

Abstract
Background
Mycobacterium abscessus is an emerging opportunistic pathogen which diversity was acknowledged by the recent description of two subspecies accommodating M. abscessus, Mycobacterium bolletii and Mycobacterium massiliense isolates.
Results
Here, genome analysis found 1–8 prophage regions in 47/48 M. abscessus genomes ranging from small prophage-like elements to complete prophages. A total of 20,304 viral and phage proteins clustered into 853 orthologous groups. Phylogenomic and phylogenetic analyses based on prophage region homology found three main clusters corresponding to M. abscessus, M. bolletii and M. massiliense. Analysing 135 annotated Tape Measure Proteins found thirteen clusters and four singletons, suggesting that at least 17 mycobacteriophages had infected M. abscessus during its evolution. The evolutionary history of phages differed from that of their mycobacterial hosts. In particular, 33 phage-related proteins have been horizontally transferred within M. abscessus genomes. They comprise of an integrase, specific mycobacteriophage proteins, hypothetical proteins and DNA replication and metabolism proteins. Gene exchanges, loss and gains which occurred in M. abscessus genomes have been driven by several mycobacteriophages.
Conclusions
This analysis of phage-mycobacterium co-evolution suggests that mycobacteriophages are playing a key-role in the on-going diversification of M. abscessus.
Reviewers
This article was reviewed by Eric Bapteste, Patrick Forterre and Eugene Koonin.
Keywords: Mycobacterium abscessus, Mycobacterium bolletii, Mycobacterium massiliense, Prophages, Mycobacteriophages
Background
Mycobacterium abscessus is a non-tuberculous species comprising emerging opportunistic pathogens [1] responsible for sporadic cases and outbreaks of skin and soft-tissue infections following surgical and cosmetic practices [2-4]; catheter-related bacteremia [5,6]; and respiratory tract infections in patients with underlying lung disorders, particularly cystic fibrosis [7-13]. M. abscessus is broadly resistant to antibiotics and the cure of localized M. abscessus infection may require surgery [14].
Previous phenotypic [3] and genetic [15-17] analyses showed diversity among collections of M. abscessus isolates acknowledged by the description of two subspecies, M. abscessus subsp. abscessus and M. abscessus subsp. bolletii[18-20]. Later taxon accommodates mycobacteria previously refered as Mycobacterium bolletii[21] and Mycobacterium massiliense[22]. M. abscessus can therefore be viewed as a complex of at least three different organisms M. abscessus, M. bolletii and M. massiliense and this nomenclature will be retained in this paper.
There are a few data regarding mycobacteriophages in M. abscessus complex including a 81-kb prophage in the reference M. abscessus genome [1]. Also, we recently resolved the electron microscopy 3D structure of a M. bolletii mycobacteriophage named Araucaria [23]. However, the repertoire of M. abscessus phages and their evolutionary history within this bacterium is unknown and no systematic exploration for prophages and mycobacteriophages has been performed among additionally available sequenced M. abscessus genomes, leaving undetermined whether these initial observations were unique to some particular isolates or were representative of the M. abscessus species.
Here, exploiting genome sequence available for 48 M. abscessus mycobacteria by original bio-informatic analyses, we explored the repertoire of M. abscessus mycobacteriophages to gain insights into their evolution history compared to that of M. abscessus hosts.
Methods
Establishing the repertoire of M. abscessus phages
The genomes of 48 M. abscessus mycobacteria available in June 2013 were downloaded from Genbank (Table 1, Additional file 1). As for 47 unfinished genomes, the contigs were reoriented based on the M. abscessus type strain genome (GenBank GCF_000069185.1) used as reference using MAUVE software [24]. The prophage regions were detected using PHAST software [25]. Protein sequences were predicted in all genomes using prodigal software [26] in order to normalize prediction. M. abscessus pan-proteome was annotated using BlastP search with a cutoff E-value < 0.001, percentage similarity > 30% and an alignment length > 50 amino-acids against a home-made database (including PHAST database, Mimivirus, Marseillevirus and additional mycobacteriophage proteins). We further analyzed the M. abscessus complex genomes for Clustered Regularly Interspaced Short Palindromic Repeats (CRISPRs) using CRISPRs finder program [27].
Table 1.
M. abscessus genomes properties and prophage regions
| Group | Strain | Genome lenght Mb | Genome GC% | N° of prophage regions |
|---|---|---|---|---|
|
M. abscessus
|
M. abscessus
.CIP104536T |
5.09 |
62.7 |
1 |
| M93 |
5.08 |
64.2 |
4 |
|
| M94 |
5.1 |
64.2 |
2 |
|
| 4S-0116-R 4S_0116_R |
4.84 |
64 |
1 |
|
| 4S-0116-S 4S_0116_S |
4.84 |
64 |
1 |
|
| 4S-0726-RA 4S_0726_RA |
4.84 |
64 |
1 |
|
| 4S-0206 M4S_0206 |
4.86 |
64 |
2 |
|
| 4S-0303 4S_0303 |
4.86 |
64 |
2 |
|
| 4S-0726-RA 4S_0726_RB |
4.86 |
64 |
1 |
|
| 3A-0930-R 3A_0930_R |
5.27 |
64 |
8 |
|
| 3A-0119-R 3A_0119_R |
5.28 |
63.8 |
7 |
|
| 3A-0810-R M3A_0810_R |
5.29 |
64 |
8 |
|
| 3A-0122-R 3A_0122_R |
5.23 |
63.9 |
5 |
|
| 3A-0122-S 3A_0122_S |
5.23 |
63.9 |
6 |
|
| 3A-0731 3A_0731 |
5.39 |
64 |
8 |
|
| 3A-0930-R 3A_0930_S |
5.25 |
64 |
8 |
|
| 6G-0728-S 6G_0728_S |
5.32 |
64.1 |
2 |
|
| 6G-0125-S 6G_0125_S |
5.33 |
64.1 |
2 |
|
| 6G-0728-R M6G_0728_R |
5.34 |
64.1 |
2 |
|
| 6G-1108 6G_1108 |
5.34 |
64.1 |
2 |
|
| 6G-0125-R 6G_0125_R |
5.14 |
64.1 |
2 |
|
| 6G-0212 M6G_0212 |
5.14 |
64.1 |
2 |
|
|
M. massiliense
|
M. massiliense
BD |
5.2 |
64.2 |
3 |
| M172 |
5.2 |
64.2 |
6 |
|
| M47J26 |
4.87 |
64.1 |
3 |
|
| M154 |
4.8 |
64.1 |
0 |
|
| M18 |
4.89 |
64.2 |
1 |
|
| 2B-0107 M2B_0107 |
4.81 |
64.2 |
2 |
|
| 2B-0307 M2B_0307 |
4.81 |
64.2 |
2 |
|
| 2B-0912-R 2B_0912_R |
4.81 |
64.2 |
2 |
|
| 2B-0912-S 2B_0912_S |
4.81 |
64.2 |
1 |
|
| 2B-0626 M2B_0626 |
4.81 |
64.2 |
2 |
|
| B-1231 M2B_1231 |
4.81 |
64.2 |
2 |
|
| 1S_51_0915 |
4.89 |
64.2 |
3 |
|
| 1S-152-0930 |
4.9 |
64.2 |
3 |
|
| 1S_152_0914 |
4.9 |
64.2 |
3 |
|
| 5S-1215 5S_1215 |
5.21 |
64.1 |
6 |
|
| 5S-0421 5S_0421 |
5.24 |
64.1 |
5 |
|
| 5S-1212 5S_1212 |
5.24 |
64.1 |
6 |
|
| 5S-0304 5S_0304 |
5.25 |
64.1 |
6 |
|
| 5S-0708 5S_0708 |
5.25 |
64.1 |
6 |
|
| 5S-0817 5S_0817 |
5.25 |
64.1 |
6 |
|
| 5S-0921 M5S_0921 |
5.25 |
64.1 |
6 |
|
| 5S-0422 5S_0422 |
5.32 |
64.1 |
6 |
|
| M159 |
4.94 |
64.2 |
1 |
|
| M115 |
4.98 |
64.1 |
3 |
|
| M. bolletii |
M. bolletii
BD
T
|
5.05 |
64.2 |
3 |
| M24 | 5.51 | 64.2 | 7 |
Determining M. abscessus phage phylogenies
M. abscessus mycobacteria tree
M. abscessus genomes were aligned using Muscle aligner implemented in Mauve software [24]. Mauve alignment generated an identity matrix which the identity scores range between 0 and 1, where 0 indicates that no identical homologous nucleotides were found, and 1 indicates that every homologous nucleotide was identical. This matrix was then used to construct M. abscessus split network using Neighbor-Net algorithm in the package SplitsTree4 [28].
