

Abstract
Proteomic profiling of serum is a powerful technique to identify differentially expressed proteins that can serve as biomarkers predictive of disease onset. In this study, we utilized 2D gel analysis followed by MALDI-TOF mass spectrometry analysis to identify putative serum biomarkers for autoimmune type 1 diabetes (T1D) in BioBreeding Diabetes Resistant (BBDR) rats induced to express disease. Treatment with toll-like receptor 3 (TLR3) ligand, polyinosinic:polycytidilic acid (pIC), plus infection with Kilham rat virus (KRV), a rat parvovirus, results in nearly 100% of young BBDR rats becoming diabetic within 11–21 days. Sera collected from pre-diabetic rats at early time points following treatment with pIC + KRV were analyzed by 2D gel electrophoresis and compared with sera from control rats treated with PBS, pIC alone, or pIC + H1, a non-diabetogenic parvovirus. None of the latter three control treatments precipitates T1D. 2D gel analysis revealed that haptoglobin, an acute phase and hemoglobin scavenger protein, was differentially expressed in the sera of rats treated with pIC + KRV relative to control groups. These results were confirmed by Western blot and ELISA studies that further validated haptoglobin levels as being differentially increased in the sera of pIC + KRV treated rats relative to controls during the first week following infection. Early elevations in serum haptoglobin were also observed in LEW1.WR1 rats that became diabetic following infection with rat cytomegalovirus (RCMV). The identification and validation of haptoglobin as a putative serum biomarker for autoimmune T1D in rats now affords us the opportunity to test the validity of this protein as a biomarker for human T1D, particularly in those situations where viral infection is believed to precede onset of disease.
Keywords: BBDR rat, biomarker, haptoglobin, KRV, type 1 diabetes
Introduction
Type 1 diabetes (T1D) is characterized by near total insulin deficiency resulting from the autoimmune destruction of insulin producing beta cells in the pancreatic islets. The disease predominantly affects young children, and imposes a significant burden on their lives stemming from its chronicity and its potentially life-threatening complications. A strong association between disease susceptibility and certain HLA genes has been shown (1, 2). Genetic susceptibility alone, however, is insufficient to explain disease penetrance since concordance in monozygotic twins ranges only between 30–50% (3). Therefore, genetic susceptibility in conjunction with environmental perturbants may be necessary to precipitate disease. The most important of these environmental perturbants may be viruses (4, 5). T1D predominates as a disease of children who tend to be exposed to a variety of infectious agents that may influence predisposition to autoimmunity during a critical period during maturation of their immune system.
Definitive proof that any given virus directly precipitates diabetes in humans is still lacking. The paucity of data supporting causal association with viral infections may be attributable to the fact that the autoimmune reaction is not clinically apparent immediately following infection. Rather, the destruction of pancreatic islets can take an indolent course, progressing for years before clinical diagnosis is established. The recent availability of novel immunomodulatory therapies such as anti-CD3, which may preserve residual beta cell mass in new onset diabetics (6), has generated a demand for noninvasive testable biomarkers that can identify the development of the autoreactive process before it becomes clinically apparent. Reliably identifying patients in the early phases of disease would provide clinicians with a window of opportunity when pharmacotherapy could be more effective in halting disease.
Currently, commonly used predictive markers for T1D include human leukocyte antigen (HLA) typing and serum autoantibodies to pancreatic islet antigens. However, these markers are most predictive in detecting familial cases of T1D, where a proband with positive antibody titers already exists, and are less predictive of disease in the general population (7, 8). Their lack of predictive power in the general population stems from the fact that high risk HLA-DQ and DR genotypes occur in only a small percentage (2%) of the population, of which only 30–40% develop T1D (9, 10). Alternatively, autoantibodies against protein tyrosine phosphatase IA2, islet cell antigen 512 (together called ICA), insulin (also known as IAA), glutamic acid decarboxylase (GAD), and zinc transporter ZnT8, have been shown to appear in the blood of diabetics many years prior to disease onset and are better predictors of disease in relatives of probands (11, 12). Ideally, the most important biomarkers would identify people at risk soon after beta cell destruction is initiated, for example, as a sequelae of a viral infection prior to the appearance of autoantibodies.
Viral infections such as cytomegalovirus, mumps, rubella, enteroviruses, and parvovirus have all been associated with human T1D (13). Indeed, the effects of diverse viruses in triggering T1D may explain the heterogeneous nature of disease onset and kinetics in the general population. In animal models of T1D, unequivocal evidence of virally-induced T1D comes from studies of BBDR rats infected with the UMass strain of rat virus (KRV-UMass), a ssDNA parvovirus (14, 15). When maintained in a viral antibody free facility (VAF), BBDR rats never develop T1D, despite having the susceptible RTIu MHC haplotype (16). However, when KRV-infected BBDR rats are pre-treated for three consecutive days with a low dose (1 μg/g) of polyinosinic:polycytidilic acid (pIC, a synthetic dsRNA moiety that serves as a ligand for TLR3 and MDA5), the conversion to diabetes among these rats is nearly 100% (16).
The exact mechanism by which treatment with KRV synergizes with pIC to induce diabetes in the BBDR rat is still unclear, but it does not appear to involve direct infection of the islets (17). However, the highly predictable time course with which the pIC + KRV treatment in BBDR rats leads to nearly 100% conversion to diabetes 11–21 days following treatment onset allows us to identify putative serum biomarkers in the earliest phase of the autoimmune process among rats that we know will develop diabetes. In this study, we used a global proteomics technique to analyze the sera of pre-diabetic rats treated with pIC plus KRV infection to identify proteins that may serve as biomarkers for T1D. Our screen of the BBDR rat serum proteome has revealed a potential biomarker, haptoglobin, an acute phase and hemoglobin scavenger protein that has a differential expression pattern in the sera of pIC + KRV treated rats as compared to control groups of BBDR rats.
Materials and methods
Animals and cell lines
Viral-antibody-free BBDR/Wor and LEW1.WR1 rats were obtained from Biomedical Research Models, Inc. (Worcester, MA). Animals were certified to be free of a panel of viruses, including KRV and RCMV. All animals were housed in a viral-antibody-free facility in microisolator cages and provided with water and commercial chow ad libitum. BBDR and LEW.1WR1 rats 21–25 days of age of either sex were used and maintained in accordance with the Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, National Research Council, National Academy of Sciences, 1996) and guidelines of the Institutional Animal Care and Use Committee of the University of Massachusetts Medical School. KRV-UMass and NRK cells were obtained from stocks maintained in our laboratories. KRV (UMass isolate) was propagated in NRK cells grown in Dulbecco’s minimal essential medium (DMEM) (18). Toolan’s H-1 virus was obtained from ATCC (Manassas, VA) and was propagated in Chang liver cells (18). RCMV was prepared as described (19).
