

Abstract
Rett syndrome is a monogenic disease due to de novo mutations in either MECP2 or CDKL5 genes. In spite of their involvement in the same disease, a functional interaction between the two genes has not been proven. MeCP2 is a transcriptional regulator; CDKL5 encodes for a kinase protein that might be involved in the regulation of gene expression. Therefore, we hypothesized that mutations affecting the two genes may lead to similar phenotypes by dysregulating the expression of common genes. To test this hypothesis we used induced pluripotent stem (iPS) cells derived from fibroblasts of one Rett patient with a MECP2 mutation (p.Arg306Cys) and two patients with mutations in CDKL5 (p.Gln347Ter and p.Thr288Ile). Expression profiling was performed in CDKL5-mutated cells and genes of interest were confirmed by real-time RT-PCR in both CDKL5- and MECP2-mutated cells. The only major change in gene expression common to MECP2- and CDKL5-mutated cells was for GRID1, encoding for glutamate D1 receptor (GluD1), a member of the δ-family of ionotropic glutamate receptors. GluD1 does not form AMPA or NMDA glutamate receptors. It acts like an adhesion molecule by linking the postsynaptic and presynaptic compartments, preferentially inducing the inhibitory presynaptic differentiation of cortical neurons. Our results demonstrate that GRID1 expression is downregulated in both MECP2- and CDKL5-mutated iPS cells and upregulated in neuronal precursors and mature neurons. These data provide novel insights into disease pathophysiology and identify possible new targets for therapeutic treatment of Rett syndrome.
Introduction
Rett syndrome (RTT, OMIM #312750) is a monogenic disorder that affects normal brain development during early childhood with an incidence of 1 in 10 000–15 000 live female births.1, 2 Patients with the classic form of RTT show apparently normal development until 6–18 months after birth when clinical signs of a mental regression start to become apparent. These include loss of speech and purposeful hand use, stereotypic hand movements, appearance of postnatal microcephaly, autistic features, ataxia, and hand apraxia.2, 3 Other clinical hallmarks include spasticity, respiratory abnormalities, and autonomic dysfunctions.4 Depending on disease severity, some variants such as Z-RTT (previously known as Preserved Speech Variant (PSV)), ‘forme fruste', congenital form, and early-onset seizures variant have been described in addition to classic RTT.2, 5
Mutations in MECP2 gene, located in Xq28, are responsible for ∼90% of classical RTT cases and a lower percentage of variant patients.6, 7 MECP2 encodes for a transcriptional regulator and epigenetic modifier. MeCP2 protein is widely expressed and its relatively higher expression in brain is spatially and developmentally regulated: its levels are low during embryogenesis and increase during postnatal neuronal maturation.8, 9 A very small number of MeCP2 target genes have been identified so far and the several transcriptional profiling studies performed on various tissues (brain from mouse models, fibroblasts and lymphocytes derived from mutated patients, and autoptic brain tissue) resulted in sets of de-regulated genes that differed for each study.10, 11, 12 Notably, analysis of postmortem brain tissue from patients carrying MECP2 mutations showed reduction in the mRNA and protein levels of many synaptic markers and a decreased number of dendritic spines in distinct cortical areas.13, 14 A significant alteration of NMDA, AMPA, and GABA receptors was demonstrated in the cortex and basal ganglia of patients, indicating that RTT is associated with abnormalities in the expression of molecules crucial for both excitatory and inhibitory synaptic transmission.15, 16
Mutations in another X-linked gene, CDKL5 (cyclin dependent kinase like 5), are responsible for the early-onset seizures variant of RTT.17, 18 Cdkl5 is a ubiquitous protein kinase particularly expressed in the brain.19, 20 It shuttles between the cytoplasm and the nucleus;21 in neurons, the shuttling is regulated by the activation of extrasynaptic NMDA receptors that induce protein relocalization from nucleus to cytoplasm, and also regulate its degradation by the proteasome.22 Moreover, neuronal Cdkl5 is enriched on the postsynaptic site of excitatory synapses where it seems to be involved in the establishment of correct spine morphology and function.23 A direct interaction of Cdkl5 with Dnmt1 has been reported, suggesting that it might also be involved in the regulation of DNA methylation and thus gene expression.24 Transcriptome analysis has been recently performed on fibroblasts from CDKL5-mutant patients revealing alterations in a few genes involved in differentiation and oxidative stress.25