M. abscessus phage tree
The annotated viral and phage proteins were classified using OrthoMCL software [29]. Only protein sequences > 50-amino acid residues were considered for further analyses. Homologous sequences were selected using the all-against-all BlastP algorithm [30] with an E value of <10−5. Then, clustering of the orthologous sequences was analyzed using the Markov Cluster algorithm [31]. The inflation index of 1.5 was used to regulate cluster tightness (granularity). The resulting orthologous groups were used to construct a whole-genome network using the Neighbor-Net algorithm based on a gene content matrix. The similarity between two species is defined as the number of phage genes in common divided by the total number of genes of the two species. [32,33]. Using this matrix, we constructed also a heatmap clusterization using R package [http://www.r-project.org/].
Detecting gene transfer events
The orthologous groups identified by OrthoMCL were submitted to PhyloPattern for the analysis and manipulation of phylogenetic trees (within the DAGOBAH framework) [34,35]. The M. abscessus tree was used as a reference to infer topologies in order to detect gene gain and lost as previously described [35]. The results were submitted to FIGENIX [36] for phylogenetic reconstruction within the DAGOBAH framework as previously described [35]. The output generated by FIGENIX was submitted to the multi-agent system DAGOBAH, in which horizontal gene transfer (HGT) events were detected using an in-house-built transfer filter called HGT agent, as previously described [35,36]. This filter uses PhyloPattern to annotate each internal duplication node of the tree with three tags, including the recipient species, the donor species and external species [34]. Then, it applies a special phyletic pattern and searches the gene tree to find recipient species that are closer to donor species than to other external species that would otherwise be placed between the recipient and donor species in the species tree. In other words, a “donor” subtree must contain only species of a specific group and not those from the “recipient” group and vice versa and there should be no common species between the donor and external groups. Using HGT agent, one can specify the name of the donor and recipient species according to their usage.
Results
The repertoire of M. abscessus phages
Among 48 analysed genomes of M. abscessus, we found that only M. abscessus M154 encodes no prophage regions whereas the other 47 M. abscessus mycobacteria genomes harbour one to eight prophage regions. A total of 171 predicted prophage regions could be separated into four types i) intact prophages encoding structural proteins, lysis proteins, integration proteins and proteins necessary for replication and recombination ii) questionable prophages iii) incomplete prophage regions iv) small prophage-like elements (Table 1, Additional file 1).
In order to estimate the number of phages infecting M. abscessus, a phylogenetic tree was constructed based on Tape Measure Proteins (TMP) (Figure 1). The TMP was selected because it is typically the longest gene in mycobacteriophage genomes and because regions within the TMP gene are conserved [37].We could annotate 135 TMPs which clustered into thirteen groups and four singletons using orthoMCL. The TMP-based phylogenetic tree was constructed using MEGA software. The tree suggested that at least 17 different mycobacteriophages had infected M. abscessus, M. bolletii and M. massiliense during their evolution.
Figure 1.
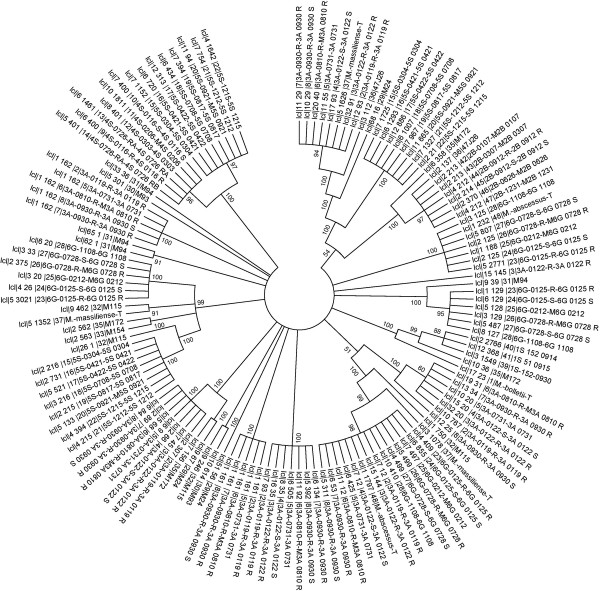
Phylogenetic tree based on annotated tape measure protein sequences using neighbour joining method.
Based on homology between prophage regions of M. abscessus genomes, the M. abscessus group could be separated into three clusters, M. abscessus, M. massiliense and M. bolletii (Figure 2). Few exceptions were observed: strains M139 and 1S_51_0915 showed prophage region homology with the M. abscessus cluster while M. bolletii M24 showed prophage region homology with the M. massiliense cluster (Figure 2). Also, a 12-kb small prophage-like element is conserved within the M. abscessus and M. massiliense clusters. Analyzing 242,067 proteins of all 48 M. abscessus proteomes found 20,304 (8.4%) proteins homologous to viral or phage proteins represented in Figure 3. These 20,304 proteins yielded 853 groups of orthologous proteins. All the species are represented in 239 groups (28.02%). Only three M. abscessus genomes have unique genes, two viral proteins in M. abscessus M94, four mycobacteriophage proteins in M. abscessus M159 and two viral proteins in M. abscessus M172. The annotation of the prophage found 44% proteins to be implicated in DNA replication and bacterial or/and phage metabolism, 37% were annotated as bacteriophage proteins (including structural, integration and terminase) and 14% proteins had no functional annotation. Interestingly, 289 proteins were annotated as holin and 75 as lysin protein. Twenty-five genomes including M. abscessus, M. bolletii and M. massiliense type strains encode endolysin-A and endolysin-B family lysin proteins. A total of 37.3% such proteins are homologous to lysin from mycobacteriophages, the other ones being homologous to lysin from phage infecting Firmicutes bacteria (Bacillus phages). Moreover, 156 proteins are repressor and anti-repressor proteins of the lambda repressor CI/C2 family (Lactobacillus phage and Staphylococcus phage), immunity repressor (Bacillus phage and Geobacillus phage) and Phage antirepressor protein KilAC domain (Rhodococcus phage). M. bolletii genome encodes only one CI/C2 repressor homologous to the CI repressor from Bacillus phage and one putative repressor located out of the Araucaria genome. All other M. abscessus encode three to nine repressors. Only M. abscessus 6G and M. abscessus type strain encode antirepressors.
Figure 2.
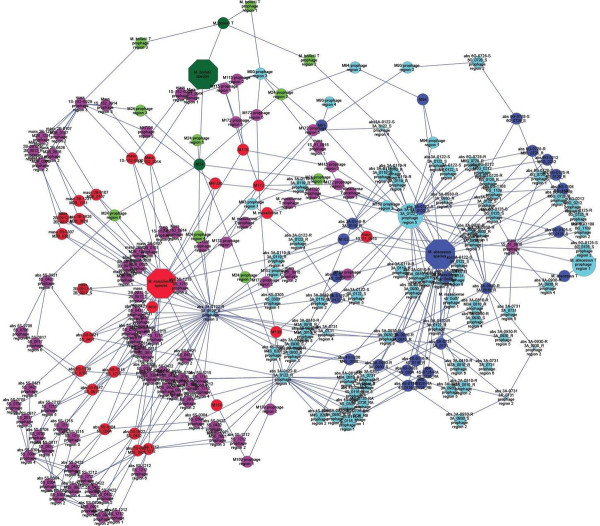
Interacting Map based on M. abscessus prophage region homology. Blue: M. abscessus species. Red: M. massiliense species. Green: M. bolletii species. The nodes represent the M. abscessus prophage regions. The node size correlates to the size of the M. abscessus prophage regions. The homologous regions were connected with edges.