Virus infection protocols
Polyinosinic:polycytidylic acid (pIC) was purchased from Sigma (St. Louis, MO), dissolved in Dulbecco’s PBS (1 mg/ml), sterile filtered, and stored at −20°C until used. The concentration of contaminating endotoxin was determined commercially (Charles River Endosafe, Charleston, SC) and was uniformly <10 units/mg. Rats were injected intraperitoneally (i.p.) with pIC (1 μg/g body weight on days −3, −2, and −1) starting at 21–25 days of age; pIC treated rats were either given no further treatment or infected i.p. with 1 × 107 PFU KRV or H-1 in a volume of 1 ml on treatment day 0. In designated experiments LEW1.WR1 rats were treated with 5 × 106 PFU of RCMV alone. In experiments designed to measure the frequency of diabetes induction, all treated rats were screened for glycosuria twice weekly. Diabetes was diagnosed on the basis of a plasma glucose concentration >250 mg/dl (11.1 mM/L).
Blood collection/serum harvesting
Blood (200–500 μl) was collected on indicated days via tail vein nicking into unheparinized tubes. On experiment termination days, rats were first euthanized in a CO2 chamber and blood was subsequently harvested by cardiac puncture. All blood samples were allowed to clot at room temperature for 1 hr, centrifuged at 4°C at 13000 × g for 10 min, and sera harvested. Sera used for proteomics experiments were additionally re-centrifuged at 4°C at 13000 × g for 10 min to remove any residual cell debris. All sera were aliquoted and frozen at −80°C until use.
Immunoaffinity removal of rat serum abundant proteins
Rat serum samples were depleted of albumin, IgG and transferrin using the Multi Affinity Removal Column (Ms-3, 4.6×100mm, Agilent Technologies). Sera were first diluted with 4 volumes of Agilent solvent A and centrifuged at 16000 × g for 1 minute through a 0.22 μm spin filter to remove particulates. Five hundred microliters of the diluted serum (1:10) was then injected onto an Applied Biosystems Vision™ Workstation liquid chromatography system with the attached Agilent Multi Affinity Removal Column at a flow rate of 0.5 ml/min. Flow through was collected in a volume of 1.5 mls, and an aliquot was analyzed by one-dimensional (1D) sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). Proteins captured by the column were removed with Agilent solvent B at a flow rate of 1 ml/min and analyzed in parallel with the flow-through samples. The column was regenerated by equilibrating with solvent A.
Native and non-native one-dimensional electrophoresis
Nondepleted serum samples were quantified using the Bradford dye-binding assay, and 3 μg of total protein from each sample was loaded onto 8–16% Tris-HCl precast gels (Bio-Rad). Gels were run in Tris glycine buffer without sodium dodecyl sulfate or any denaturing agents at a constant voltage of 200 V for 50 min. Depleted samples were concentrated on Amicon Ultra 4 10kDa cutoff ultracentrifugation columns (Millipore). Protein quantitation of the concentrated sample was achieved using the Bradford dye-binding assay. Four micrograms of the concentrated samples were reduced with 50mM DTT and subjected to electrophoresis on 4–12% Bis-Tris SDS NuPage gels (Invitrogen) according to the manufacturer’s protocol. Gels were run with the MOPS SDS running buffer (Invitrogen) at a constant voltage of 200V for 50 min. Both native and non-native one dimensional gels were stained overnight with Coomassie blue and destained with milliQ water prior to imaging.
Two-dimensional electrophoresis
Prior to isoelectric focusing (IEF), samples were solubilized in 40mM Tris, 7M urea, 2M thiourea and 2% CHAPS, reduced with tri-butylphosphine, and alkylated with 10mM acrylamide for 90 min at room temperature. Protein was precipitated from each sample by mixing it with nine times the sample volume of acetone for 30 minutes at room temperature. Following precipitation, the pellet was again solubilized and buffer exchanged in 7M urea, 2M thiourea, and 2% CHAPS until conductivity was < 150 μS/cm, and 100μg protein was subjected to IEF on 11 cm pH 4–7 and pH 6–11 immobilized pH gradient (IPG) strips (Amersham). Following IEF, IPG strips were equilibrated in 6M urea, 2% SDS, 50mM Tris-acetate buffer (pH 7.0), and 0.01% bromophenol blue and subjected to SDS-PAGE on 8–16% Tris-HCL Gel (Bio-Rad). All gels were stained in Sypro® Ruby (Molecular Probes, Eugene, OR) and imaged by a charge-coupled device camera on a fluorescent imager (Bio-Rad Gel-Doc).
Axima QIT MALDI MS analysis
Gel spots excised with an automatic gel cutter (Bio-Rad) were washed twice for 30 min with 200 μl of 25mM ammonium bicarbonate in 50% acetonitrile (ACN). ACN (50 μl) was added to shrink the gel, the excess removed and the gel dried using a SpeedVac. Digestion was performed by the addition of 30 μl of 2 ng/μl trypsin (Sigma Proteomics Grade) in 25mM ammonium bicarbonate with incubation at 37°C overnight. This solution was transferred to a clean tube and the gel was further extracted with 30 μl of 80% ACN:0.1% formic acid. The combined extracts were dried using a SpeedVac to a 10μl volume and acidified with 1–2 μl of 1% trifluoroacetic acid (TFA). Samples were loaded on a μC18 Zip Tip (Millipore) pre-equilibrated with 0.1% TFA. After washing with 2 × 10 μl aliquots of 0.1% TFA, samples were deposited directly onto the MALDI sample target using 1 μl of matrix solution (15 mg/ml of 2,5-dihydroxybenzoic acid; MassPrep DHB, Waters Corp., Mildford, MA) in 50:50 ACN:0.1% TFA. Sample plates were allowed to air dry prior to insertion into the mass spectrometer. Analysis was performed on a Kratos Axima QIT (Shimadzu Instruments) matrix-assisted-laser desorption/ionization (MALDI-TOF) mass spectrometer. Peptides were analyzed in positive ion, mid mass range mode (m/z 700–3000). The instrument was externally calibrated with Angiotensin II (MH+ at m/z 1046.54), P14R (MH+ at m/z 1533.86) and ACTH 18–39 (MH+ at m/z 2465.20). Precursors were selected based on signal intensity at a mass resolution width of 250 for collision-induced dissociation (CID) fragmentation using argon as the collision gas. Database searches were performed with Mascot (Matrix Sciences, Ltd.) using the rat International Protein Index (IPI) database and Peptide Mass Fingerprint program for MS data and the MS/MS Ion Search program for CID data. Since low abundance samples typically provide an insufficient number of peptides to make an identification based solely on MS information, all identifications were confirmed or established with CID (MS/MS) data.
Western blot analysis
The Bradford assay was used to quantify protein levels from rat sera. Three micrograms of each serum sample were reduced by 2-mercaptoethanol, loaded onto 4–20% precast Tris-glycine gradient gels (Invitrogen) along with purified haptoglobin (Life Sciences) and molecular weight marker (Gibco), and run at 150V for 1 hour. Gels were transferred onto PVDF membranes, blocked with 5% milk for at least 1 hour, and incubated with primary antibody (chicken anti-rat haptoglobin, Abcam) for 2 hours. Membranes were washed three times with TBS+tween, and incubated with secondary antibody (rabbit anti-chicken HRP, Abcam) for 1 hour. Membranes were washed two times with TBS+tween, once with TBS alone, and developed with ECL (Gibco). Membranes were imaged using Kodak chemiluminescent film.
