3, DMF, room temp.
2.2. Structure–activity relationships
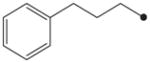
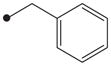
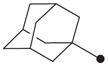
In order to first investigate whether the oxyoxalamide function can act as an effective primary pharmacophore to inhibit the human sEH, a series of substituted oxyoxalamides were synthesized. Our previous studies show that potent inhibition of the target enzyme is obtained with the substitution of relatively hydrophobic alkyl or cycloalkyl groups and substituted alkyl or aromatic groups on the left side and the right side of urea or amide pharmacophores, respectively (1 and 2 in Fig. 1).12,14,15,21,23 Thus, hydrophobic alkyls and substituted alkyl or aryl groups were incorporated in the both sides of the diketo of oxyoxalamide moiety, and inhibition potency of the oxyoxalamide derivatives for the human sEH was evaluated (Table 1 and Fig. 2A). As seen in compounds 3 and 4, 3-phenylpropyl (3) and 1-adamantyl (4) groups in the left side of the diketo, which are useful for making potent inhibitors with urea or amide functions, were not effective for producing inhibition activity for the target enzyme with oxyoxalamide function. However, in replacing the substituent in the left side of the diketo of compounds 3 and 4 by an adamantylmethyl group (5), moderate inhibition was gained. Moreover, interestingly, introduction of a 2-adamantyl group (6) in the left side of the diketo moiety afforded a high improvement (16-fold) in inhibition potency compared to that of compound 5. Comparing compound 6 with 2-adamantane to compound 4 with 1-adamantane, improvement in inhibition activity was dramatic, indicating that the 2-adamantyl group is particularly suitable for being the left side substituent of oxyoxalamide function to yield significant inhibition potency. It is also implied that slight changes in orientation or size of the alkyl groups in the left side of oxyoxalamide function as shown in compounds 3–6 result in a large variation in inhibition for the target enzyme, which is different from the results that similarly potent inhibition is observed in urea or amide derivatives substituted with the alkyl groups.14,15,22,23
Figure 1.

Substituents of urea and amide primary pharmacophores (1) which yield potent inhibitors of the human sEH, and examples of potent urea and amide compounds with a secondary pharmacophore (2): n = 0–10, 1 pharmacophore = primary pharmacophore; 2 pharmacophore = secondary pharmacophore. The IC50 of urea and amide compounds in structure 2 for the human enzyme is 14 and 9.1 nM, respectively.
Figure 2.
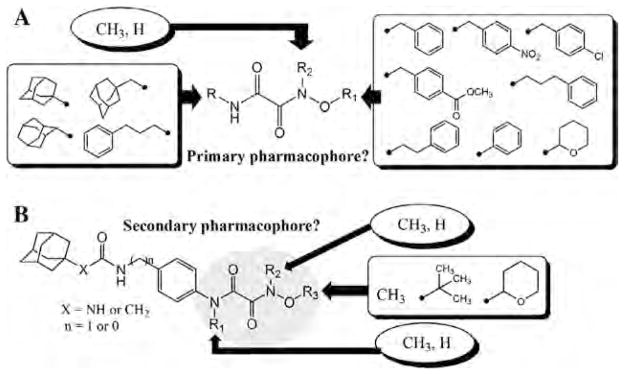
Structure–activity relationships of various oxyoxalamide derivatives as inhibitors of the human sEH were explored. In order to first investigate whether the substituted oxyoxalamides can be an effective primary pharmacophore to inhibit the target enzyme, various substituents were introduced into the oxyoxalamide function as shown in A (Tables 1 and 2). In addition, in order to see if it can be an effective secondary pharmacophore to improve inhibition and/or solubility of amide- or urea-based inhibitors, the substituted oxyoxalamides with a variety of groups were incorporated to amide and urea inhibitors as seen in B (Tables 3 and 4).
Based on the above results, the 2-adamantyl was fixed on the left side of the diketo moiety of the oxyoxalamide, and then a benzyl group in the right side of the diketo of compound 6 was further modified with phenyl and several arylalkyl groups. Because aryl containing groups in the right side of urea or amide pharmacophores (e.g., 2 in Fig. 1) provide much higher binding activity than aliphatic alkyl groups,15,21,23 compounds with aryl substituent (7–13) were synthesized. As seen in compound 7, replacement of the benzyl group of compound 6 by a phenyl group resulted in no inhibition. This implies that the methylene benzyl carbon in the right side of the oxyoxalamide is necessary for producing inhibition of the target enzyme. When a functional group such as a methyl ester (8), nitro (9), or chloro (10) was incorporated on the 4-position of the benzyl group of compound 6, >300-fold drop in inhibition was also observed, indicating that these substituents on the benzyl group of compound 6 are not effective for increasing inhibition potency. These results are not consistent with previous observations with urea or amide primary pharmacophores.21,23 Because we previously showed that a longer alkyl chain like an ethyl or a propyl between primary amide pharmacophore and benzene ring in the right side of the amide function provides improved inhibition for the target enzyme,23 2-phenylethyl and 3-phenylpropyl were introduced instead of the benzyl group of compound 6. As seen in compounds 11 and 12, a propyl chain (12) resulted in approximately 8-fold better inhibition than an ethyl chain (11). However, this still gave approximately 10-fold drop in inhibition when compared to the potency of compound 6, supporting that one methylene carbon of the benzyl group of compound 6 is important for yielding significant inhibition potency. N-Substitution of compound 6 with a methyl group (13) dropped inhibition potency approximately 60-fold. Inhibition was further lost in the presence of a dimethyl or a polar substituent as observed in compounds 14 and 15, respectively, indicating that a benzyl group next to an unsubstituted NH function is effective as a right side substituent for yielding promising inhibitors with an oxyoxalamide function as a primary pharmacophore. Furthermore, when one carbonyl group of compound 6 was deleted as seen in compound 16 in Table 2, no inhibition was observed, suggesting that the carbonyl group of oxyoxalamide function plays an important role in inhibiting the target human enzyme like that of urea or amide functions.12 The inhibition potency of compound 6 was approximately 15-fold lower when compared to that of a very potent urea-based inhibitor (AUDA),13 indicating that the oxyoxalamide 6 can be used as a new lead structure for structure-activity relationship studies to further produce as potent oxyoxalamide inhibitors as urea-based compounds. Overall, the results in Tables 1 and 2 show that the substituted oxyoxalamides are promising novel functionalities as primary pharmacophores to yield inhibitors of significant potency for the human sEH.