Figure 3.
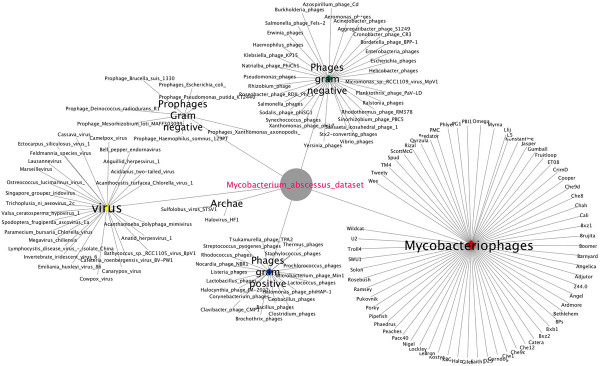
M. abscessus viral and phage proteins annotation.
Twelve genomes including M. abscessus type strain encode no CRISPRs, seven genome including M. bolletii type strain encode one possible CRISPR, twelve genomes encode two possible CRISPRs, twelve genomes encode three possible CRISPRs and four genomes encodes four CRISPRs including M154, M115, M172 and M18 strains (Additional file 2).
Phylogenomic and phylogenetic analyses
M. abscessus tree
The split network based on whole-genome content of M. abscessus shows the separation of M. abscessus strains into three main clusters respectively comprising M. abscessus, M. massiliense and M. bolletii genomes (Figure 4A). M. abscessus clusters comprise seven splits of a set of 22 strains. M. massiliense cluster comprises two sub-clusters; one sub-cluster forming the strains M159 and M115 and one sub-cluster forming 22 other strains. The M. massiliense cluster comprises 12 splits of a set of 24 strains. M. bolletii cluster comprises of two strains M. bolletii type strain and strain M24.
Figure 4.
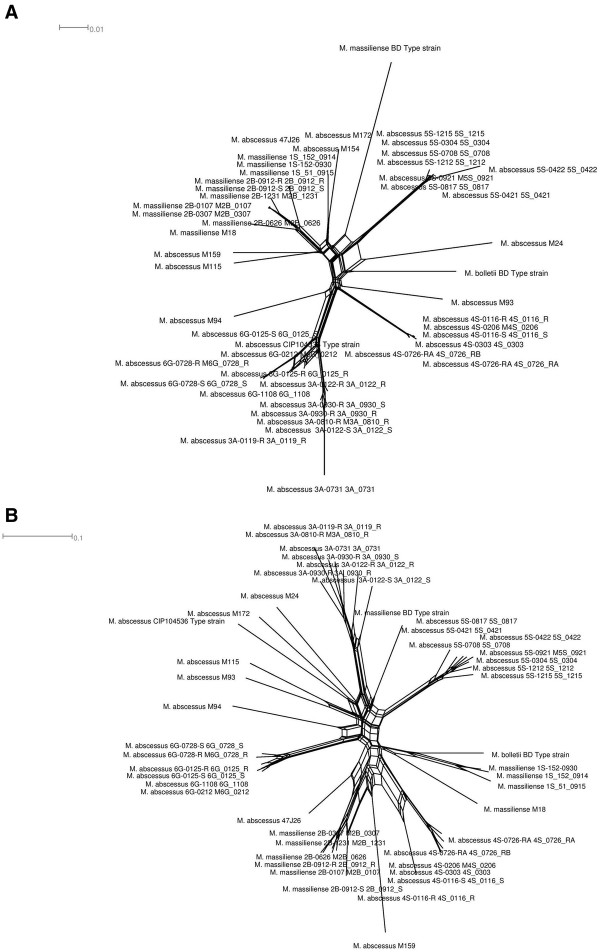
M. abscessus phylogeny. A- M. abscessus species split network. B- M. abscessus phage split network based on phage gene content matrix. Every edge is associated with a split of the taxa, but there may be a number of parallel edges associated with each split. The edges separate taxa on one side of the split from the taxa on the other side of the split. The length of an edge in the network is proportional to the weight of the associated split. This is analogous to the length of a branch in a phylogenetic tree.
M. abscessus phage tree
The split network based on prophage gene content (i.e., the presence or absence of orthologous proteins) showed an organization that differed from that of the M. abscessus tree (Figure 4B). The organization of splits in the M. abscessus phage split network differs from M. abscessus split network. M. massiliense type strain clusters with M. abscessus, while M. abscessus 4S strain clusters with M. massiliense strains 2B and M159. The phage split network shows clusterization of M. massiliense 1S strains with M. bolletii while M. abscessus 6G strains forming a different cluster from M. abscessus. This phylogenomic analysis showed that M. abscessus viral and phage gene repertoires have different evolutionary histories. Also, a heatmap clusterization was constructed using a matrix of presence/absence of orthologous proteins. The heatmap clusterization showed a species organization different from that of the M. abscessus tree suggesting that M. abscessus may have been infected by several phages during their evolution (Figure 5). Likewise, using the tree based on whole-genome content, individual phylogenetic analysis for the different orthologous proteins groups revealed many topologies that differed from that of the M. abscessus tree. These results suggested that gene loss and HGT are relevant for all gene functions. Interestingly, Araucaria TMP clusters with M. massiliense strains 1S and M172, suggesting that mycobacteriophages infecting M. massiliense mycobacteria may have features similar to Araucaria.
Figure 5.
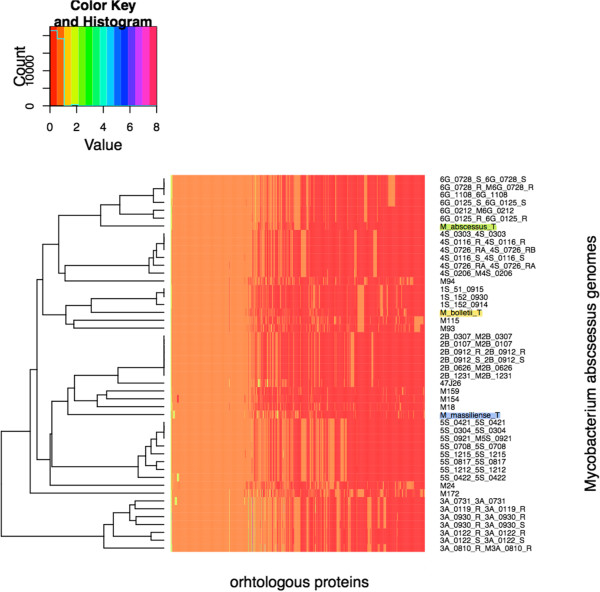
Heatmap clusterization based on protein presence absence matrix.
Detection of HGT cases
Among the 853 orthologous groups, phylogenetic trees were successfully reconstructed for 213 (25%) of the cases, 156 transfer events (Additional file 3) were detected out of which 33 cases were associated with strong boostrap support for HGT. A 45.45% proportion of the transferred proteins are homologous to mycobacteriophages proteins, 12.12% homologous to viral proteins, 21.21% to proteins of phages infecting gram-positive bacteria and 21.21% to proteins of phages infecting gram-negative bacteria. The probable sources are environmental bacteria in 33 cases, M. abscessus in 13 cases, M. bolletii in 6 cases and M. massiliense in 4 cases.