ELISA
Haptoglobin ELISA kit was purchased from Immunology Consultants Lab (Newberg, OR) and used according to the manufacturer’s instructions. Standard curves were generated utilizing haptoglobin standards supplied in the kit. All test samples were quantified in duplicate, and any values out of range were appropriately diluted and reanalyzed.
Statistical analysis
Two-tailed Student’s T tests were performed to detect differences in spot values of 2D gels for serum collected on the first day following KRV treatment (day +1) for each combination of the three treatment groups, pIC + KRV, pIC alone, and PBS alone. ANOVA analysis with the Bonferroni test for a posteriori contrasts (GraphPad Prism 4.0, San Diego, CA) was used to compare ELISA data on serum haptoglobin levels. P values of less than 0.05 were considered to be significant.
Results
2D gel and MALDI-TOF-MS/MS analysis identifies haptoglobin as an early putative biomarker for virus-induced autoimmune diabetes
To screen for early biomarkers of virus-induced autoimmune diabetes, BBDR rats were pre-treated with pIC on days −3, −2, and −1 followed by KRV infection on day 0. Control rats (which will not become diabetic) were injected with PBS or pIC alone on the same days, but not infected with KRV. Blood was collected from all rats for analysis on day +1. Serum was obtained from each sample, depleted of the three most highly abundant proteins (albumin, immunoglobulin and transferrin), and subjected to 2D gel electrophoresis followed by image analysis of differentially expressed proteins. Proteins whose levels were significantly different on day +1 between the rats in each of the treatment groups were selected for identification by MALDI-TOF mass spectrometry. Table 1 contains proteins that are differentially expressed in sera from rats treated with pIC + KRV or pIC alone versus PBS, and reflect the effects of pIC treatment +/− KRV. In contrast, the proteins in Table 2 show proteins that are differentially expressed in sera from rats treated with pIC + KRV versus those treated with pIC alone. Of the proteins identified, most were either acute phase proteins such as α2 macroglobulin or complement component 3, or proteins involved in hemolysis and the heme scavenging pathway such as hemoglobin, biliverdin reductase B, and hemopexin. However, the serum protein whose levels were most affected by treatment was haptoglobin (~9-fold increase in pIC + KRV or pIC alone treated rats compared to PBS controls).
Table 1.
Differentially expressed proteins in sera of rats treated with pIC + KRV or pIC alone vs PBS
| Protein ID | Function | Measured pl | Measured MW (kDa) | Fold Difference | P Value |
|---|---|---|---|---|---|
| haptoglobin | acute phase heme scavenger hemolysis | 5.694 5.230 5.363 5.114 |
41.240 43.719 42.562 44.196 |
9.642 3.212 2.488 2.049 |
0.0003603 0.0009308 0.0007297 0.002 |
| alpha and beta globin | hemolysis | 7.700 | 12.491 | 6.179 | 0.007 |
| alpha 2 macroglobulin | acute phase hemolysis serpin | 6.309 6.414 6.319 6.723 5.420 5.457 5.386 |
32.089 42.516 30.937 30.564 132.628 129.799 132.628 |
4.336 4.059 3.258 2.693 2.480 2.206 2.032 |
0.003 0.017 0.007 2.59E-05 3.04E-05 0.0002132 0.023 |
| biliverdin reductase B | acute phase heme degradation | 6.553 | 23.304 | 2.970 | 0.008 |
| major beta hemoglobin | hemolysis | 7.312 7.004 |
13.037 12.199 |
2.904 2.822 |
4.92E-07 0.011 |
| complement component 3 | acute phase | 8.635 5.834 |
61.733 17.071 |
2.841 −2.375 |
0.000105 0.046 |
| plasminogen protein | plasmin precursor | 7.095 | 166.928 | 2.399 | 0.001 |
| hemopexin | acute phase heme degradation | 6.685 6.685 |
50.229 46.493 |
2.112 2.016 |
6.86E-05 0.0001485 |
| alpha 1 inhibitor III | serpin | 5.156 4.497 4.916 |
38.697 19.921 23.870 |
−2.324 −2.340 −2.674 |
8.64E-08 0.008 0.001 |
| Spin 2a | serpin | 4.623 | 17.290 | −2.437 | 0.0001016 |
Positive values indicate the fold increase in serum protein levels while negative values indicate the fold decrease in pIC + KRV and pIC alone treated rats compared to PBS treated controls.
Table 2.
Differentially expressed proteins in sera of rats treated with pIC + KRV vs pIC alone
| Protein ID | Measured pl | Measured MW (kDa) | Fold Difference | P Value |
|---|---|---|---|---|
| preprohaptoglobin | 5.902 5.174 |
29.093 32.225 |
−3.579 −3.156 |
0.008 0.00001633 |
| haptoglobin precursor | 4.994 5.360 |
12.967 12.695 |
−2.854 −2.624 |
0.014 0.007 |
| haptoglobin precursor or hatoglobin alpha 1s |
5.871 |
30.625 |
−2.501 |
0.001 |
| bal-647 | 5.202 5.388 |
30.411 29.891 |
−3.060 −2.245 |
0.039 0.022 |
| complement component 3 | 7.921 | 231.297 | −2.115 | 4.81E-06 |
Negative values indicate the fold decrease in serum protein levels in pIC + KRV treated rats relative to those treated with pIC alone.
Haptoglobin is an acute phase and hemoglobin scavenger protein that normally circulates in the serum of uninfected rats. Haptoglobin is translated as a single polypeptide, preprohaptoglobin, which contains a signal sequence as well as the α- and ß-subunit regions. Prohaptoglobin (lacking the signal sequence) is glycosylated in the endoplasmic reticulum, dimerized, and a portion is cleaved into the α- and ß-components. In the rat, haptoglobin is secreted as both an α2ß2 tetramer and also as dimerized prohaptoglobin (20, 21). It is the α2ß2 tetrameric form that sequesters free hemoglobin, which may be released into the circulation as a result of trauma or inflammation. Such trauma or inflammation can additionally stimulate increased synthesis of haptoglobin precursor protein from hepatocytes, with subsequent cleavage by serum proteases to the active α2ß2 tetrameric structure (20, 21). We observed that several modified variants of the activated haptoglobin protein were identified by MALDI-TOF mass spectrometry (Table 1). These isoforms ranged in pI from 5.1 to 5.6 and in molecular weight from 41 – 44 kDa, and all isoforms appeared to be significantly elevated (between 2- and 9-fold) in the sera of rats treated with pIC + KRV or pIC alone, when compared with rats treated with PBS alone. Interestingly, the other significantly elevated protein (~6-fold) in the sera of pIC + KRV or pIC alone treated rats compared with rats treated with PBS alone included the alpha and beta globin chains of hemoglobin, targets of haptoglobin binding. In addition, it is intriguing that the preprohaptoglobin was lower in pIC + KRV treated rats than those receiving pIC alone (Table 2), consistent with an increased conversion of precursor haptoglobin to its active form in the pIC + KRV treated rats. Thus, as early as one day after virus infection, significant differences were found in the sera of rats that did, or did not, progress to diabetes.