A functional group incorporated on around the 5th to 7th atom from the carbonyl of primary pharmacophores such as ureas or amides can play a role as a secondary pharmacophore to modify inhibition potency and/or physical properties of urea or amide inhibitors (Fig. 2B).12,14,22,23 In general, functions such as amides, esters, ketones, ethers, sulfonamides, sulfoxides, and sulfones are useful secondary pharmacophores for improving physical properties without decreasing inhibition potency of urea inhibitors.12,14,23 In order to investigate whether the oxyoxalamide function can act as an effective secondary pharmacophore for improving inhibition potency and/or physical properties, a series of amide and urea derivatives with substituted oxyoxalamide groups were synthesized as shown in Tables 3 and 4. We previously showed that incorporation of a functional group like an ester as a secondary pharmacophore on a benzene ring in the right side of amide inhibitors like compound 17 in Table 3 is highly useful for enhancing inhibition potency for the target enzyme (2 in Table 2).23 Thus, the oxyoxalamide function was introduced to compound 17 to see if it works as an effective secondary pharmacophore for improving inhibition potency or the physical properties of amide inhibitor. When an oxyoxalamide group was substituted on the 4-position of the benzene ring of compound 17 as seen in compound 18, at least a 3-fold enhancement in inhibition was gained, indicating that the oxyoxalamide function is also a useful secondary pharmacophores for improving inhibitory potency of amide compounds. Because a polar functional group like an oxyoxalamide inserted as a secondary pharmacophore plays an important role in improving physical properties (e.g. water solubility) of hydrophobic inhibitors as well, oxyoxalamides with relatively less hydrophobic groups were incorporated on the amide inhibitors. As seen in compound 19, substitution of a t-butyl group on the oxygen atom of the oxyoxalamide instead of a methyl group of compound 18 lowered inhibition a 1.5-fold, showing that a smaller alkyl group on the oxygen atom is better for inhibition. In addition, oxyoxalamides with N,O-dimethyl (20) or N,N,O-trimethyl (21) yielded a further improved potency (1.5-fold) compared to that of O-methyl derivative (18), implying that the N-substitution of the oxyoxalamide is useful for enhancing inhibition for the target enzyme. On the other hand, an oxyoxalamide with a polar group on the oxygen atom (22) led to a complete loss in inhibition, suggesting that non-polar small alkyl groups such as a methyl group are optimal substituents for the oxyoxalamide function to produce significant inhibition potency. Interestingly, when the adamantyl group in the left side of the amide primary pharmacophore of compounds 20 and 21 was modified by an adamantylmethyl group (23 and 24), inhibition potency was enhanced up to 25-fold. In addition, replacement of the benzyl group in the right side of the amide function of compounds 23 and 24 with a phenyl group (25 and 26) improved inhibition potency approximately 2-fold. This is approximately 50-fold enhanced inhibition compared to that of compound 21, indicating that optimizing around the amide pharmacophore is a useful approach for yielding potent amideoxyoxalamide inhibitors. Production of highly potent amideoxyoxalamide inhibitors depends on the structure of the primary pharmacophore more than that of secondary pharmacophore. It also was found that N-methylation of the both nitrogen atoms of the oxyoxalamide is especially important for affording potent amide-oxyoxalamide derivatives as shown by comparing compound 24 to 23. Compound 26 with N,N,O-trimethylated oxyoxalamide had an 8-fold better inhibition than compound 25 with N,O-dimethylated oxyoxalamide, again showing the effectiveness of N-substitution of the both nitrogen atoms of the oxyoxalamide for the production of further improved amide–oxyoxalamide inhibitors. When water solubility of the potent compounds (23–26) was measured, a 2–15-fold enhancement was obtained in comparison with that of the amide inhibitor with no oxyoxalamide function (17). Furthermore, the solubility was comparable to those of previously reported soluble amide-(2)23 and urea-based (IK950)14 inhibitors. It was also found that inhibitions obtained from compounds 24 and 26 were similar to those of potent inhibitors 2 and IK950. This implies that the incorporation of the substituted oxyoxalamide function as a secondary pharmacophore is effective for not only improving inhibition potency, but also significantly increasing water solubility of amide compounds. Comparing compound 24 to 23, it was found that N-methylation of the both nitrogen atoms of the oxyoxalamide function provides better water solubility. A similar result was also observed between compounds 25 and 26, suggesting that the trimethylated oxyoxalamide is especially effective for improving solubility in water.
Table 4.
Inhibition of human sEH by urea derivatives substituted with oxyoxalamide function
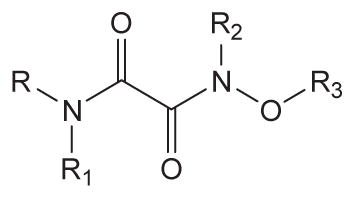
| ||||||
|---|---|---|---|---|---|---|
| No. | R | R1 | R2 | R3 | Human sEH IC50 a (nM) | Solubilityb (μM) |
| 27c |
|
16 | 20 | |||
| 28 |
|
H | H | CH3 | 6.6 | 312 |
| 29 |
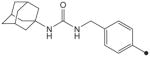
|
H | H |
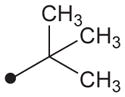
|
19 | 39 |
| 30 |
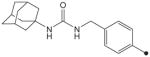
|
H | CH3 | CH3 | 5.0 | 312 |
| 31 |
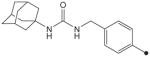
|
CH3 | CH3 | CH3 | 1.2 | 625 |
| 32 |
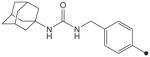
|
H | H |
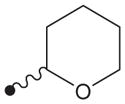
|
19 | 78 |
| AUDAd | 3.2 | 63 | ||||
| IK950d | 14 | 625 | ||||
Test compounds prepared in DMSO was reacted with human sEH (1 nM) for 10 min in 25 mM Bis–Tris/HCl buffer (202 μL; pH 7.0) at 30 °C. The fluorescent substrate (CMNPC; [S] = 5 μM) was then introduced to the incubation mixture. Inhibition potency against the human sEH was determined by measuring the appearance of the 6-methoxy-2-naphthaldehyde with an excitation wavelength of 330 nm and an emission wavelength of 465 nm for 10 min on a fluorometer. Results are averages of three separate measurements. See the Supplementary data for the detailed procedures.
Water solubility was determined by adding a variety of concentrations of a test compound prepared in DMSO to 0.1 M sodium phosphate buffer (pH 7.4) in a final ratio of 5:95 (v/v). The turbidity of the water solution was measured at 650 nm to determine solubility in water. Results are the average of triplicate determinations.
Urea inhibitor with no substitution by oxyoxalamide function, which was synthesized by the reaction of 1-adamantyl isocyanate with benzyl amine in DMF in 100% yield.13
The results in Table 3 show that attachment of a substituted oxyoxalamide as a secondary pharmacophore to amide inhibitors results in significant enhancements in inhibition potency and water solubility. Next, in order to see if the incorporation of the oxyoxalamide function to urea inhibitors affects their inhibition and water solubility, several urea-oxyoxalamide derivatives were synthesized as shown in Table 4 and Fig. 2B. As seen in compound 28, an O-methyl-oxyoxalamide substitution resulted in a 2.5-fold better inhibitor when incorporated to urea compound 27, indicating that the oxyoxalamide function is useful for further improving inhibition potency of potent urea compounds. On the other hand, O-t-butyl-oxyoxalamide (29) did not provide an increase in inhibition, implying that a smaller alkyl group like a methyl on the oxygen atom is better than a relatively bulky t-butyl group for yielding improved urea inhibitors, which is similar to that observed in the corresponding amide derivatives 18 and 19 in Table 3. N,O-Dimethyl (30) and N,N,O-trimethyl (31) oxyoxalamides made a 3-fold and a 13-fold improvements in inhibition, respectively, in comparison with that of compound 27. Interestingly, N-methylation of the both nitrogen atoms of the oxyoxalamide function was also found to be important for yielding highly potent inhibition in urea-oxyoxalamide derivatives as seen in compound 31, which is the same result as that observed in the corresponding amide derivative (26). On the other hand, a polar substitution in the oxyoxalamide (32) was not effective for making an improved urea inhibitor, which is also similar to that gained with amide derivative 22 in Table 3. Furthermore, approximately 2–30-fold enhancement in water solubility was gained from the substituted urea compounds (28–32) when compared to that of compound 27, making the substituted oxyoxalamide function an effective secondary pharmacophore for improving both inhibition and solubility. Among the derivatives tested, the most potent compound 31 with N,N,O-trimethyl-oxyoxalamide was most soluble in water, which is a 10-fold higher solubility than that of a urea inhibitor, AUDA. Moreover, the solubility of compound 31 was same as that of IK950 known as a soluble urea inhibitor useful for various in vivo studies. In addition, a 3–12-fold enhancement in inhibition was observed in compound 31 when compared to those of AUDA and IK950. These results suggest that N-methylation of the both nitrogen atoms of the oxyoxalamide function is highly effective for both of inhibition and solubility of urea compounds as well, as that observed in the corresponding amide derivatives 24 and 26.