Discussion
Analysing 171 prophage regions in 47 M. abscessus complex genomes indicated that M. abscessus complex has been infected by at least 17 different mycobacteriophages, including Araucaria, the sole available mycobacteriophage that we recently isolated from M. bolletii[23]. Noteworthy, M. abscessus M154, an isolate from Malaysia [38] is the only strain lacking any evidence for phage. Our previous analysis revealed no unique genes in this strain [39], which nevertheless encodes four possible CRISPRs, one cmr1 family and one cmr4 family, with potential immunity against phage infection [40]. Alternatively, no phage was detected in this strain because of database limitations. For example, Araucaria did not yield significant homology with any other M. abscessus complex phage, as confirmed by phylogenetic studies using whole viral and phage proteins clusterization and TMP protein sequence based tree. Sequencing additional mycobacteriophages may next reveal prophage regions in M. abscessus M154.
We further observed that some M. abscessus phage proteins had homology to other mycobacteriophages and to phages infecting environmental bacteria. M. abscessus complex mycobacteria are opportunistic pathogens, but these observations suggest that environments, rather than host microbiota, are sources of evolution for M. abscessus complex mycobacteriophages. Noteworthy, potential sources are living in amoeba (Additional file 4) where M. abscessus complex mycobacteria including M. massiliense[22] and M. bolletii[41] are also residing. Amoeba are a place for DNA exchanges between sympatric organisms and the amoeba themselves [42-48]. Data here reported suggest that amoeba are a likely place for mycobacteriophage exchanges and therefore, may be a place to look at for the discovery of new mycobacteriophages.
Accordingly, a striking feature of M. abscessus complex phage genomes is their pervasive mosaicism, a previously reported hallmark of mycobacteriophages [49,50]. Our phylogenomic and phylogenetic analyses revealed a different split network topology between the hosts and the phages. This probably reflects reciprocal genome evolution through a dynamic co-evolutionary process [51]. M. abscessus complex was infected by at least 17 phages and these infections contributed to differentiate M. abscessus complex into several clusters of mycobacteria. Widespread occurrence of phage sequences in almost all studied M. abscessus complex isolates suggests that the rate of prophage invasion is faster than the rate of mutation, implying rapid evolution of M. abscessus. Also, in M. abscessus complex a total of 6/33 (19%) gene transfers occurred between a set of donor species (at least two donors) and a recipient species and 27/33 (81%) gene transfers occurred between a single species donor and a recipient species, clearly indicating it is an on-going process.
Conclusions
Excluding the prophage-free strain M154, phages account for only 6.7-9.6% of M. abscessus complex genomic content, but they profoundly impact their hosts, participating to their on-going diversification.
Reviewers’ comments
We appreciate the reviewer’s comments from Dr. Eric Bapteste (UPMC, Institut de Biologie Paris Seine, France), Dr. Patrick Forterre (Institut de Génétique Microbiologie, 91405 Orsay Cedex, France Institut Pasteur) and Dr. Eugene Koonin (National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, MD, USA). We have revised the manuscript according to your comments and suggestions.
Reviewer 1: Dr. Eric Bapteste (UPMC, Institut de Biologie Paris Seine, France)
The general topic of this research and the reported findings are very relevant for Biology Direct, however the current version of this MS is certainly not yet ready for publication.
Even though I am willing to trust the authors about their main conclusions, I strongly recommend major revisions, because it is currently hardly possible to evaluate most of the evidence on which they based their observations.
In short:
- The trees supporting lateral gene transfers should be presented in an organized fashion in a Supp. Mat.
Authors’ response: The reviewer is right, all 605 trees are now provided as Additional File and trees supporting LGT are highlighted.
- Many current figures are not of sufficient quality to be printed in a journal (gene/taxon names are impossible to read, etc.)
Authors’ response: Authors improved the quality of documents.
- The methodology used for the tree reconstruction is not sufficiently detailed: crucial information such as the number of positions retained or the substitution models used are lacking.
Authors’ response: Methodology has been expanded (Lines 87–92; lines 100–102; lines 112–122).
- Many figures presented in the text are under-interpreted, and not critically discussed.
Elements requiring significantly more details:
p.4. l. 86: The authors report that ‘M. abscessus proteomes were aligned using Mauve software [24]’. I am not familiar with ProgressiveMauve, but does this software really align proteomes, or is it rather a tool to align genomes based on their content and gene order? If so, the next sentence in the MS is hard to understand
Authors’ response: M. abscessus genomes were aligned using Mauve software. Progressive Mauve uses Muscle or clustlW to perform alignment. Here we used Muscle. The authors corrected and explained the methodology (P.4 line 86).
‘Then M. abscessus tree was constructed using Neighbor-Net algorithm in the package SplitsTree’. What distance matrix was provided to Splitstree? What was this distance reflecting?
Authors’ response: Mauve alignment generates an identity matrix file which calculates the identity score range between 0 and 1; 0 indicates no homologous nucleotides and 1 indicates that every homologous nucleotide was identical. The authors explained this part in the text (P.5 line 88).
If the proteomes alignments evoked above were ‘classic’ protein alignments, then what happened to poorly aligned sites? How many positions were retained? For how many proteins? The material and methods must be much more detailed for the reader to really understand the analysis and the results. Please note that this criticism also applies for the trees that show some LGT.
Authors’ response: The authors corrected that M. abscessus genomes was aligned not proteomes in the text (P.4 line 86).
p.5. l, 95–96: The analysis described here faces a potential pitfall if ‘presence’ and ‘absence’ were treated in a symmetric fashion, especially if there were lots of ‘absences’ in this matrix. While ‘presences’ can be used to group genomes sharing some features, more caution is required in the use of shared ‘absences’.
If the groupings of genomes are firstly caused by the lack of shared features, then these groupings can be very artefactual (sharing ‘losses’ is different from sharing nothing. The potential problem here is that two genomes that have positively nothing, or not much in common, may still be grouped due to their lack of genes, while sometimes it is a better idea not to group genomes that share nothing in common!). How did the authors address this potential issue?
Authors’ response: Here we constructed a matrix based on the similarity between two species which is defined as the number of genes that they have in common divided by their total number of genes (P.5 line l00-104).
p.5, l. 98–108: Incongruence between trees is considered to be synonym of lateral gene transfer, and the possibility of tree reconstruction artefacts is not discussed. Since the tree reconstruction methods are poorly described, it is difficult to evaluate this part of the work.
Authors’ response: The methods of tree reconstruction are now described in the materials and methods section. (P.5 line 105–124).
p.6., l. 119: The authors write that ‘a phylogenetic tree was constructed based on Tape Measure Proteins (TMP)’ What are ‘Tape Measure Proteins’? Why this marker? What methods/positions/models were used to reconstruct this tree?
Authors’ response: The TMP was selected because it is typically the longest gene in mycobacteriophage genomes and because regions within TMP gene are conserved (P.6 line 125).p.7, l. 156. Figure 4A: I have not been able to see this figure, or if it refers to the split network, then its description in the text must be expanded and be more critical. What do the proportion/size/presence of splits indicate? What is their biological meaning? Also for example, what does the position of M. massiliense BD type strain suggest? Same question for the position of M. abscessus 47 J26?
Authors’ response: The authors performed a better figure quality and more description in the text. The length of an edge in a split network is analogous to the length of a branch in a phylogenetic tree.p.7, l.160: same problem with Figure 4B.
Authors’ response: The authors performed a better figure quality and more description in the text (P.7 lines 158–160).Overall, the exploitation of these 2 figures is a bit vague. The authors only write about it that: ‘A phylogenomic tree based on prophage gene content (i.e., the presence or absence of orthologous proteins) showed an organization that differed from that of the M. abscessus tree (Figure 4B).’ Please, increase the descriptions of what these differences are (or use a metrics to compare these two split networks).
Authors’ response: The authors performed a better figure quality and more description in the text (P.8 line 186–190).p.8, l.165: Figure 5 is neither described nor exploited in a way that allows to make sense of the main text about it. Please, give more time to a careful critical description of the figure.
Authors’ response: The authors clarified this point (P.8 line 174).
p.8, l. 172: The 214 phylogenetic trees mentioned here (reconstructed how, please precise) should be logically classified and presented as Supp. Mat, or made available somewhere. Currently, it is simply impossible to review this part of the MS without being able to look at the evidence.