Both precursor and activated forms of haptoglobin remain elevated in pIC + KRV treated rats compared to those treated with pIC +/− non-diabetogenic H1 virus
To next 1) validate the results obtained in the 2D gel experiments using a different assay and 2) analyze serum haptoglobin levels in the context of disease outcome, Western blot analysis of undepleted serum samples was performed. BBDR rats were randomized to one of four treatment groups: PBS alone, pIC alone, pIC + KRV, or pIC + H-1 virus. KRV and H-1, both parvoviruses, share >80% sequence homology within their structural proteins, and 100% homology within their nonstructural proteins. H-1, however, when injected alone or with pIC, does not cause diabetes in BBDR rats (22). In one study, blood was collected by tail bleed. Because we were concerned about the extent to which the method of bleeding rats might have promoted hemolysis in our samples, thereby contributing to the selective identification of heme associated proteins, we also performed a second study using cardiac puncture. In our experience, this method is least likely to rupture red blood cells and promote hemolysis.
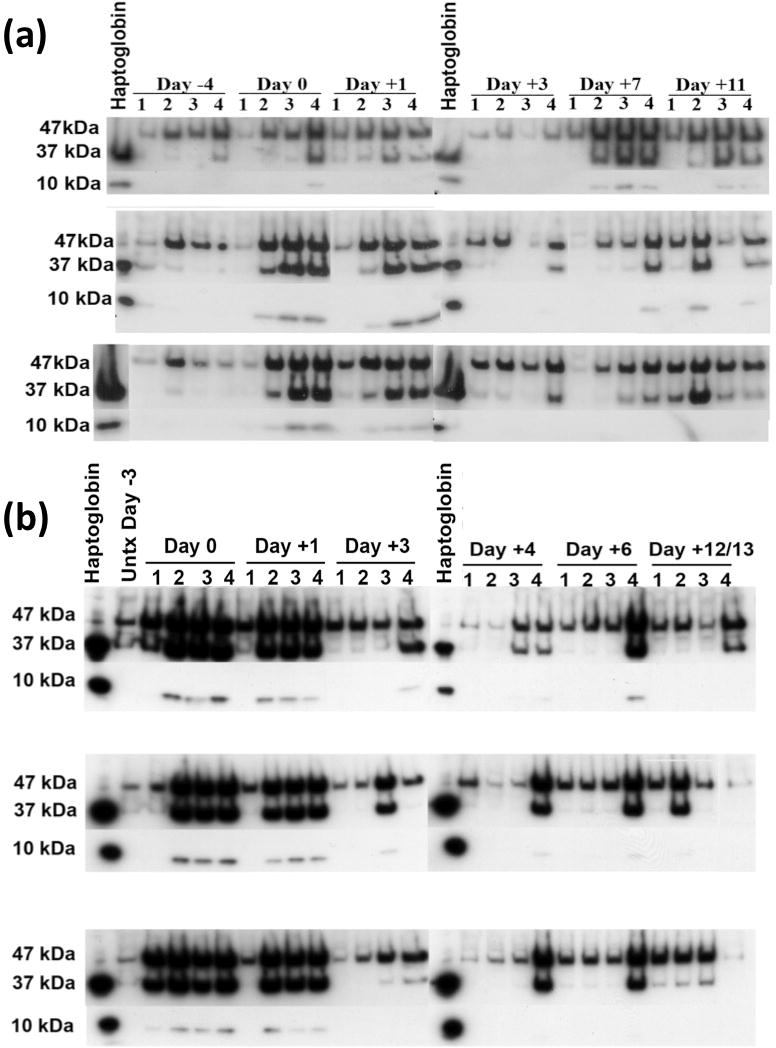
Blood was collected from BBDR rats over a ~2 week time course (where day −4 is pretreatment). As above, rats were treated with pIC (or PBS) on days −3, −2, and −1 followed by KRV (or H1) infection on day 0. Representative Western blot data from the tail bleed and cardiac puncture studies are shown in Figure 1a and 1b, respectively. Three haptoglobin-specific bands were typically found in each sample: (1) prohaptoglobin (47kDa), the uncleaved protein that normally circulates in the body even in the absence of infection, and the (2) beta (37kDa) and (3) alpha (10kDa) chain of the cleaved, activated haptoglobin molecule. On day 0 and day +1, in both the tail bleed and cardiac puncture experiments, Western blot analysis demonstrated an increase in all three bands in the pIC treated group with or without concomitant virus infection of rats compared with rats treated with PBS alone. The intensity of all three bands decreased subsequent to day +1, although levels remained highest in the pIC + KRV group. This decrease in prohaptoglobin and activated haptoglobin in the serum appears to correspond with the serum clearance of pIC, which is undetectable by 24–48 hrs following i.p. injection (Dr. Danny Zipris, personal communication).
Figure 1.
Western blot analysis of undepleted sera from rats treated with PBS alone (1), pIC alone (2), pIC + H-1 (3) or pIC + KRV (4). Protein concentration was assessed by the Bradford assay and equal protein was loaded in each well. Rats were tail bled (a) or cardiac bled (b) at indicated time points. Purified rat haptoglobin was used as a positive control; molecular weight of precursor prohaptoglobin is 47 kD and cleaved, activated haptoglobin is 37 kD (β chain) and 10 kD (α chain). In (a), serum was collected from a single rat in each group for the time points indicated; shown are data from 3 rats per group. In (b), each lane represents serum from a single rat.
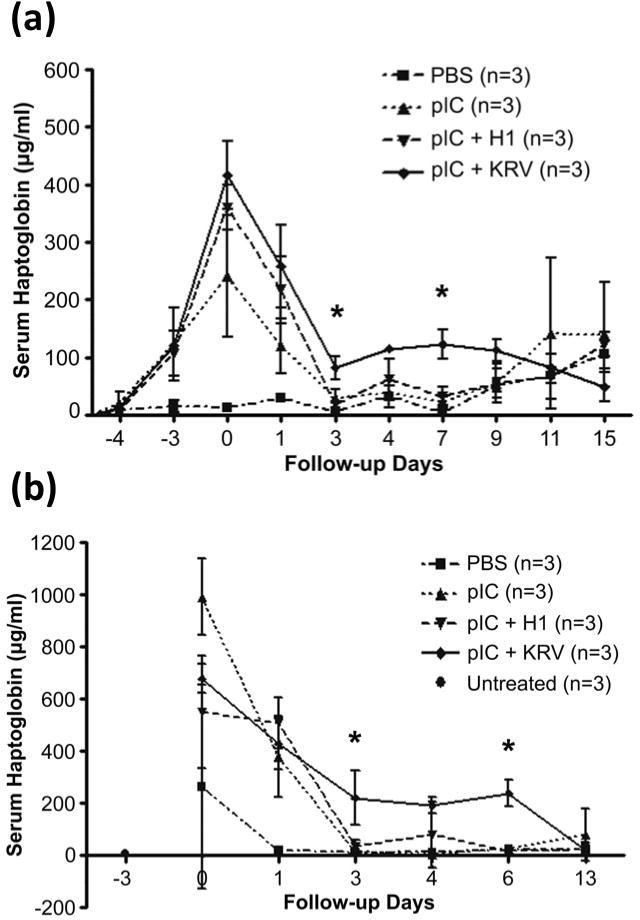
ELISA quantitation of serum haptoglobin levels distinguish pIC + KRV treated rats from those treated with pIC alone or with concomitant infection with the non-diabetogenic H1 virus
The above studies show that serum haptoglobin is increased in virus-induced diabetes, but to have utility as a biomarker it must also be quantifiable. Therefore, we next used ELISA analysis to quantify serum haptoglobin and correlate the levels with diabetes outcome. In sera from tail bleeds or cardiac punctures of rats treated with PBS alone, pIC alone, pIC + KRV or pIC + H-1, we observed similar trends in serum haptoglobin levels quantified by ELISA (Figure 2) as observed by Western blot (Figure 1). Although the total amount of haptoglobin was lower in the sera harvested from tail blood than cardiac blood, perhaps due to increased clotting and consumption of free serum haptoglobin, the overall changes in haptoglobin levels over time in each of the treatment groups appeared remarkably similar.