3. Conclusions
This study investigated whether the oxyoxalamide function works as an effective primary and/or secondary pharmacophore to inhibit the human sEH. In order to first see its potential to be a primary pharmacophore, a series of oxyoxalamides substituted with alkyl, cycloalkyl, aryl, or substituted aryl groups were synthesized (Table 1 and Fig. 2A). The inhibition results indicated that a 2-adamantyl group (6) is the most effective left side substituent of the oxyoxalamide function for producing significant inhibition potency for the target human enzyme. Interestingly, the 1-adamantyl group (4) or other hydrophobic groups (3 and 5), which are highly useful for yielding potent inhibition in amide or urea inhibitors, led to dramatically reduced or total lack of inhibition potency with the oxyoxalamide function. When the right side of the oxyoxalamide function was optimized with aryl containing substituents effective for affording potent amide or urea inhibitors, significant inhibition potency was gained from a non-substituent benzyl group (6). In the presence of a functional group on the benzene ring (8–10) or a longer alkyl group (11 and 12) between the benzene ring and the oxyoxalamide function, which are reported as necessary elements for the production of potent amide or urea inhibitors, a dramatic loss in inhibition was induced with the oxyoxalamide pharmacophore. In addition, N-substitution of the oxyoxalamide function of compound 6 was not effective for improving inhibition for the target enzyme (13 and 14). Overall structure–activity relationship (SAR) results demonstrate that the substituted oxyoxalamide function is a promising primary pharmacophore for human sEH inhibitors and the structural requirements for producing significant inhibition from oxyoxalamide pharmacophore are different from those for other primary pharmacophores such as amides and ureas. Furthermore, compound 6 found in the present study can be a novel lead structure for the development of further improved oxyoxalamide and other related derivatives. Next, a series of substituted oxyoxalamides were incorporated into inhibitors with an amide or urea primary pharmacophore to investigate whether the oxyoxalamide function acts as an effective secondary pharmacophore for improving inhibition potency and/or water solubility of them. The SAR results in Tables 3 and 4 and Fig. 2B indicated that substituted oxyoxalamide function is a useful secondary pharmacophore for enhancing inhibition potency of amide and urea inhibitors. Substitution of the oxyoxalamide with smaller alkyl groups such as a methyl group was highly useful for improving inhibition potency (18, 20–21, 23–26, 28, 30, and 31). However, oxyoxalamides with a larger alkyl group (19 and 29) or a polar group (22 and 32) led to a significant loss in inhibition. These data also indicate that a N,N,O-trimethylated oxyoxalamide is especially effective for making potent amide- or urea–oxyoxalamide derivatives (24, 26, and 31). In compounds with N,O-dimethylated (23, 25, and 30) or O-mono-methylated (28) oxyoxalamide, reduced inhibition of up to a 10-fold was observed, suggesting that the incorporation of three methyl groups on the oxyoxalamide is important for yielding highly potent amide- or urea–oxyoxalamide compounds. In addition, the potent compounds with oxyoxalamide function in Tables 3 and 4 had a 2–20-fold enhanced water solubility. Especially, the highest increase in water solubility was gained from compounds with the trimethylated oxyoxalamide function, suggesting that substituted oxyoxalamides incorporated into amide or urea inhibitors as a secondary pharmacophore are useful for improving not only inhibition but also solubility. The resulting compounds found in the present study will be the basis for the design of selective amide- or urea–oxyoxalamide inhibitors with improved physical properties, which will be useful for the development of intravenous or orally available compounds for hypertension, vascular inflammation, and other cardiovascular disorders related by endogenous mediators, including EETs and other epoxyfatty acids.
4. Experimental section
4.1. Chemistry
Unless otherwise noted, all materials were purchased from commercial suppliers and used without further purification. Purity and characterization of compounds were established by a combination of TLC, LC–MS, melting point, and NMR analysis described below. All melting points were determined with a Stuart SMP3 apparatus (A.H. Thomas Co.) and are uncorrected. 1H NMR spectra were recorded on a Digital Avance 400 MHz spectrometer (Bruker Analytik GmbH), using tetramethylsilane (TMS) as an internal standard. 13C NMR spectra were recorded on a JEOL JNM-EX400 spectrometer (JEOL Ltd, Japan), using TMS as an internal standard. High resolution mass spectra were measured by LC-MS (Xevo Q-TOFMS; Waters, UK) using positive mode electrospray ionization. Thin-layer chromatography (TLC) was performed on EMD precoated silica gel 60 F254 plates, and spots were visualized with UV light and stained with basic KMnO4. The purity of all final compounds was determined to be greater than 95% unless otherwise indicated. Synthetic methods are described for representative compounds.
4.1.1. N1-(Adamant-2-yl)-N2-(benzyloxy)oxalamide (6)
To a solution of adamant-2-ylamine hydrochloride (1.00 g, 5.53 mmol) and triethylamine (1.47 g, 10.6 mmol) in dichloromethane (20 mL) was added ethyl (chlorocarbonyl)formate (0.73 g, 5.32 mmol) in dichloromethane (2 mL) at 0 °C. After stirring overnight at room temperature, the product was extracted with diethyl ether (50 mL), washed with an aqueous solution of 1 N HCl (30 mL) and water (50 mL × 2), and dried over MgSO4. The ether solution was evaporated to dryness and the residue was used for the next reaction without purification. An aqueous solution of 1 N NaOH (2 mL) was added dropwise to a solution of the above residue in ethanol (10 mL) and the reaction mixture was stirred for 30 min at room temperature. After the reaction was acidified to pH 2 by adding an aqueous solution of 1 N HCl, the acid product was extracted with dichloromethane (50 mL × 2). The combined organic solution was washed with water (50 mL × 2), dried over MgSO4, and evaporated to dryness. To the residue (0.19 g, 0.85 mmol) in dichloromethane (20 mL) was added 4-dimethylaminopyridine (DMAP; 0.10 g, 0.85 mmol) and benzyloxyamine hydrochloride (0.14 g, 0.85 mmol) at room temperature. After the reaction was stirred for 5 min, 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide (EDCI; 0.16 g, 0.85 mmol) was added to the reaction mixture at room temperature. After stirring overnight, the product was extracted with diethyl ether (50 mL). The organic layer was washed with an aqueous solution of 1 N HCl (30 mL × 2) and water (50 mL × 2), dried over MgSO4, and evaporated. The residue was purified by using silica gel column chromatography (hexane/ethyl acetate = 3:1) to afford compound 6 as a solid in 85% yield. 1H NMR δ (CDCl3): 1.65–1.68 (3H, m), 1.84–1.88 (9H, m), 1.94 (2H, s), 3.99 (1H, s), 4.97 (2H, s), 7.37–7.43 (5H, m), 7.70 (1H, s), 9.64 (1H, s). 13C NMR δ (CDCl3): 26.9, 31.6, 36.9, 37.3, 53.8, 78.7, 128.7, 129.1, 129.2, 134.3, 156.9, 157.7. Purity: >90%. HRMS (ESI) m/z calcd for C19H24N2O3 [M+H]+ 329.1865, found [M+H]+ 329.1862, mp137 °C.