Authors’ response: The phylogenetic trees are provided in Additional file4.
p.10. l-210-211: ‘Also, M. abscessus complex phages further shuttled gene transferts, 16/29 (55%) of which occurred between different M. abscessus complex clusters but 13/29 (45%) of which occurred between strains of the same cluster, clearly indicating it is an on-going process’. Where do these numbers come from? How were they obtained? Where is the evidence?
Authors’ response: The authors clarified this point in the text (P.10 line 241–246).p.19, l. 379: Figure 2 legend: What is ‘an interacting map’? How is one supposed to read such a map? What are the nodes? What are the edges? More descriptions are required.
Authors’ response: The authors clarified this point (P.19 line 422).
There are also some minor typos/issues:
p.3, l. 56 ‘three different organisms’: do you really mean organisms, or species, or strains here?
Authors’ response: We mean organisms.p.4, l.86 (and in some other places in the text): the authors refer to the splitsnetwork as the ‘M. abscessus tree’. Elswhere, as in the legend of Figure 4, they call this type of graphs ‘network trees’. This wording is confusing. Is it a network or is it a tree? To me, each of this graph should be called a split network.
Authors’ response: The authors corrected network tree to split network (P.5 line 91).
p.5., l. 95: Likewise, what the authors call ‘a whole-genome phylogenetic tree’ looks very much like a network.
Authors’ response: The authors corrected phylogenetic tree to network (P.5 line 100).Figure 2: ‘Few exceptions were observed: strains M139 and 1S_51_0915 showed prophage region homology with the M. abscessus cluster while M. bolletii M24 showed prophage region homology with the M. massiliense cluster’. Please help the reader more to see this, it is impossible to guess where the strains discussed here are in this map, add some arrows.Figure 3 is likely too large in its current format for publication.
Authors’ response: The authors improved the quality of Figures.
p.6, l.136: ‘Interestingly”. Why? Please explain why it is interesting.
Authors’ response: The authors clarified this point (P.7 line 137).SI 1 & 2: ‘porphages’ should be prophages Figure legends: Figure 1: ‘Phylogentic’ must be fixed + see problems with Figure 4 and Figure 2 legends discussed above.
Authors’ response: The authors corrected this point.
p.34. Table three: what is the difference between a ‘parent species’ and a ‘donor species?’ (‘donor’ takes only 1 ‘n’). What does the column ‘Nb Duplications before parent’ refers to?
Authors’ response: The parent species is the node which contains the two sub-trees: recipient and donor species and the number of duplication before parent refers to the number of gene duplication before the HGT event.
Second revision requested be the Reviewer 1: Dr. Eric Bapteste (UPMC, Institut de Biologie Paris Seine, France)
The revised version of the MS by Sassi et al. is improved. I am still uncertain whether the quantification of HGT using gene trees means much biologically. I suppose this is because I doubt that trees alone convey that kind of evidence anyway. I am more convinced by studies of synteny showing prophages with similar genes inserted at various positions of Mycobacterium genomes. I remain also unconvinced (to be honest somewhat skeptical) about the quality of the figures. I suppose this latter possible issue would be addressed by the publisher then.
p.9. l. 195. The content of Additional file 4 is useful, also not yet perfect for its purpose: both trees with and without candidate HGT are present in this file (i.e. there are around 214 trees in it, not 156 trees), making it difficult to evaluate the trees with HGT only. The legend for this file (p.27, l.614) is confusing as it seems to announce 214 trees with candidate HGT. When one looks at some of these tree files however, one finds the following associated description: ‘None horizontal gene transfert event’. (So some of these trees should be removed from Additional file 4, and the English of this final description could be improved).
Authors’ response: In the additional file4we changed the report by trees as figures and it presents 75 trees representing a total of 156 transfer events. The HGT is represented by yellow squares in the figures. The legend is corrected accordingly to the reviewer's comment.
p.9. l.199-201: When discussing the sources of HGT, I find it strange that the category ‘unknown’ is not quantified, nor discussed. In the few trees I have looked at from additional file 4, ‘unknown’ was the major HGT donor…
Authors’ response: The category “unknown” in the report files represents the taxonomy used in the project which is not that same used by NCBI. As the species name is too long we removed it and only the strain name is presented, meaning that the program we run does not recognize the names we gave in data. Here we present as additional file4the figures of the trees to avoid any confusion.
p.10. l. 227. ‘a different topology’, sure, but a topology of what? I suppose of split networks. Indeed, the networks look different, but note that their difference is still not assessed by any formal distance computed between them, nor by any statistical test. Some might find that this aspect of the study would have deserved to be improved.
Authors’ response: Indeed a different split network topology. This is corrected in the text Page10, line 227.
All the minor comments were corrected.
Reviewer 2: Dr. Patrick Forterre (Institut de Génétique Microbiologie, 91405 Orsay Cedex, France Institut Pasteur)
The authors have analyzed the proviruses integrated in 48 strains of the Mycobacterium abscessus complex. Interestingly, this analysis allowed detecting six new families of mycobacterioviruses, in addition to the previously described virus Araucaria. The authors observe that these viruses roughly co-evolved with their hosts since they can be divided in three clusters corresponding to the three Mycobacterium abscessus sub-complexes. However, they also noticed many incongruence between various tree topologies that are interpreted as horizontal gene transfer (HGT). It is unclear for me which of these transfer correspond to independent gain and/or loss of proviruses in different lineages of M. absessus and which ones are due to real transfer of viruses from one lineage to the other. It is also not clear why the authors concluded that viral infection contributes to the differentiation of the M. abscessus complex.
The presentation of Figures and Table could be improved. The Tables 1 and 2 could be placed in supplementary material and important information about the proviruses summarized in Figure (diagram) and/or Table (how many genomes have 0, 1, 2….7,8 integrated elements, size distribution, main features of the four classes proposed). The trees/networks are also difficult to interpret.
Table 2.