Figure 2.
Serum haptoglobin levels determined by ELISA in BBDR rats treated with PBS alone, pIC alone, pIC + H-1 and pIC + KRV. Serum samples were obtained from rats that were (a) tail bled or (b) cardiac bled at indicated time points, and were run in duplicate against a standard curve of purified rat haptoglobin. *P < 0.05 between pIC + KRV and pIC + H-1 groups.
In both tail and cardiac bled rats, haptoglobin levels were dramatically elevated on days 0 (after pIC treatment but prior to virus infection) and +1 (one day after virus infection) in all groups that received pIC relative to rats treated with PBS alone. Haptoglobin levels declined thereafter, perhaps as a consequence of clearance of pIC from the bloodstream. In contrast, serum haptoglobin levels remained significantly elevated in the pIC + KRV treated rats when compared with the pIC + H-1 treated rats on days +3 and day +7 in tail bled rats (Figure 2a, P < 0.05), and on day +3 and day +6 in rats that were bled via the cardiac route (Figure 2b, P < 0.05). Both tail and cardiac bled rats also had higher haptoglobin levels on day +4 in the pIC + KRV group, although this elevation was not statistically significant. This rapid increase and decline in total haptoglobin levels, with a more sustained elevation only in the pIC + KRV treated animals, was also observed in the Western blot analysis (Figure 1).
Identification of hemoglobin:haptoglobin complexes in sera of BBDR rats treated with pIC +/− KRV
Our 2D and MALDI-TOF mass spectrometry analysis identified a significant increase in both haptoglobin and hemoglobin alpha and beta chains in pIC alone and pIC + KRV treated rats (Table 1). Since haptoglobin is known to complex with hemoglobin, we investigated the potential formation of haptoglobin:hemoglobin or other protein complexes by native gel. We subjected day +1 sera from rats treated with PBS alone, pIC alone or pIC + KRV, to native gel analysis (without denaturation). Bands that appeared to be different between treatment groups by gross visualization were excised, tryptically digested, and peptides identified by MALDI-TOF mass spectrometry. Interestingly, the most prominent band that appeared in the native gel of serum samples from day +1 of pIC alone and pIC + KRV treated rats was identified as a complex of hemoglobin alpha chain and haptoglobin (Figure 3). This band disappeared in the sera from rats treated with pIC alone on days +3 and +6, but persisted in the sera from rats treated with pIC + KRV on those same days.
Figure 3.
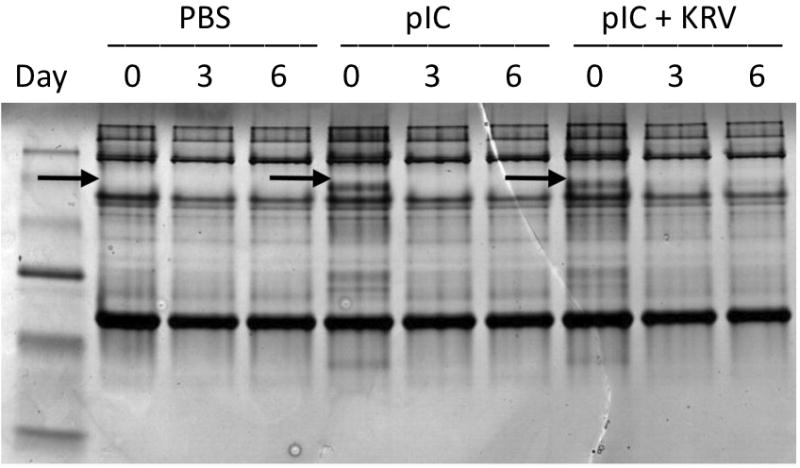
Native gel of sera from BBDR rats treated with PBS alone, pIC alone or pIC+KRV on indicated days. Protein concentration was quantified using the Bradford assay and equivalent concentrations of proteins were loaded into each well. Protein bands (indicated by arrow) that differed between groups were excised from individual lanes using a spot cutter; a molecular weight marker is shown in lane 1. Gel plugs from each lane were tryptically digested and peptides identified by MALDI-TOF/MS/MS.
Serum haptoglobin levels do not distinguish diabetes outcome in BBDR rats treated with KRV alone
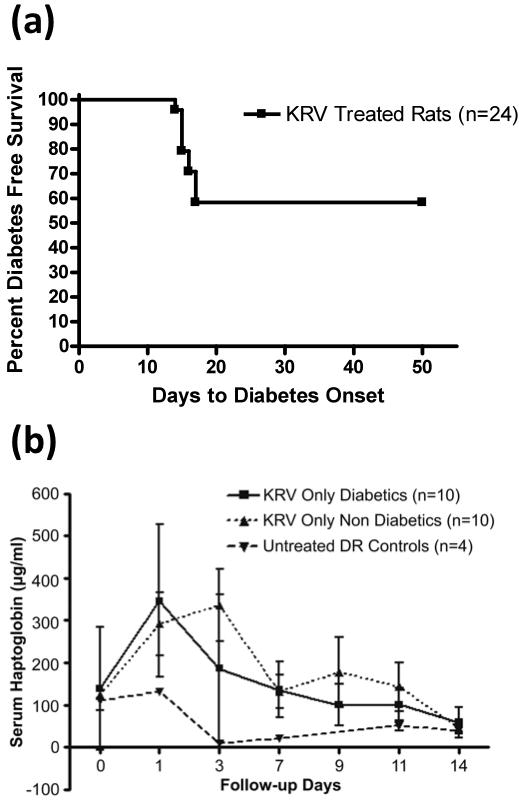
Validation of haptoglobin as a biomarker of T1D requires testing the predictive value of the marker in a variety of experimental settings where diabetes penetrance is not complete. For the next study, we again employed BBDR rats infected with KRV, but without pIC pre-treatment. BBDR rats treated with KRV alone develop T1D with a cumulative frequency of ~30% within 21 days (18). Because haptoglobin levels by Western blot and ELISA analyses gave concordant results, these subsequent studies utilized ELISA alone.