Compounds 3–5 and 7–15 were synthesized in the same procedure used for the preparation of compound 6 by using a corresponding alkyl- or cycloalkyl-amine and a substituted oxyamine instead of adamant-2-ylamine and benzyloxyamine hydrochloride.
4.1.2. N1-(Benzyloxy)-N2-(3-phenylpropyl)oxalamide (3)
1H NMR δ (CDCl3): 1.89 (2H, t, J = 7.3 Hz), 2.66 (2H, t, J = 7.3 Hz), 3.32 (2H, q, J = 7.3 Hz), 4.96 (2H, s), 7.16–7.26 (4H, m), 7.27–7.31 (3H, m), 7.37–7.40 (5H, m), 9.56 (1H, s). HRMS (ESI) m/z calcd for C18H20N2O3 [M+H]+ 313.1552, found [M+H]+ 313.1543, mp 123 °C.
4.1.3. N1-(Adamant-1-yl)-N2-(benzyloxy)oxalamide (4)
1H NMR δ (CDCl3): 1.68 (6H, s), 2.00 (6H, s), 2.10 (3H, s), 4.86 (2H, s), 7.10 (1H, s), 7.37–7.42 (5H, m), 9.58 (1H, s). HRMS (ESI) m/z calcd for C19H24N2O3 [M+H]+ 329.1865, found [M+H]+ 329.1855, mp 178 °C.
4.1.4. N1-(Adamant-1-ylmethyl)-N2-(benzyloxy)oxalamide (5)
1H NMR δ (CDCl3): 1.49 (6H, br s), 1.73 (6H, br s), 1.99 (3H, br s), 2.97 (2H, d, J = 6.8 Hz), 4.97 (2H, s), 7.38–7.42 (5H, m), 9.60 (1H, s). HRMS (ESI) m/z calcd for C20H26N2O3 [M+H]+ 343.2022, found [M+H]+ 343.2023, mp 195 °C.
4.1.5. N1-(Adamant-2-yl)-N2-(phenyloxy)oxalamide (7)
1H NMR δ (CDCl3): 1.66–1.69 (3H, m), 1.87–1.90 (9H, m), 1.98 (2H, s), 4.04 (1H, s), 7.07–7.11 (3H, m), 7.31–7.35 (2H, m), 7.70 (1H, s), 10.14 (1H, s). HRMS (ESI) m/z calcd for C18H22N2O3 [M+H]+ 315.1709, found [M+H]+ 315.1711, mp 187 °C.
4.1.6. N1-(Adamant-2-yl)-N2-(4-methoxycarbonylbenzyloxy)oxalamide (8)
1H NMR δ (CDCl3): 1.63–1.68 (3H, m), 1.86–1.89 (9H, m), 1.94 (2H, s), 3.93 (3H, s), 4.01 (1H, s), 5.02 (2H, s), 7.50 (2H, d, J = 8.3 Hz), 7.71 (1H, s), 8.06 (2H, d, J = 8.3 Hz), 9.70 (1H, s). HRMS (ESI) m/z calcd for C21H26N2O5 [M+H]+ 387.1920, found [M+H]+ 387.1916, mp 175 °C.
4.1.7. N1-(Adamant-2-yl)-N2-(4-nitrobenzyloxy)oxalamide (9)
1H NMR δ (CDCl3): 1.64–1.67 (3H, m), 1.86–1.88 (9H, m), 1.94 (2H, s), 4.00 (1H, s), 5.09 (2H, s), 7.52 (2H, d, J = 8.3 Hz), 7.76 (1H, s), 8.23 (2H, d, J = 8.3 Hz), 11.74 (1H, s). HRMS (ESI) m/z calcd for C19H23N3O5 [M+H]+ 374.1716, found [M+H]+ 374.1714, mp >210 °C.
4.1.8. N1-(Adamant-2-yl)-N2-(4-chlorobenzyloxy)oxalamide (10)
To a solution of 4-chlorobenzyl bromide (4.00 g, 0.01947 mol) and N-hydroxyphthalimide (3.18 g, 0.01947 mol) in DMF (40 mL) was added triethylamine (3.94 g, 0.03894 mol) at 0 °C. After stirring overnight at room temperature, the product was extracted with diethyl ether (80 mL × 2). The organic layer was washed with water (80 mL × 2), dried over MgSO4, and evaporated to dryness. To the residue in 10% methanol in trichloromethane (30 mL) was added hydrazine hydrate (0.93 g, 0.02912 mol) at room temperature. The reaction was stirred overnight and the product was extracted with diethyl ether (80 mL × 2). The combined organic layer was washed with water (80 mL × 2), dried over MgSO4, and evaporated. The residue was purified by using silica gel column chromatography (hexane/ethyl acetate = 5:1) to give (4-chlorobenzyl) oxyamine in 65% yield. Then, compound 10 was prepared in the same manner used for the preparation of compound 6 by using this oxyamine. 1H NMR δ (CDCl3): 1.65–1.68 (3H, m), 1.83–1.89 (9H, m), 1.94 (2H, s), 4.00 (1H, s), 4.93 (2H, s), 7.26 (2H, s), 7.36 (2H, s), 7.68 (1H, s), 9.60 (1H, s). HRMS (ESI) m/z calcd for C19H23ClN2O3 [M+H]+ 363.1475, found [M+H]+ 363.1472, mp 173 °C.
4.1.9. N1-(Adamant-2-yl)-N2-(2-phenylethyloxy)oxalamide (11)
(2-Phenylethyl)oxyamine was prepared in the same procedure used for the preparation of compound 10 by using 2-phenylethyl bromide instead of 4-chlorobenzyl bromide. Compound 11 was then synthesized in the same manner described in the preparation of compound 6 by using (2-phenylethyl)oxyamine. 1H NMR δ (CDCl3): 1.65–1.68 (3H, m), 1.83–1.89 (9H, m), 1.95 (2H, s), 3.02 (2H, t, J = 7.1 Hz), 4.01 (1H, s), 4.20 (2H, t, J = 7.1 Hz), 7.22–7.24 (3H, m), 7.26–7.33 (2H, m), 7.70 (1H, s), 9.71 (1H, s). Purity: >90%. HRMS (ESI) m/z calcd for C20H26N2O3 [M+H]+ 343.2022, found [M+H]+ 343.2015, mp >210 °C.