M. abscessus HGT cases
| Putative HGT | Homology | Parent species | Recipient species | Donor species | Nb duplications before parent |
|---|---|---|---|---|---|
| 1 |
PHAGE_Mycoba_Peaches-gi|282598664|ref|YP_003358761.1|gp58[Mycobacterium_phage_Peaches] |
[M93] |
[6G-0125-R] |
[M93] |
5 |
| 2 |
PHAGE_Plankt_PaV_LD-gi|371496158|ref|YP_004957306.1|ABCtransporter[PlanktothrixphagePaV-LD] |
[PSEUDO ~ −5S-0421] |
[M94] |
[M93 M115] |
4 |
| 3 |
PHAGE_Mycoba_LeBron-gi|304360967|ref|YP_003857149.1|gp18[Mycobacterium_phage_LeBron] |
[M115] |
[M172] |
[M115] |
2 |
| 4 |
PHAGE_Mycoba_Giles-gi|160700672|ref|YP_001552352.1|gp23[Mycobacterium_phage_Giles] |
[M94] |
[6G-0728-R] |
[M94] |
5 |
| 5 |
PHAGE_Mycoba_Che9c-gi|29566118|ref|NP_817687.1|gp10[Mycobacterium_phage_Che9c] |
[M18] |
[3A-0122_S] |
[M18] |
2 |
| 6 |
PHAGE_Mycoba_Pukovnik-gi|192824238|ref|YP_001994879.1|gp62[Mycobacterium_phage_Pukovnik] |
[M154] |
[M115] |
[M154] |
5 |
| 7 |
PHAGE_Tricho_2c-gi|116326757|ref|YP_803294.1|hypotheticalproteinTNAV2c_gp071[Trichoplusia_ni_ascovirus_2c] |
[5S-0921] |
[4S-0726] |
[5S-0921] |
4 |
| 8 |
PHAGE_Salmon_PVP_SE1-gi|363539742|ref|YP_004894027.1|hypotheticalprotein[SalmonellaphagePVP-SE1] |
[M. massiliense T] |
[M24] |
[M. massiliense T] |
5 |
| 9 |
PHAGE_Rhodoc_REQ3-gi|372449972|ref|YP_005087193.1|phageintegrase[RhodococcusphageREQ3] |
[M172] |
[3A_0930_S] |
[M172] |
3 |
| 10 |
PHAGE_Salmon_PVP_SE1-gi|363539618|ref|YP_004893903.1|phosphoribosylpyrophosphatesynthetase[SalmonellaphagePVP-SE1] |
[3A-0122_S1] |
[M. massiliense T] |
[3A-0122_S1] |
5 |
| 11 |
PHAGE_Mycoba_Myrna-gi|203454746|ref|YP_002225062.1|gp183[Mycobacterium_phage_Myrna] |
[M172] |
[M94] |
[M172] |
5 |
| 12 |
PHAGE_Mycoba_Omega-gi|29566822|ref|NP_818386.1|gp85[Mycobacterium_phage_Omega] |
[M115] |
[M24] |
[M115] |
5 |
| 13 |
PHAGE_Mycoba_Pacc40-gi|206600097|ref|YP_002241602.1|gp18[Mycobacterium_phage_Pacc40] |
[M24] |
[M94] |
[M24] |
6 |
| 14 |
PHAGE_Mycoba_Pacc40-gi|206600097|ref|YP_002241602.1|gp18[Mycobacterium_phage_Pacc40] |
[PSEUDO ~ − M159] |
[M24] |
[M172 3A-0122_S6 3A-0122_S2 3A-0122_S4 47 J26] |
1 |
| 15 |
PHAGE_Acanth_mimivirus-gi|311977570|ref|YP_003986690.1|DNAtopoisomerase1b[Acanthamoebapolyphagamimivirus] |
[3A-0122_S7] |
[3A-0122_S5] |
[3A-0122_S7] |
2 |
| 16 |
PHAGE_Rhodoc_RER2-gi|372449922|ref|YP_005087145.1|hypotheticalprotein[RhodococcusphageRER2] |
[M24] |
[3A-0731] |
[M24] |
3 |
| 17 |
PHAGE_Aeromo_31-gi|66391812|ref|YP_238737.1|hypotheticalproteinPHG31p8[Aeromonas_phage_31] |
[4S-0726] |
[4S-0303] |
[4S-0726] |
6 |
| 18 |
PHAGE_Lactoc_P087-gi|229605000|ref|YP_002875699.1|putativecysteinesynthase[Lactococcus_phage_P087] |
[47 J26] |
[5S-0708] |
[47 J26] |
6 |
| 19 |
PHAGE_Mycoba_Myrna-gi|203454746|ref|YP_002225062.1|gp183[Mycobacterium_phage_Myrna] |
[M172] |
[M94] |
[M172] |
5 |
| 20 |
PHAGE_Plankt_PaV_LD-gi|371496158|ref|YP_004957306.1|ABCtransporter[PlanktothrixphagePaV-LD] |
[3A-0122_S5] |
[M94] |
[3A-0122_S5] |
2 |
| 21 |
PHAGE_Acanth_mimivirus-gi|311977513|ref|YP_003986633.1|putativedTDP-D-glucose4,6-dehydratase[Acanthamoebapolyphagamimivirus] |
[3A-0122_S4] |
[4S-0726-RA] |
[3A-0122_S4] |
4 |
| 22 |
PHAGE_Bacill_36-gi|156564011|ref|YP_001429750.1|PcrAhelicase[Bacillus_phage_0305phi8_36] |
[3A-0122_S7] |
[3A-0119-R] |
[3A-0122_S7] |
3 |
| 23 |
PHAGE_Mycoba_Che9c-gi|29566174|ref|NP_817745.1|gp68[Mycobacterium_phage_Che9c] |
[M18] |
[M94] |
[M18] |
3 |
| 24 |
PHAGE_Mycoba_Che8-gi|29565783|ref|NP_817355.1|gp17[Mycobacterium_phage_Che8] |
[M24] |
[M172 3A-0122_S4 3A-0122_S2 47 J26] |
[M24] |
2 |
| 25 |
PHAGE_Tricho_2c-gi|116326757|ref|YP_803294.1|hypotheticalproteinTNAV2c_gp071[Trichoplusia_ni_ascovirus_2c] |
[M115] |
[4S-0116_S] |
[M115] |
3 |
| 26 |
PHAGE_Microm_MpV1-gi|313768434|ref|YP_004062114.1|hypotheticalprotein[Micromonassp.RCC1109virusMpV1] |
[M115] |
[M94] |
[M115] |
3 |
| 27 |
PHAGE_Mycoba_Pipefish-gi|109521870|ref|YP_655307.1|gp30[Mycobacterium_phage_Pipefish] |
[M18] |
[M172] |
[M18] |
1 |
| 28 |
PHAGE_Plankt_PaV_LD-gi|371496158|ref|YP_004957306.1|ABCtransporter[PlanktothrixphagePaV-LD] |
[M. bolletii T] |
[4S-0726-RA] |
[M. bolletii T] |
7 |
| 29 |
PHAGE_Lactoc_P087-gi|229605000|ref|YP_002875699.1|putativecysteinesynthase [Lactococcus_phage_P087] |
[M24] |
[M. bolletii T] |
[M24] |
7 |
| 30 |
PHAGE_Mycoba_Omega-gi|29566768|ref|NP_818332.1|gp31[Mycobacterium_phage_Omega] |
[M94] |
[6G-1108] |
[M94] |
3 |
| 31 |
PHAGE_Burkho_phi1026b-gi|38707948|ref|NP_945089.1|gp58[Burkholderia_phage_phi1026b] |
[PSEUDO ~ −M154] |
[M. bolletii T] |
[M. massiliense T M154 M172 M159] |
6 |
| 32 |
PHAGE_Mycoba_Cjw1-gi|29565933|ref|NP_817504.1|gp55[Mycobacterium_phage_Cjw1] |
[PSEUDO ~ −M159] |
[M. bolletii T] |
[M172 3A-0122_S0 1S-152-0930 3A-0122_S1] |
2 |
| 33 | PHAGE_Brocho_BL3-gi|327409421|ref|YP_004301563.1|gp29[BrochothrixphageBL3] | [M115] | [M24] | [M115] | 5 |
There are several minor points Lane 123; define prophage region homology.
Authors’ response: This methodology section has been re-written (Lines 112–123).
Lane 128: 242,067 proteins (which proteins?) in general be more precise
Authors’ response: The authors clarified this point (P.7 line 139).
Lane 140 and elsewhere, gram-positive bacteria is no more a valid taxonomic grouping, better to indicate Firmicutes
Authors’ response: The authors corrected this point (P.7 line 151).
Lane 168: Auracaria should be Araucaria
Authors’ response: The authors corrected this point (P.8 line 182).
Lane 193: gram-negative and gram positive bacteria! i.e. all bacteria except mycobacteria?? So environmental bacteria is sufficient.
Authors’ response: The authors corrected this point (P.9 line 206).
Reviewer 3: Dr. Eugene Koonin (National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, MD, USA)
The importance of bacteriophage contribution to the evolution of bacterial genomes is increasingly recognized. Here Sassi and coworkers conclude that bacteriophages drive the evolution of the Mycobacterium abscessus complex. I find this appealing and credible idea but fail to see how the data presented in the manuscript, even assuming that the identification of prophages is accurate (no specific evidence of that is provided), support such a strong conclusion. I can agree that the authors demonstrate differences in the prophage content between the bacteria in the complex. Then, I suppose, the argument would be that the trees of the bacteria and phages are different, suggesting that there has been some exchange of prophages and individual genes. The robustness of the trees is a concern because the trees for phages can be notoriously difficult. But, even assuming they are correct, this argument seems to fall far short of the ambitious claim of the paper. It could be very helpful if the authors made an effort to carefully present their logic.
Authors’ response: The authors revised the manuscript in light of the reviewer comments to further discuss the impact of mycobacteriophages on the on-going diversification of this group of mycobacteria. (Lines 245–250).