Following treatment with a single i.p. dose of KRV alone, 42% (10 of 24) of BBDR rats became diabetic within 17 days (Figure 4a). Haptoglobin levels of KRV treated rats, regardless of diabetes status, were always higher than untreated controls for all time points following infection, indicating that KRV alone promotes an increase in serum haptoglobin levels (Figure 4b). However, at any given time point, there was no statistically significant difference in serum haptoglobin levels between rats that became diabetic compared with those that remained diabetes free.
Figure 4.
Diabetes incidence and serum haptoglobin levels in BBDR rats treated with KRV alone. (a) Kaplan Meier survival curve depicts the percent of rats exhibiting diabetes free survival; n = 24 KRV treated rats, 10 developed diabetes. (b) Rats were tail bled on the indicated days and serum haptoglobin levels determined by ELISA. For any given time point, there were no significant differences in serum haptoglobin levels between KRV treated rats that became diabetic and those that remained diabetes-free.
Serum haptoglobin levels trend higher in pre-diabetic compared to diabetes-free LEW1.WR1 rats treated with RCMV alone
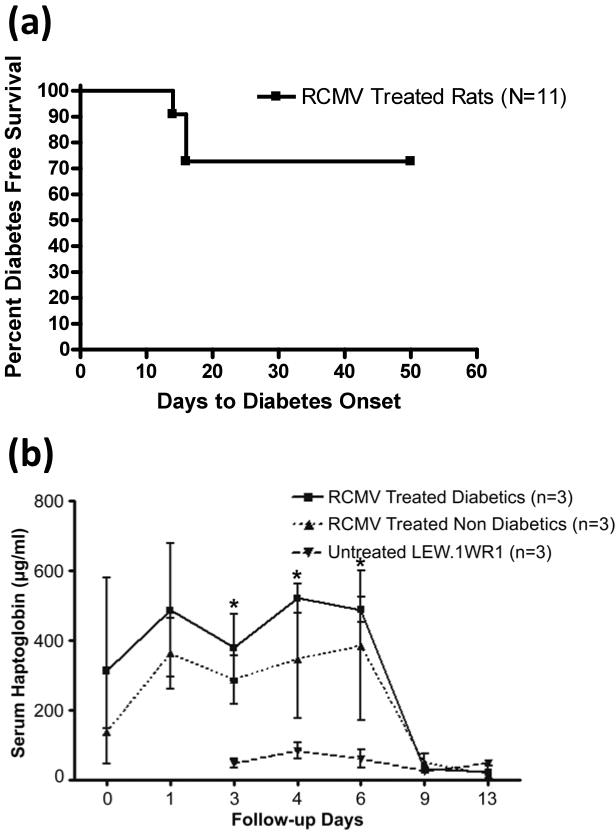
Because multiple viruses have been implicated in the initiation of human T1D, we next wished to investigate whether serum haptoglobin levels associate with diabetes outcome in a different rodent model of virally induced diabetes. For these studies we used LEW1.WR1 rats, which develop T1D in response to rat cytomegalovirus (RCMV) infection with the same frequency and time course as BBDR rats in response to KRV (23). In our studies we found that 22% (3 of 11) of LEW1.WR1 rats treated with a single dose of RCMV developed diabetes within 16 days (Figure 5a). RCMV treated rats that went on to become diabetic (but not those that remained diabetes-free) had significantly higher haptoglobin levels compared with untreated controls (Figure 5b, P <0.05).
Figure 5.
Diabetes incidence and serum haptoglobin levels in LEW1.WR1 rats treated with RCMV alone. (a) Kaplan Meier survival curve depicts the percent of rats exhibiting diabetes free survival; n = 11 RCMV treated rats, 3 developed diabetes. (b) Rats were tail bled on the indicated days and serum haptoglobin levels were determined by ELISA; n = 3 for each group (3 of 8 serum samples from RCMV treated non-diabetic rats were analyzed). Serum haptoglobin levels were significantly higher (*P < 0.05) in diabetic RCMV treated rats compared to untreated rats.
Discussion
The need to identify a biomarker that can predict virus-induced T1D in humans prompted us to search for such a marker in the serum proteome of a virus-induced rodent model of the disease. Causal association between viruses and T1D has been definitively established in rodent models, including the BBDR rat that becomes diabetic following infection with KRV. Importantly, T1D induced by pIC + KRV in BBDR rats shares several key similarities with human T1D, including lack of a gender bias among diabetics, and the risk of life threatening disease complications, including diabetic ketoacidosis. In this report, we used 2-D gel electrophoresis in conjunction with mass spectrometry to identify haptoglobin as an early biomarker in BBDR rat sera of pIC + KRV induced T1D. As confirmed by both Western and ELISA analyses, haptoglobin remained elevated in pIC + KRV treated rats, but not in those that remained diabetes-free following treatment with pIC alone or in conjunction with a non-diabetogenic virus.
In addition to haptoglobin, our initial 2-D gel analysis identified many acute phase proteins known to be involved in heme degradation, including hemopexin and biliverdin reductase. Alpha and beta hemoglobin were also identified, pointing to the engagement of the hemolytic pathway in our samples. Other proteins identified included alpha 1 inhibitor III, spin 2a, and alpha 2 macroglobulin. These proteins belong to the serine protease inhibitor (serpin) superfamily and play key roles in controlling inflammation (24). Of the identified proteins, the most significant differences in protein quantity occurred in the variants of mature haptoglobin, which increased up to 9-fold in both pIC alone and pIC + KRV treated rats relative to rats treated with PBS alone. Yet, when comparing the pIC alone and pIC + KRV treated groups, rats that were co-treated with KRV had significant decreases in the serum protein levels of haptoglobin precursors, which may reflect an enhanced cleavage of haptoglobin to the activated α- and ß-components. Indeed, this is consistent with the enhanced expression of the cleaved α- and ß-forms of haptoglobin shown by Western analysis (Figure 1) in most of the pIC + KRV treated rats.
A variety of infections, including parvoviral infection, can induce RBC hemolysis (25), releasing potentially toxic free hemoglobin into the serum. Activation of haptoglobin from its pro form allows free hemoglobin molecules to be sequestered in hemoglobin:haptoglobin complexes and cleared from the circulation (26). Our native gel data demonstrate that the elevation in serum haptoglobin in response to pIC treatment observed on day 0 corresponds with an elevation in the hemoglobin:haptoglobin complex formation in the serum, with a subsequent disappearance of this complex corresponding with the predicted clearance of pIC from the serum. However, in pIC + KRV treatment, the elevation in serum hemoglobin:haptoglobin complexes persisted at least through day +6, the time point when it was last tested. These results suggest that the observed increase in serum haptoglobin levels with pIC + KRV treatment, as quantified by ELISA, has a functional consequence of sequestering free hemoglobin in the serum. Whether subsequent KRV infection generated significantly more hemoglobin:haptoglobin complex formation, or prevented its clearance, is not known. However, lack of clearance of hemoglobin:haptoglobin complexes may suggest some monocyte dysfunction that prevents adequate clearance or, perhaps, saturation of the binding sites.