4.1.10. N1-(Adamant-2-yl)-N2-(3-phenylpropyloxy)oxalamide (12)
(3-Phenylpropyl)oxyamine was prepared in the same procedure used for the preparation of compound 10 by using 3-phenylpropyl bromide instead of 4-chlorobenzyl bromide. Compound 12 was then synthesized in the same manner described in the preparation of compound 6 by using (3-phenylpropyl)oxyamine. 1H NMR δ (CDCl3): 1.66–1.69 (3H, m), 1.83–1.89 (9H, m), 1.98 (2H, s), 2.08 (2H, quint, J = 7.3 Hz), 2.74 (2H, t, J = 7.3 Hz), 4.06 (1H, s), 4.29 (2H, t, J = 7.3 Hz), 7.18–7.26 (3H, m), 7.28–7.30 (2H, m), 7.42 (1H, s), 9.70 (1H, s). HRMS (ESI) m/z calcd for C21H28N2O3 [M+H]+ 357.2178, found [M+H]+ 357.2178, mp 53 °C.
4.1.11. N1-(Adamant-2-yl)-N2-(benzyloxy)-N2-methyloxalamide (13)
A mixture of compound 6 (0.51 g, 1.55 mmol), potassium carbonate (0.43 g, 3.10 mmol), and iodomethane (0.33 g, 2.32 mmol) in DMF (20 mL) was stirred overnight at room temperature. The product was extracted with diethyl ether (80 mL). The organic solution was washed with water (80 mL × 2), dried over MgSO4, and evaporated. The residue was purified by using silica gel column chromatography (hexane/ethyl acetate = 4:1) to afford compound 13 as a solid in 55% yield. 1H NMR δ (CDCl3): 1.61–1.66 (3H, m), 1.84–1.88 (9H, m), 1.92 (2H, s), 4.01 (1H, s), 4.10 (3H, s), 5.06 (2H, s), 6.93 (1H, s), 7.35–7.38 (5H, m). HRMS (ESI) m/z calcd for C20H26N2O3 [M+H]+ 343.2022, found [M+H]+ 343.2016, mp 110 °C.
4.1.12. N1-(Adamant-2-yl)-N2-methyl-N2-(methyloxy)oxalamide (14)
1H NMR δ (CDCl3): 1.64–1.67 (3H, m), 1.86–1.88 (9H, m), 1.96 (2H, s), 3.76 (3H, s), 3.79 (3H, s), 4.00 (1H, s), 6.81 (1H, s). Purity: >90%. HRMS (ESI) m/z calcd for C14H22N2O3 [M+H]+ 267.1709, found [M+H]+ 267.1707, mp 148 °C.
4.1.13. N1-(Adamant-2-yl)-N2-(tetrahydro-2H-pyran-2-yloxy)oxalamide (15)
1H NMR δ (CDCl3): 1.62–1.70 (8H, m), 1.81–1.88 (10H, m), 1.95 (2H, s), 3.67 (1H, s), 3.99–4.04 (2H, m), 5.04 (1H, s), 7.72 (1H, s), 9.96 (1H, s). HRMS (ESI) m/z calcd for C17H26N2O4 [M+H]+ 323.1971, found [M+H]+ 323.1973, mp 137 °C.
4.1.14. N-(Benzyloxy)-2-(adamant-2-ylamino)acetamide (16)
A mixture of methyl 2-bromoacetate (1.22 g, 7.99 mmol), adamant-2-ylamine hydrochloride (1.50 g, 7.99 mmol), and potassium carbonate (2.21 g, 15.9 mmol) in DMF (30 mL) was stirred overnight at room temperature. The product was extracted with diethyl ether (80 mL × 2). The organic solution was washed with water (100 mL × 2), dried over MgSO4, and evaporated to dryness. To the residue in methanol (15 mL) was added an aqueous solution of 1 N NaOH (4 mL) at room temperature, and the reaction was stirred for 1 h. After evaporating the reaction mixture to dryness, the residue was used for the next reaction without further purification. A mixture of the acid residue (0.79 g, 3.75 mmol), DMAP (0.46 g, 3.75 mmol), and benzyloxyamine hydrochloride (0.60 g, 3.75 mmol) in dichloromethane (30 mL) was stirred for 5 min at room temperature. To this reaction mixture was added EDCI (0.72 g, 3.75 mmol) at room temperature. After stirring overnight, the product was extracted with diethyl ether (80 mL × 2). The combined organic solution was washed with an aqueous solution of 1 N HCl (40 mL) and water (80 mL × 2), dried over MgSO4, and evaporated. The residue was purified by using silica gel column chromatography (hexane/ethyl acetate = 1:1) to give compound 16 as a yellowish oil in 70% yield. 1H NMR δ (CDCl3): 1.44–1.47 (2H, m), 1.61–1.72 (10H, m), 1.83–1.89 (4H, m), 2.53 (1H, s), 3.33 (2H, s), 4.95 (2H, s), 7.35–7.42 (5H, m). Purity: >90%. HRMS (ESI) m/z calcd for C19H26N2O2 [M+H]+ 315.2072, found [M+H]+ 315.2071.
4.1.15. N-(4-(N2-Methyloxyoxalamido)benzyl)adamantanecarboxamide (18)
1H NMR δ (CDCl3): 1.68–1.71 (6H, m), 1.88 (6H, s), 2.05 (3H, s), 3.88 (3H, s), 4.42 (2H, s), 5.88 (1H, s), 7.27 (2H, d, J = 10 Hz), 7.57 (2H, d, J = 10 Hz), 9.09 (1H, s), 9.82 (1H, s). HRMS (ESI) m/z calcd for C21H27N3O4 [M+H]+ 386.2080, found [M+H]+ 386.2076, mp >210 °C.
4.1.16. N-(4-(N2-t-Butyloxyoxalamido)benzyl)adamantanecarboxamide (19)
1H NMR δ (CDCl3): 1.35 (9H, s), 1.68–1.72 (6H, m), 1.88 (6H, s), 2.04 (3H, s), 4.43 (2H, s), 5.88 (1H, s), 7.29 (2H, d, J = 10 Hz), 7.57 (2H, d, J = 10 Hz), 9.16 (1H, s), 9.42 (1H, s). HRMS (ESI) m/z calcd for C24H33N3O4 [M+H]+ 428.2549, found [M+H]+ 428.2546, mp >210 °C.
4.1.17. N-(4-(N2-Methyl- N2-(methyloxy)oxalamido)benzyl)adamantanecarboxamide (20)
1H NMR δ (CDCl3): 1.66–1.76 (6H, m), 1.88 (6H, s), 2.04 (6H, s), 3.29 (3H, s), 3.83 (3H, s), 4.41 (2H, s), 7.25 (2H, d, J = 8.3 Hz), 7.55 (2H, d, J = 8.3 Hz), 9.34 (1H, s). HRMS (ESI) m/z calcd for C22H29N3O4 [M+H]+ 400.2236, found [M+H]+ 400.2239, mp 181 °C.