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MS, PG and OC performed the analyses. MS and MD designed the study. MS, PP and MD interpreted data and wrote the draft. All authors read and approved the final manuscript.
Supplementary Material
Mycobacterium abscessus genome properties and their prophage regions.
Mycobacterium abscessus CRISPRs and the correlation with number of prophage regions.
The reconstructed trees for HGT events. Each tree contains one to six HGT events. The yellow squares represent the HGT event.
Environmental bacteria hosting homologous M. abscessus phage proteins and evidence for bacteria-amoeba interaction.
Contributor Information
Mohamed Sassi, Email: mohamedsassi08@gmail.com.
Philippe Gouret, Email: philippe.gouret@univ-provence.fr.
Olivier Chabrol, Email: olivier.chabrol@univ-provence.fr.
Pierre Pontarotti, Email: Pierre.Pontarotti@univ-provence.fr.
Michel Drancourt, Email: michel.drancourt@univ-amu.fr.
Acknowledgments
MS was financially supported by Infectiopole Sud Foundation.
References
- Ripoll F, Pasek S, Schenowitz C, Dossat C, Barbe V, Rottman M, Macheras E, Heym B, Herrmann JL, Daffé M, Brosch R, Risler JL, Gaillard JL. Non mycobacterial virulence genes in the genome of the emerging pathogen Mycobacterium abscessus. PLoS One. 2009;4:e5660. doi: 10.1371/journal.pone.0005660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuya EY, Paez A, Srinivasan A, Cooksey R, Augenbraun M, Baron M, Brudney K, Della-Latta P, Estivariz C, Fischer S, Flood M, Kellner P, Roman C, Yakrus M, Weiss D, Granowitz EV. Outbreak of Mycobacterium abscessus wound infections among "lipotourists" from the United States who underwent abdominoplasty in the Dominican Republic. Clin Infect Dis. 2008;46:1181–1188. doi: 10.1086/529191. [DOI] [PubMed] [Google Scholar]
- Viana-Niero C, Lima KV, Lopes ML, Rabello MC, Marsola LR, Brilhante VC, Durham AM, Leão SC. Molecular characterization of Mycobacterium massiliense and Mycobacterium bolletii in isolates collected from outbreaks of infections after laparoscopic surgeries and cosmetic procedures. J Clin Microbiol. 2008;46:850–855. doi: 10.1128/JCM.02052-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh WJ, Jeon K, Lee NY, Kim BJ, Kook YH, Lee SH, Park YK, Kim CK, Shin SJ, Huitt GA, Daley CL, Kwon OJ. Clinical significance of differentiation of Mycobacterium massiliense from Mycobacterium abscessus. Am J Respir Crit Care Med. 2011;183:405–410. doi: 10.1164/rccm.201003-0395OC. [DOI] [PubMed] [Google Scholar]
- Petrini B. Mycobacterium abscessus: an emerging rapid-growing potential pathogen. APMIS. 2006;114:319–328. doi: 10.1111/j.1600-0463.2006.apm_390.x. [DOI] [PubMed] [Google Scholar]
- Hayes D Jr. Mycobacterium abscessus and other nontuberculous mycobacteria: evolving respiratory pathogens in cystic fibrosis: a case report and review. South Med J. 2005;98:657–661. doi: 10.1097/01.SMJ.0000163311.70464.91. [DOI] [PubMed] [Google Scholar]
- Griffith DE, Girard WM, Wallace RJ Jr. Clinical features of pulmonary disease caused by rapidly growing mycobacteria. An analysis of 154 patients. Am Rev Respir Dis. 1993;147:1271–1278. doi: 10.1164/ajrccm/147.5.1271. [DOI] [PubMed] [Google Scholar]
- Griffith DE. Emergence of nontuberculous mycobacteria as pathogens in cystic fibrosis. Am J Respir Crit Care Med. 2003;167:810–812. doi: 10.1164/rccm.2301001. [DOI] [PubMed] [Google Scholar]
- Olivier KN, Weber DJ, Wallace RJ Jr, Faiz AR, Lee JH, Zhang Y, Brown-Elliot BA, Handler A, Wilson RW, Schechter MS, Edwards LJ, Chakraborti S, Knowles MR. Nontuberculous mycobacteria. I: multicenter prevalence study in cystic fibrosis. Am J Respir Crit Care Med. 2003;167:828–834. doi: 10.1164/rccm.200207-678OC. [DOI] [PubMed] [Google Scholar]
- Pierre-Audigier C, Ferroni A, Sermet-Gaudelus I, Le Bourgeois M, Offredo C, Vu-Thien H, Fauroux B, Mariani P, Munck A, Bingen E, Guillemot D, Quesne G, Vincent V, Berche P, Gaillard JL. Age-related prevalence and distribution of nontuberculous mycobacterial species among patients with cystic fibrosis. J Clin Microbiol. 2005;43:3467–3470. doi: 10.1128/JCM.43.7.3467-3470.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalermskulrat W, Sood N, Neuringer IP, Hecker TM, Chang L, Rivera MP, Paradowski LJ, Aris RM. Non-tuberculous mycobacteria in end stage cystic fibrosis: implications for lung transplantation. Thorax. 2006;61:507–513. doi: 10.1136/thx.2005.049247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson BE, Gilljam M, Lindblad A, Ridell M, Wold AE, Welinder-Olsson C. Molecular epidemiology of Mycobacterium abscessus, with focus on cystic fibrosis. J Clin Microbiol. 2007;45:1497–1504. doi: 10.1128/JCM.02592-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy I, Grisaru-Soen G, Lerner-Geva L, Kerem E, Blau H, Bentur L, Aviram M, Rivlin J, Picard E, Lavy A, Yahav Y, Rahav G. Multicenter cross-sectional study of nontuberculous mycobacterial infections among cystic fibrosis patients, Israel. Emerg Infect Dis. 2008;14:378–384. doi: 10.3201/eid1403.061405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith DE, Aksamit T, Brown-Elliott BA, Catanzaro A, Daley C, Gordin F, Holland SM, Horsburgh R, Huitt G, Iademarco MF, Iseman M, Olivier K, Ruoss S, von Reyn CF, Wallace RJ Jr, Winthrop K. ATS Mycobacterial Diseases Subcommittee. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367–416. doi: 10.1164/rccm.200604-571ST. [DOI] [PubMed] [Google Scholar]
- Macheras E, Roux AL, Bastian S, Leão SC, Palaci M, Sivadon-Tardy V, Gutierrez C, Richter E, Rüsch-Gerdes S, Pfyffer G, Bodmer T, Cambau E, Gaillard JL, Heym B. Multilocus sequence analysis and rpoB sequencing of Mycobacterium abscessus (sensu lato) strains. J Clin Microbiol. 2011;49:491–499. doi: 10.1128/JCM.01274-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassi M, Ben Kahla I, Drancourt M. Mycobacterium abscessus multispacer sequence typing. BMC Microbiol. 2013;13:3. doi: 10.1186/1471-2180-13-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant JM, Grogono DM, Greaves D, Foweraker J, Roddick I, Inns T, Reacher M, Haworth CS, Curran MD, Harris SR, Peacock SJ, Parkhill J, Floto RA. Whole-genome sequencing to identify transmission of Mycobacterium abscessus between patients with cystic fibrosis: a retrospective cohort study. Lancet. 2013;381:1551–1560. doi: 10.1016/S0140-6736(13)60632-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leao SC, Tortoli E, Viana-Niero C, Ueki SY, Lima KV, Lopes ML, Yubero J, Menendez MC, Garcia MJ. Characterization of mycobacteria from a major Brazilian outbreak suggests that revision of the taxonomic status of members of the Mycobacterium chelonae-M. abscessus group is needed. J Clin Microbiol. 2009;47:2691–2698. doi: 10.1128/JCM.00808-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leao SC, Tortoli E, Euzéby JP, Garcia MJ. Proposal that Mycobacterium massiliense and Mycobacterium bolletii be united and reclassified as Mycobacterium abscessus subsp. bolletii comb. nov., designation of Mycobacterium abscessus subsp. abscessus subsp. nov. and emended description of Mycobacterium abscessus. Int J Syst Evol Microbiol. 2011;61:2311–2313. doi: 10.1099/ijs.0.023770-0. [DOI] [PubMed] [Google Scholar]