The further evaluation of haptoglobin as a biomarker was of particular interest due to its biological properties. Haptoglobin, an acute phase reactant, has been reported to inhibit hemoglobin-induced oxidative damage in cells, and possesses anti-inflammatory and immunomodulatory properties. It has been demonstrated to inhibit prostaglandin synthesis, dampen the inflammatory capacity of neutrophils, monocytes and macrophages (27), and interfere with B- and T-cell mitogenic responses in vitro (28). Additionally, in humans, mutations in the haptoglobin gene are associated with increased risk of diabetic complications such as retinopathy, nephropathy and cardiovascular disease (29, 30). Therefore, we were interested to determine if elevations in haptoglobin in our diabetes induction model might be a reliable marker for the autoreactive inflammatory response.
We performed several validation studies to determine if haptoglobin levels were sensitive in detecting T1D development, including in virus models in which T1D expression is variable. In our study, KRV alone (without pre-treatment with a TLR ligand) caused diabetes in 42% of infected BBDR rats. However, although serum haptoglobin levels were higher in KRV treated rats than in untreated controls, the absolute serum levels of haptoglobin did not distinguish diabetes outcome within KRV treated animals. In contrast, in the RCMV model, serum haptoglobin levels trended higher in LEW1.WR1 rats that became diabetic compared to those that remained diabetic-free, although this difference was not statistically significant. However, we did find a statistically significant difference when comparing the serum haptoglobin levels of pre-diabetic (but not diabetes-free) RCMV treated rats with untreated controls.
In this report, we provide evidence of haptoglobin as a biomarker for pIC + KRV-induced T1D, and its sustained serum elevations in this model may reveal insights into the pathogenesis of the disease. KRV is known to be strong inducer of pro-inflammatory cytokines such as IFN-γ and IL-12, and has been reported to alter lymphocyte responses and cytotoxic lymphocyte activity in vitro (22). A closely related virus, H-1, despite sharing 80–90% sequence homology, does not induce pro-inflammatory cytokines to the same degree (18). Five days following infection, serum IL-20p40 levels, for example, are highest in KRV-treated rats when compared with rats treated with pIC alone or H-1 virus alone (18). The kinetics of cytokine secretion corresponds with the kinetics with which we observed significant increases in the serum haptoglobin levels in pIC + KRV treated rats.
In addition to T and B cells, KRV is known to infect hepatocytes, and both KRV and H-1 are particularly pathogenic to the developing liver and cerebellum (31, 32). Haptoglobin is an acute phase response protein secreted primarily by the liver in response to proinflammatory cytokines (33), and perhaps, in the KRV model, in response to direct infection with KRV, although this remains to be demonstrated. Sustained elevations in serum haptoglobin in the pIC + KRV treated rats may be attempts by the body, albeit ineffective, to guard against excessive inflammation and cytotoxicity generated by this perturbation. Indeed, the length of time and degree to which haptoglobin levels are elevated may reveal a chronic inflammatory response to an environmental perturbant that can eventually precipitate autoimmunity in a susceptible host. Identification of differentially expressed serum proteins such as haptoglobin in the early phases of virus-induced diabetes in the BBDR rat might uncover novel biomarkers that may be used in conjunction with autoantibodies to provide a more robust predictor of environmentally initiated T1D in genetically susceptible children.
Acknowledgments
We would like to thank Michael Bates, Elaine Norowski, Linda Leehy, and Erich Lidstone for their technical assistance in animal handling and other laboratory techniques. This work was supported by a grant from the American Diabetes Association, and grants AI073871 and an institutional Diabetes Endocrinology Research Center (DERC) grant DK32520 from the National Institutes of Health. The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Footnotes
The authors have no conflict of interest to report.
Author contributions: All authors participated in planning the study and preparing the manuscript. AJK and CY designed experiments, collected serum samples, and performed ELISA analyses. KY prepared virus, and AJK, SWT, DK, JEE, KMG, and JL performed proteomics analyses and analyzed data. AJK, RB and DLG wrote the manuscript, and JPM, KY, DLG, AAR, and RB contributed to design of the studies and reviewed the manuscript.
References
- 1.She JX. Susceptibility to type I diabetes: HLA-DQ and DR revisited. Immunol Today. 1996;17:323–329. doi: 10.1016/0167-5699(96)10014-1. [DOI] [PubMed] [Google Scholar]
- 2.Field LL. Genetic linkage and association studies of Type I diabetes: challenges and rewards. Diabetologia. 2002;45:21–35. doi: 10.1007/s125-002-8241-7. [DOI] [PubMed] [Google Scholar]
- 3.Rossini AA, Handler ES, Mordes JP, Greiner DL. Human autoimmune diabetes mellitus: lessons from BB rats and NOD mice–Caveat emptor. Clin Immunol Immunopathol. 1995;74:2–9. doi: 10.1006/clin.1995.1002. [DOI] [PubMed] [Google Scholar]
- 4.von Herrath M. Can we learn from viruses how to prevent type 1 diabetes?: the role of viral infections in the pathogenesis of type 1 diabetes and the development of novel combination therapies. Diabetes. 2009;58:2–11. doi: 10.2337/db08-9027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jun HS, Yoon JW. The role of viruses in type I diabetes: two distinct cellular and molecular pathogenic mechanisms of virus-induced diabetes in animals. Diabetologia. 2001;44:271–285. doi: 10.1007/s001250051614. [DOI] [PubMed] [Google Scholar]
- 6.Chatenoud L, Bluestone JA. CD3-specific antibodies: a portal to the treatment of autoimmunity. Nat Rev Immunol. 2007;7:622–632. doi: 10.1038/nri2134. [DOI] [PubMed] [Google Scholar]
- 7.Decochez K, De Leeuw IH, Keymeulen B, Mathieu C, Rottiers R, Weets I, Vandemeulebroucke E, Truyen I, Kaufman L, Schuit FC, Pipeleers DG, Gorus FK. IA-2 autoantibodies predict impending type I diabetes in siblings of patients. Diabetologia. 2002;45:1658–1666. doi: 10.1007/s00125-002-0949-8. [DOI] [PubMed] [Google Scholar]