4.1.18. N-(4-(N1-Methyl-N2-methyl- N2-(methyloxy)oxalamido)- benzyl)adamantanecarboxamide (21)
1H NMR δ (CDCl3): 1.68–1.72 (6H, m), 1.88 (6H, s), 2.04 (6H, s), 2.96 (3H, s), 3.31 (3H, s), 3.79 (3H, s), 4.43 (2H, s), 5.92 (1H, s), 7.24–7.26 (4H, m). HRMS (ESI) m/z calcd for C23H31N3O4 [M+H]+ 414.2393, found [M+H]+ 414.2386, mp 170 °C.
4.1.19. N-(4-(N2-(Tetrahydro-2H-pyran-2-yloxy)oxalamido)benzyl) adamantanecarboxamide (22)
1H NMR δ (CDCl3): 1.61–1.65 (6H, m), 1.68–1.72 (6H, m), 1.88 (6H, s), 2.04 (6H, s), 3.66–3.70 (2H, m), 4.02 (1H, t, J = 6.8 Hz), 4.42 (2H, s), 5.90 (1H, s), 7.25 (2H, d, J = 8.3 Hz), 7.57 (2H, d, J = 8.3 Hz), 9.15 (1H, s), 9.98 (1H, s). Purity: >90%. HRMS (ESI) m/z calcd for C25H33N3O5 [M+H]+ 456.2498, found [M+H]+ 456.2495, mp 195 °C.
4.1.20. N1-(4-((2-Adamantylacetamido)methyl)phenyl)-N2-methyl- N2-methyloxyoxalamide (23)
1H NMR δ (CDCl3): 1.62–1.71 (12H, m), 1.96–1.97 (5H, s), 3.29 (3H, s), 3.83 (3H, s), 4.42 (2H, s), 5.58 (1H, s), 7.27 (2H, d, J = 8.3 Hz), 7.56 (2H, d, J = 8.3 Hz), 9.33 (1H, s). HRMS (ESI) m/z calcd for C23H31N3O4 [M+H]+ 414.2393, found [M+H]+ 414.2390, mp 203 °C.
4.1.21. N1-(4-((2-Adamantylacetamido)methyl)phenyl)-N1-methyl- N2-methyl-N2-methyloxyoxalamide (24)
1H NMR δ (CDCl3): 1.63–1.72 (12H, m), 1.96–1.97 (5H, s), 2.95 (3H, s), 3.33 (3H, s), 3.80 (3H, s), 4.43 (2H, s), 5.65 (1H, s), 7.23–7.30 (4H, m). 13C NMR δ (CDCl3): 28.6, 31.2, 32.8, 35.7, 36.7, 42.6, 42.9, 51.7, 62.2, 126.3, 128.8, 138.6, 140.7, 164.3, 165.6, 170.9. HRMS (ESI) m/z calcd for C24H33N3O4 [M+H]+ 428.2549, found [M+H]+ 428.2545, mp 97 °C.
4.1.22. N1-(4-(2-Adamantylacetamido)phenyl)-N2-methyl-N2-(methyloxy)oxalamide (25)
After a mixture of adamant-1-ylacetic acid (1.87 g, 9.60 mmol), DMAP (1.17 g, 9.60 mmol), and 4-(tert-butyloxycarbonyl)aminoaniline (2.00 g, 9.60 mmol) in dichloromethane (30 mL) was stirred for 10 min at room temperature, EDCI (1.84 g, 9.60 mmol) was added portionwise to the reaction mixture. The reaction was stirred overnight and the product was then extracted with ethyl acetate (80 mL ×2). The organic solution was washed with an aqueous solution of 1 N HCl (50 mL) and water (80 mL ×2), dried over MgSO4, and evaporated. To the residue washed with diethyl ether (20 mL) in dichloromethane (30 mL) was added an aqueous solution of 4 N HCl (10 mL) at room temperature. After stirring overnight, the product was extracted with ethyl acetate (80 mL ×2). The organic layer was washed with water (80 mL ×2), dried over MgSO4, and evaporated to dryness. The residue was used for the next reaction without further purification. To a solution of the residue (1.42 g, 5.27 mmol) and triethylamine (0.80 g, 7.90 mmol) in dichloromethane (30 mL) was added ethyl (chlorocarbonyl)formate (0.72 g, 5.27 mmol) at 0 °C. After stirring overnight at room temperature, the product was extracted with ethyl acetate (80 mL ×2). The organic solution was washed with an aqueous solution of 1 N HCl (40 mL) and water (80 mL ×2), dried over MgSO4, and evaporated to dryness. An aqueous solution of 1 N NaOH (5 mL) was added to a solution of the residue in ethanol (10 mL) at room temperature. After the reaction was stirred overnight at room temperature and acidified to pH 2 by adding an aqueous solution of 1 N HCl, the product was extracted with ethyl acetate (80 mL ×2). The organic solution was washed with water (80 mL ×2), dried over MgSO4, and evaporated to dryness. Continuously, to a solution of the residue (1.22 g, 3.42 mmol), DMAP (0.42 g, 5.13 mmol), methoxymethylamine hydrochloride (0.50 g, 5.13 mmol), and triethylamine (0.52 g, 5.13 mmol), which was stirred for 10 min, was added EDCI (0.98 g, 5.13 mmol) at room temperature. After stirring overnight, the product was extracted with ethyl acetate (80 mL ×2). The organic solution was washed with water (80 mL ×2), dried over MgSO4, and evaporated. The residue was purified by using silica gel column chromatography (hexane/ethyl acetate = 1:1) to afford compound 25 in 45% yield. 1H NMR δ (CDCl3): 1.63–1.69 (12H, m), 2.04 (3H, s), 2.09 (2H, s), 3.28 (3H, s), 3.82 (3H, s), 7.06 (1H, s), 7.51–7.57 (4H, m), 9.33 (1H, s). HRMS (ESI) m/z calcd for C22H29N3O4 [M+H]+ 400.2236, found [M+H]+ 400.2239, mp 208 °C.
Compounds 18–23, 28–30 and 32 were prepared in the same manner used for the synthesis of compound 25 by using 4-aminobenzylamine, adamant-1-ylcarboxylic acid or adamant-1-yl isocyanate, and a corresponding substituted oxyamine instead of 4-(tert-butyloxycarbonyl)-aminoaniline and methoxymethylamine hydrochloride.