- Cho YJ, Yi H, Chun J, Cho SN, Daley CL, Koh WJ, Jae Shin S. The genome sequence of 'Mycobacterium massiliense' strain CIP 108297 suggests the independent taxonomic status of the Mycobacterium abscessus complex at the subspecies level. PLoS One. 2013;8:e81560. doi: 10.1371/journal.pone.0081560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adekambi T, Berger P, Raoult D, Drancourt M. rpoB gene sequence- based characterization of emerging non-tuberculous mycobacteria with descriptions of Mycobacterium bolletii sp. nov., Mycobacterium phocaicum sp. nov. and Mycobacterium aubagnense sp. nov. Int J Syst Evol Microbiol. 2006;56:133–143. doi: 10.1099/ijs.0.63969-0. [DOI] [PubMed] [Google Scholar]
- Adekambi T, Reynaud-Gaubert M, Greub G, Gevaudan MJ, La Scola B, Raoult D, Drancourt M. Amoebal coculture of “Mycobacterium massiliense” sp. nov. from the sputum of a patient with hemoptoic pneumonia. J Clin Microbiol. 2004;42:5493–5501. doi: 10.1128/JCM.42.12.5493-5501.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassi M, Bebeacua C, Drancourt M, Cambillau C. The First Structure of a Mycobacteriophage, the Mycobacterium abscessus subsp. bolletii Phage Araucaria. J Virol. 2013;87:8099–8109. doi: 10.1128/JVI.01209-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling AE, Mau B, Perna NT. ProgressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One. 2010;5:e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. PHAST: a fast phage search tool. Nucleic Acids Res. 2011;39:W347–W352. doi: 10.1093/nar/gkr485. Web Server issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grissa I, Vergnaud G, Pourcel C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007;35:W52–W57. doi: 10.1093/nar/gkm360. Web Server issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson DH, Bryant D. Application of Phylogenetic Networks in Evolutionary Studies. Mol Biol Evol. 2006;23:254–267. doi: 10.1093/molbev/msj030. [DOI] [PubMed] [Google Scholar]
- Li L, Stoeckert J, Christian J, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dongen S. Graph clustering by flow simulation. The Netherlands: PhD thesis. University of Utrecht; 2000. [Google Scholar]
- Snel B, Bork P, Huynen MA. Genome phylogeny based on gene content. Nat Genet. 1999;1:108–111. doi: 10.1038/5052. [DOI] [PubMed] [Google Scholar]
- Georgiades K, Merhej V, El Karkouri K, Raoult D, Pontarotti P. Gene gain and loss events in Rickettsia and Orientia species. Biol Direct. 2011;6:6. doi: 10.1186/1745-6150-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouret P, Thompson JD, Pontarotti P. PhyloPattern: regular expressions to identify complex patterns in phylogenetic trees. BMC Bioinformatics. 2009;10:298. doi: 10.1186/1471-2105-10-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le PT, Ramulu HG, Guijarro L, Paganini J, Gouret P, Chabrol O, Raoult D, Pontarotti P. An automated approach for the identification of horizontal gene transfers from complete genomes reveals the rhizome of Rickettsiales. BMC Evol Biol. 2012;12:243. doi: 10.1186/1471-2148-12-243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouret P, Vitiello V, Balandraud N, Gilles A, Pontarotti P, Danchin EG. FIGENIX: intelligent automation of genomic annotation: expertise integration in a new software platform. BMC Bioinformatics. 2005;6:198. doi: 10.1186/1471-2105-6-198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KC, Castro-Nallar E, Fisher JN, Breakwell DP, Grose JH, Burnett SH. Phage cluster relationships identified through single gene analysis. BMC Genomics. 2013;14:410. doi: 10.1186/1471-2164-14-410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo SW, Wong YL, Tan JL, Ong CS, Wong GJ, Ng KP, Ngeow YF. Annotated genome sequence of Mycobacterium massiliense strain M154, belonging to the recently created taxon Mycobacterium abscessus subsp. bolletii comb. nov. J Bacteriol. 2012;194:4778. doi: 10.1128/JB.01043-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassi M, Drancourt M. Genome analysis reveals three genomospecies in Mycobacterium abscessus. BMC Genomics. 2014;15:359. doi: 10.1186/1471-2164-15-359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV, Makarova KS. CRISPR-Cas: evolution of an RNA-based adaptive immunity system in prokaryotes. RNA Biol. 2013;10:679–686. doi: 10.4161/rna.24022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adékambi T, Ben Salah S, Khlif M, Raoult D, Drancourt M. Survival of environmental mycobacteria in Acanthamoeba polyphaga. Appl Environ Microbiol. 2006;72:5974–5981. doi: 10.1128/AEM.03075-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer M, Yutin N, Pagnier I, Barrassi L, Fournous G, Espinosa L, Robert C, Azza S, Sun S, Rossmann MG, Suzan-Monti M, La Scola B, Koonin EV, Raoult D. Giant Marseillevirus highlights the role of amoebae as a melting pot in emergence of chimeric microorganisms. Proc Natl Acad Sci U S A. 2009;106:21848–21853. doi: 10.1073/pnas.0911354106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penz T, Horn M, Schmitz-Esser S. The genome of the amoeba symbiont "Candidatus Amoebophilus asiaticus" encodes an afp-like prophage possibly used for protein secretion. Virulence. 2010;1:541–545. doi: 10.4161/viru.1.6.13800. [DOI] [PubMed] [Google Scholar]
- Filée J, Chandler M. Gene exchange and the origin of giant viruses. Intervirology. 2010;53:354–361. doi: 10.1159/000312920. [DOI] [PubMed] [Google Scholar]
- Colson P, Raoult D. Gene repertoire of amoeba-associated giant viruses. Intervirology. 2010;53:330–343. doi: 10.1159/000312918. [DOI] [PubMed] [Google Scholar]
- Thomas V, Greub G. Amoeba/amoebal symbiont genetic transfers: lessons from giant virus neighbours. Intervirology. 2010;53:254–267. doi: 10.1159/000312910. [DOI] [PubMed] [Google Scholar]
- Bertelli C, Greub G. Lateral gene exchanges shape the genomes of amoeba-resisting microorganisms. Front Cell Infect Microbiol. 2012;2:110. doi: 10.3389/fcimb.2012.00110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slimani M, Pagnier I, Raoult D, La Scola B. Amoebae as battlefields for bacteria, giant viruses, and virophages. J Virol. 2013;87:4783–4785. doi: 10.1128/JVI.02948-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatfull GF, Hendrix RW. Bacteriophages and their genomes. Curr Opin Virol. 2011;1:298–303. doi: 10.1016/j.coviro.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatfull GF. The secret lives of mycobacteriophages. Adv Virus Res. 2012;82:179–288. doi: 10.1016/B978-0-12-394621-8.00015-7. [DOI] [PubMed] [Google Scholar]
- Lindell D, Jaffe JD, Coleman ML, Futschik ME, Axmann IM, Rector T, Kettler G, Sullivan MB, Steen R, Hess WR, Church GM, Chisholm SW. Genome-wide expression dynamics of a marine virus and host reveal features of co-evolution. Nature. 2007;449:83–86. doi: 10.1038/nature06130. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Mycobacterium abscessus genome properties and their prophage regions.
Mycobacterium abscessus CRISPRs and the correlation with number of prophage regions.
The reconstructed trees for HGT events. Each tree contains one to six HGT events. The yellow squares represent the HGT event.
Environmental bacteria hosting homologous M. abscessus phage proteins and evidence for bacteria-amoeba interaction.