- 8.Barker JM, Barriga KJ, Yu L, Miao D, Erlich HA, Norris JM, Eisenbarth GS, Rewers M. Prediction of autoantibody positivity and progression to type 1 diabetes: Diabetes Autoimmunity Study in the Young (DAISY) J Clin Endocrinol Metab. 2004;89:3896–3902. doi: 10.1210/jc.2003-031887. [DOI] [PubMed] [Google Scholar]
- 9.Powers A. Diabetes Mellitus. In: Kasper DLBE, Fauci AS, Hauser SL, Longo DL, Jameson JL, Loscalzo J, editors. Harrison’s Principles of Internal Medicine. New York: McGraw-Hill Medical Publishing Division; 2008. [Google Scholar]
- 10.Zipris D. Epidemiology of type 1 diabetes and what animal models teach us about the role of viruses in disease mechanisms. Clin Immunol. 2009 doi: 10.1016/j.clim.2008.12.011. [DOI] [PubMed] [Google Scholar]
- 11.Rewers M, Norris JM, Eisenbarth GS, Erlich HA, Beaty B, Klingensmith G, Hoffman M, Yu L, Bugawan TL, Blair A, Hamman RF, Groshek M, McDuffie RS., Jr Beta-cell autoantibodies in infants and toddlers without IDDM relatives: diabetes autoimmunity study in the young (DAISY) J Autoimmun. 1996;9:405–410. doi: 10.1006/jaut.1996.0055. [DOI] [PubMed] [Google Scholar]
- 12.Verge CF, Gianani R, Kawasaki E, Yu L, Pietropaolo M, Jackson RA, Chase HP, Eisenbarth GS. Prediction of type I diabetes in first-degree relatives using a combination of insulin, GAD, and ICA512bdc/IA-2 autoantibodies. Diabetes. 1996;45:926–933. doi: 10.2337/diab.45.7.926. [DOI] [PubMed] [Google Scholar]
- 13.Filippi C, von Herrath M. How viral infections affect the autoimmune process leading to type 1 diabetes. Cell Immunol. 2005;233:125–132. doi: 10.1016/j.cellimm.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 14.Guberski DL, Thomas VA, Shek WR, Like AA, Handler ES, Rossini AA, Wallace JE, Welsh RM. Induction of type I diabetes by Kilham’s rat virus in diabetes-resistant BB/Wor rats. Science. 1991;254:1010–1013. doi: 10.1126/science.1658938. [DOI] [PubMed] [Google Scholar]
- 15.Thomas VA, Woda BA, Handler ES, Greiner DL, Mordes JP, Rossini AA. Altered expression of diabetes in BB/Wor rats by exposure to viral pathogens. Diabetes. 1991;40:255–258. doi: 10.2337/diab.40.2.255. [DOI] [PubMed] [Google Scholar]
- 16.Whalen BJ, Mordes JP, Rossini AA. The BB rat as a model of human insulin-dependent diabetes mellitus. Curr Protoc Immunol. 2001:13. doi: 10.1002/0471142735.im1503s19. Chapter 15:Unit 15. [DOI] [PubMed] [Google Scholar]
- 17.Brown DW, Welsh RM, Like AA. Infection of peripancreatic lymph nodes but not islets precedes Kilham rat virus-induced diabetes in BB/Wor rats. J Virol. 1993;67:5873–5878. doi: 10.1128/jvi.67.10.5873-5878.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zipris D, Lien E, Xie JX, Greiner DL, Mordes JP, Rossini AA. TLR activation synergizes with Kilham rat virus infection to induce diabetes in BBDR rats. J Immunol. 2005;174:131–142. doi: 10.4049/jimmunol.174.1.131. [DOI] [PubMed] [Google Scholar]
- 19.Tirabassi RS, Guberski DL, Blankenhorn EP, Leif JH, Woda BA, Liu Z, Winans D, Greiner DL, Mordes JP. Infection with viruses from several families triggers autoimmune diabetes in LEW*1WR1 rats: prevention of diabetes by maternal immunization. Diabetes. 2010;59:110–118. doi: 10.2337/db09-0255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hanley JM, Haugen TH, Heath EC. Biosynthesis and processing of rat haptoglobin. J Biol Chem. 1983;258:7858–7869. [PubMed] [Google Scholar]
- 21.Haugen TH, Hanley JM, Heath EC. Haptoglobin. A novel mode of biosynthesis of a liver secretory glycoprotein. J Biol Chem. 1981;256:1055–1057. [PubMed] [Google Scholar]
- 22.Zipris D, Hillebrands JL, Welsh RM, Rozing J, Xie JX, Mordes JP, Greiner DL, Rossini AA. Infections that induce autoimmune diabetes in BBDR rats modulate CD4+CD25+ T cell populations. J Immunol. 2003;170:3592–3602. doi: 10.4049/jimmunol.170.7.3592. [DOI] [PubMed] [Google Scholar]
- 23.Blankenhorn EP, Cort L, Greiner DL, Guberski DL, Mordes JP. Virus-induced autoimmune diabetes in the LEW.1WR1 rat requires Iddm14 and a genetic locus proximal to the major histocompatibility complex. Diabetes. 2009;58:2930–2938. doi: 10.2337/db09-0387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lucas A, Liu L, Dai E, Bot I, Viswanathan K, Munuswamy-Ramunujam G, Davids JA, Bartee MY, Richardson J, Christov A, Wang H, Macaulay C, Poznansky M, Zhong R, Miller L, Biessen E, Richardson M, Sullivan C, Moyer R, Hatton M, Lomas DA, McFadden G. The serpin saga; development of a new class of virus derived anti-inflammatory protein immunotherapeutics. Adv Exp Med Biol. 2009;666:132–156. doi: 10.1007/978-1-4419-1601-3_11. [DOI] [PubMed] [Google Scholar]
- 25.Chambers LA, Rauck AM. Acute transient hemolytic anemia with a positive Donath-Landsteiner test following parvovirus B19 infection. J Pediatr Hematol Oncol. 1996;18:178–181. doi: 10.1097/00043426-199605000-00017. [DOI] [PubMed] [Google Scholar]
- 26.Fabriek BO, Dijkstra CD, van den Berg TK. The macrophage scavenger receptor CD163. Immunobiology. 2005;210:153–160. doi: 10.1016/j.imbio.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 27.Oh SK, Pavlotsky N, Tauber AI. Specific binding of haptoglobin to human neutrophils and its functional consequences. J Leukoc Biol. 1990;47:142–148. doi: 10.1002/jlb.47.2.142. [DOI] [PubMed] [Google Scholar]
- 28.Baseler MW, Burrell R. Purification of haptoglobin and its effects on lymphocyte and alveolar macrophage responses. Inflammation. 1983;7:387–400. doi: 10.1007/BF00916303. [DOI] [PubMed] [Google Scholar]
- 29.Costacou T, Ferrell RE, Orchard TJ. Haptoglobin genotype: a determinant of cardiovascular complication risk in type 1 diabetes. Diabetes. 2008;57:1702–1706. doi: 10.2337/db08-0095. [DOI] [PubMed] [Google Scholar]
- 30.Nakhoul FM, Miller-Lotan R, Awaad H, Asleh R, Levy AP. Hypothesis–haptoglobin genotype and diabetic nephropathy. Nat Clin Pract Nephrol. 2007;3:339–344. doi: 10.1038/ncpneph0467. [DOI] [PubMed] [Google Scholar]
- 31.Baringer JR, Nathanson N. Parvovirus hemorrhagic encephalopathy of rats. Electron microscopic observations of the vascular lesions. Lab Invest. 1972;27:514–522. [PubMed] [Google Scholar]
- 32.Margolis G, Kilham L, Ruffolo PR. Rat virus disease, an experimental model of neonatal hepatitis. Exp Mol Pathol. 1968;8:1–20. doi: 10.1016/0014-4800(68)90002-6. [DOI] [PubMed] [Google Scholar]
- 33.Castell JV, Gomez-Lechon MJ, David M, Andus T, Geiger T, Trullenque R, Fabra R, Heinrich PC. Interleukin-6 is the major regulator of acute phase protein synthesis in adult human hepatocytes. FEBS Lett. 1989;242:237–239. doi: 10.1016/0014-5793(89)80476-4. [DOI] [PubMed] [Google Scholar]