4.1.23. N1-(4-(2-Adamantylacetamido)phenyl)-N1-methyl-N2- methyl-N2-(methyloxy)oxalamide (26)
A mixture of compound 25 (0.30 g, 0.75 mmol), potassium carbonate (0.16 g, 1.12 mmol), and iodomethane (0.11 g, 1.12 mmol) in DMF (15 mL) was stirred overnight at room temperature. The product was extracted with diethyl ether (80 mL). The organic solution was washed with water (80 mL ×2), dried over MgSO4, and evaporated. The residue was purified by using silica gel column chromatography (hexane/ethyl acetate = 1:1) to give compound 26 in 60% yield. 1H NMR δ (CDCl3): 1.64–1.69 (12H, m), 1.99 (3H, s), 2.09 (2H, s), 2.98 (3H, s), 3.30 (3H, s), 3.80 (3H, s), 7.18 (2H, d, J = 8.3 Hz), 7.48 (2H, d, J = 8.3 Hz). 13C NMR δ (CDCl3): 28.6, 31.3, 33.3, 35.7, 36.6, 42.5, 52.7, 62.2, 120.2, 126.8, 136.9, 137.9, 164.2, 165.7, 169.6. Purity: >90%. HRMS (ESI) m/z calcd for C23H31N3O4 [M+H]+ 414.2393, found [M+H]+ 414.2391, mp 179 °C.
Compounds 21, 24, and 31 were synthesized in the same manner used for the preparation of compound 26 by using compound 20, 23, or 30, respectively, instead of compound 25.
4.1.24. 1-(4-(N2-Methyloxyoxalamido)benzyl)-3-adamantylurea (28)
Compound 28 was prepared in the same methods described in the syntheses of compounds 25 and 30. 1H NMR δ (CDCl3): 1.65 (6H, s), 1.95 (6H, s), 2.06 (3H, s), 3.83 (3H, s), 4.09 (1H, s), 4.30 (2H, s), 4.44 (1H, s), 7.25 (2H, d, J = 8.3 Hz), 7.53 (2H, d, J = 8.3 Hz), 9.12 (1H, s), 9.35 (1H, s). HRMS (ESI) m/z calcd for C21H28N4O4 [M+H]+ 401.2189, found [M+H]+ 401.2187, mp 177 °C.
4.1.25. 1-(4-(N2-t-Butyloxyoxalamido)benzyl)-3-adamantylurea (29)
Compound 29 was prepared in the same methods described in the syntheses of compounds 25 and 30. 1H NMR δ (CDCl3): 1.56 (9H, s), 1.66 (6H, s), 1.95 (6H, s), 2.05 (3H, s), 4.08 (1H, s), 4.30 (2H, s), 4.43 (1H, s), 7.30 (2H, d, J = 8.3 Hz), 7.57 (2H, d, J = 8.3 Hz), 9.16 (1H, s), 9.45 (1H, s). HRMS (ESI) m/z calcd for C24H34N4O4 [M+H]+ 443.2658, found [M+H]+ 443.2654, mp >210 °C.
4.1.26. 1-(4-(N2-Methyl-N2-(methyloxy)oxalamido)benzyl)-3-adamantylurea (30)
To a solution of adamant-1-yl isocyanate (0.73 g, 4.09 mmol) in DMF (15 mL) was added dropwise 4-aminobenzylamine (0.50 g, 4.09 mmol) in DMF (2 mL) at 0 °C. After stirring overnight at room temperature, the product was extracted with diethyl ether (80 mL ×2). The organic layer was washed with water (80 mL ×2), dried over MgSO4, and evaporated to give 1-(adamant-1-yl)-3-(4-aminobenzyl) urea in 100% yield. Compound 30 was synthesized in the same method described in the preparation of compound 25 by using 1-(adamant-1-yl)-3-(4-aminobenzyl)urea instead of the amide-amine intermediate. 1H NMR δ (CDCl3): 1.66 (6H, s), 1.95 (6H, s), 2.04 (3H, s), 3.28 (3H, s), 3.82 (3H, s), 4.09 (1H, s), 4.30 (2H, s), 4.40 (1H, s), 7.30 (2H, d, J = 8.3 Hz), 7.55 (2H, d, J = 8.3 Hz), 9.33 (1H, s). HRMS (ESI) m/z calcd for C22H30N4O4 [M+H]+ 415.2345, found [M+H]+ 415.2347, mp 202 °C.
4.1.27. 1-(4-(N1-Methyl-N2-methyl-N2-(methyloxy)oxalamido)-benzyl)-3-adamantylurea (31)
Compound 31 was prepared in the same methods described in the syntheses of compounds 25 and 30. 1H NMR δ (CDCl3): 1.66 (6H, s), 1.95 (6H, s), 2.04 (3H, s), 2.95 (3H, s), 3.30 (3H, s), 3.80 (3H, s), 4.08 (1H, s), 4.31 (2H, s), 4.44 (1H, s), 7.22–7.29 (4H, m). 13C NMR δ (CDCl3): 29.5, 31.2, 35.7, 36.4, 42.4, 43.5, 51.0, 62.2, 126.1, 128.3, 139.9, 140.4, 157.0, 164.3, 165.7. HRMS (ESI) m/z calcd for C23H32N4O4 [M+H]+ 429.2502, found [M+H]+ 429.2505, mp >210 °C.
4.1.28. 1-(4-(N2-(Tetrahydro-2H-pyran-2-yloxy)oxalamido)benzyl)-3-adamantylurea (32)
Compound 32 was prepared in the same methods described in the syntheses of compounds 25 and 30. 1H NMR δ (CDCl3): 1.61–1.64 (12H, m), 1.93 (6H, s), 2.04 (3H, s), 3.69 (1H, s), 4.09–4.14 (2H, m), 4.25 (2H, s), 4.70 (1H, s),5.19 (1H, s), 7.17 (2H, d, J = 8.3 Hz), 7.49 (2H, d, J = 8.3 Hz), 9.36 (1H, s), 10.56 (1H, s). HRMS (ESI) m/z calcd for C25H34N4O5 [M+H]+ 471.2607, found [M+H]+ 471.2608, mp 202 °C.
5. Biology
5.1. Enzyme preparation
Recombinant human sEH was prepared by using baculovirus expression system as previously reported.25 Briefly, Sf9 insect cells were infected by recombinant baculovirus harboring human sEH gene fused with a 6xHis tag. At 72 h post-infection, the infected cells were homogenized and the recombinant protein was purified by immobilized metal affinity chromatography. After removing the 6xHis tag using the tobacco etch virus protease, human sEH was further purified by anion-exchange chromatography.
5.2. IC50. assay conditions
Standard solutions of compounds in Tables 1–4 were prepared in DMSO. Fluorescent assays were performed by using a substrate (cyano-(2-methoxynaphthalen-6-yl)-methyl trans-(3-phenyl-oxyran-2-yl)-methyl carbonate; CMNPC; [S] = 5 μM) to determine IC50 values of the derivatives.26 Inhibition activity against human sEH (1 nM) was determined by measuring the appearance of the 6-methoxy-2-naphthaldehyde with an excitation wavelength of 330 nm and an emission wavelength of 465 nm for 10 min on a fluorometer (Victor3; PerkinElmer).25,26 The IC50 values were gained by regression of at least six datum points with a minimum of three points in a linear region of the curve. IC50 results are averages of three separate measurements. 12-(3-Adamantan-1-yl-ureido) dodecanoic acid (AUDA)13 in Table 2 was used as a positive control for the inhibition assay in the present study.
5.3. Solubility
Water solubility of amide and urea derivatives in Tables 3 and 4 was determined experimentally by light scattering method in sodium phosphate buffer at 25 ± 1.5 °C. In brief, aqueous solubility was determined by adding varying concentrations of a test compound prepared in DMSO to 0.1 M sodium phosphate buffer (pH 7.4) in a final ratio of 5:95 (v/v). Insolubility of the compound was shown by the increase in turbidity of the water solution. The turbidity was measured as optical density at 650 nm on a SH-8000 microplate reader (Corona Electric, Ibaraki, Japan) at 25 ± 1.5 °C. Results are averages of three separate measurements.
Supplementary Material
Acknowledgments
This work was supported by Hyundai Pharm Research Grant (HOB-024). Partial support was from NIEHS Grant R01 ES002710 and a Grant-in-aid for Young Scientists (B) 23710042 from Japan Society for the Promotion of Science.
Abbreviations
- EETs
epoxyeicosatrienoic acids
- sEH
soluble epoxide hydrolase
- EDCI
1-[3-(dimethylamino)propyl]-3-ethyl-carbodiimide
- DMAP
4-dimethylaminopyridine
- DMF
N,N-dimethylformamide
- CMNPC
cyano-(2-methoxynaphthalen-6-yl)-methyl trans-(3-phenyl-oxyran-2-yl)-methyl carbonate
Footnotes
Supplementary data (1H NMR of compounds 6, 26, and 31, LC–MS analyses, preparation of human sEH, and inhibition assays) associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmc.2013.12.027.
References and notes
- 1.Robinson DR. Am J Med. 1983;75:26. doi: 10.1016/0002-9343(83)90325-x. [DOI] [PubMed] [Google Scholar]
- 2.Miller SB. Semin Arthritis Rheum. 2006;36:37. doi: 10.1016/j.semarthrit.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 3.Peters-Golden M, Gleason MM, Togias A. Clin Exp Allergy. 2006;36:689. doi: 10.1111/j.1365-2222.2006.02498.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pfister SL, Gauthier KM, Campbell WB. Adv Pharmacol. 2010;60:27. doi: 10.1016/B978-0-12-385061-4.00002-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Imig JD, Zhao X, Capdevila JH, Morisseau C, Hammock BD. Hypertension. 2002;39:690. doi: 10.1161/hy0202.103788. [DOI] [PubMed] [Google Scholar]
- 6.Zhao X, Yamamoto T, Newman JW, Kim IH, Watanabe T, Hammock BD, Stewart J, Pollock JS, Pollock DM, Imig JD. J Am Soc Nephrol. 2004;15:1244. [PubMed] [Google Scholar]
- 7.Jung O, Brandes RP, Kim IH, Schweda F, Schmidt R, Hammock BD, Busse R, Fleming I. Hypertension. 2005;45:759. doi: 10.1161/01.HYP.0000153792.29478.1d. [DOI] [PubMed] [Google Scholar]
- 8.Imig JD, Zhao X, Zaharis CZ, Olearczyk JJ, Pollock DM, Newman JW, Kim IH, Watanabe T, Hammock BD. Hypertension. 2006;46:975. doi: 10.1161/01.HYP.0000176237.74820.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schmelzer KR, Kubala L, Newman JW, Kim IH, Eiserich JP, Hammock BD. Proc Natl Acad Sci USA. 2005;102:9772. doi: 10.1073/pnas.0503279102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Inceoglu B, Wagner K, Schebb NH, Morisseau C, Jinks SL, Ulu A, Hegedus C, Rose T, Brosnan R, Hammock BD. Proc Natl Acad Sci USA. 2011;108:5093. doi: 10.1073/pnas.1101073108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu D, Li N, He Y, Timofeyev V, Lu L, Tsai HJ, Kim IH, Tuteja D, Mateo RKP, Singapuri A, Davis BB, Low R, Hammock BD, Chiamvimonvat N. Proc Natl Acad Sci USA. 2006;103:18733. doi: 10.1073/pnas.0609158103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim IH, Morisseau C, Watanabe T, Hammock BD. J Med Chem. 2004;47:2110. doi: 10.1021/jm030514j. [DOI] [PubMed] [Google Scholar]
- 13.Kim IH, Nishi K, Tsai HJ, Bradford T, Koda Y, Watanabe T, Morisseau C, Blanchfield J, Toth I, Hammock BD. Bioorg Med Chem. 2007;15:312. doi: 10.1016/j.bmc.2006.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim IH, Tsai HJ, Nishi K, Kasagami T, Morisseau C, Hammock BD. J Med Chem. 2007;50:5217. doi: 10.1021/jm070705c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kasagami T, Kim IH, Tsai HJ, Nishi K, Hammock BD, Morisseau C. Bioorg Med Chem Lett. 2009;19:1784. doi: 10.1016/j.bmcl.2009.01.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones PD, Tsai HJ, Do Z, Moresseau C, Hammock BC. Bioorg Med Chem Lett. 2006;16:5212. doi: 10.1016/j.bmcl.2006.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hwang SH, Tsai HJ, Liu JY, Morisseau C, Hammock BD. J Med Chem. 2007;50:3825. doi: 10.1021/jm070270t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anandan SK, Gless RD. Bioorg Med Chem Lett. 2010;20:2740. doi: 10.1016/j.bmcl.2010.03.074. [DOI] [PubMed] [Google Scholar]
- 19.Shen HC, Ding FX, Wang S, Xu S, Chen HS, Tong X, Tong V, Mitra K, Kumar S, Zhang X, Chen Y, Zhou G, Pai LY, Alonso-Galicia M, Chen X, Zhang B, Tata JR, Berger JP, Colletti S. Bioorg Med Chem Lett. 2009;19:3398. doi: 10.1016/j.bmcl.2009.05.036. [DOI] [PubMed] [Google Scholar]
- 20.Lo HY, Man CC, Fleck RW, Farrow NA, Ingraham RH, Kukulka A, Proudfoot JR, Betageri R, Kirrane T, Patel U, Sharma R, Hoermann MA, Kabcenell A, Lombaert SD. Bioorg Med Chem Lett. 2010;20:6379. doi: 10.1016/j.bmcl.2010.09.095. [DOI] [PubMed] [Google Scholar]
- 21.Kim IH, Nishi K, Kasagami T, Morisseau C, Liu JY, Tsai HJ, Hammock BD. Bioorg Med Chem Lett. 2012;22:5889. doi: 10.1016/j.bmcl.2012.07.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim IH, Heirtzler FR, Morisseau C, Nishi K, Tsai HJ, Hammock BD. J Med Chem. 2005;48:3621. doi: 10.1021/jm0500929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim IH, Park YK, Hammock BD, Nishi K. J Med Chem. 2011;54:1752. doi: 10.1021/jm101431v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eldrup AB, Soleymanzadeh F, Taylor SJ, Muegge I, Farrow NA, Joseph D, McKellop K, Man CC, Kukulka A, Lombaert SD. J Med Chem. 2009;52:5880. doi: 10.1021/jm9005302. [DOI] [PubMed] [Google Scholar]
- 25.Nishi K, Kim IH, Ma SJ. Protein Expr Purif. 2010;69:34. doi: 10.1016/j.pep.2009.09.013. [DOI] [PubMed] [Google Scholar]
- 26.Morisseau C, Hammock BD. Current Protocols in Toxicology. John Wiley & Sons; New Jersey: 2007. pp. 4.23.1–4.23.18. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.