The overlapping phenotype of MECP2- and CDKL5-mutated patients, the similar expression pattern during postnatal mouse brain development, and the interaction of both proteins with Dnmt1 suggest that Cdkl5 and MeCP2 might participate in a common neuronal pathway.24 Studies investigating the possibility of a direct functional interaction between the two proteins gave conflicting results. Mari et al26 suggested that: (1) CDKL5 binds and phosphorylates MeCP2 in vitro and in vivo; (2) CDKL5 is able to autophosphorylate itself and to phosphorylate MeCP2 in vitro; and (3) this capability is lost in mutant MeCP2 proteins.27 Opposite results were obtained by Lin et al20 who showed that MeCP2 binds CDKL5 but is not a direct target of its phosphorylation. More recently, a role for MeCP2 in the regulation of CDKL5 expression levels in brain has been demonstrated.28
A revolutionary approach for the creation of patient-specific human cellular models for neurologic and neurodevelopmental disorders has been developed in 2007 when Yamanaka and colleagues29 demonstrated that adult somatic cells can be reverted to induced pluripotent stem (iPS) cells, an immature status analogous to embryonic stem cells. The iPS cells are similar to human embryonic stem cells (hESCs) for gene expression, morphology, proliferation, and pluripotency.29, 30 Thanks to the capacity of these cells to differentiate into neurons, many neurodegenerative (amyotrophic lateral sclerosis (ALS), spinal muscular atrophy (SMA), Huntington's disease (HD), familial dysautonomia (FD), Parkinson's disease (PD), Alzheimer's disease (AD)) and neurodevelopmental diseases (Fragile X syndrome (FXS), schizophrenia (SCZD), Prader–Willi/Angelman's syndrome (PW/AS)) have been modeled with patient-specific iPS cells.31, 32, 33 This technology has also been employed for the study of RTT. In particular, different groups generated MECP2-mutated iPS lines and characterized the resulting neurons reporting morphological and electrophysiological defects corresponding to those found in both mouse brain and patient autoptic material.34, 35, 36 In 2010, we generated iPS lines from two patients with CDKL5 mutations, one female and one male.37 For the female patient, isogenic clones expressing either the wild-type or the mutant CDKL5 allele were obtained. In order to test whether CDKL5 could be involved in the regulation of gene expression and to define the molecular mechanisms involved in CDKL5-associated disease, we now searched for alterations in global gene expression profiles in CDKL5-mutant iPS cells. This analysis identified six dysregulated genes of which GRID1 was dysregulated in both MECP2-mutated and CDKL5-mutated iPS cells.
Materials and methods
See Supplementary Online Data for full methods.
iPS clones
Previously characterized fully reprogrammed iPS clones derived from one male and one female CDKL5-mutated patient were used.37 The female patient harbors a truncating mutation c.1039C>T (p.(Gln347Ter)), whereas the male patient has a missense change c.863C>T (p.(Thr288Ile)). For the female patient, three iPS clones differing for CDKL5 allelic expression were used: clone 20 that exclusively expresses the wild-type CDKL5 allele and clone 46 and clone 33 with exclusive expression of the mutated CDKL5 allele. For the male patient, one clone was used (clone 58) and a clone derived from normal male fibroblasts (BJ) was used as control. One MECP2 mutant clone c.916C>T (p.(Arg306Cys)) and BJ control clone were obtained from James Ellis laboratory (Toronto, ON, Canada).38 Mutation numbering is based on the following reference sequences: NM_004992.3 for MECP2 and NM_003159.2 for CDKL5.
Clones maintenance and neuronal differentiation
The iPS cells were maintained and differentiated as previously described (see Supplementary Data).37 At day 18, neural precursor cells (NPCs) were isolated using Anti-PSA-NCAM MicroBeads (30-092-966, Miltenyi Biotec, Bologna, Italy) according to the manufacturer's protocol. Sorted cells were fluorescently stained with an anti-IgM antibody conjugated with PE dye (130-095-908; Miltenyi Biotec) and analysed by flow cytometry using a GUAVA EasyCyte 6-2L instrument (Merk-Millipore group, Billerica, MA, USA) to assess purity of the recovered population. Unlabelled cells were used as negative control. Cultures were harvested at day 70 for analysis of mature neurons. To obtain a more pure population of neurons for qRT-PCR, mature cultures were sorted using a CD24 MicroBead Kit (130-095-951, Miltenyi Biotec).39 Sorted neurons were plated on poly-ornithine and laminin-coated slides and allowed to attach for 48 h before being stained for β-III-Tubulin (Chemicon; Merk-Millipore group) to confirm their identity.
RNA isolation
Total RNA was extracted from iPS clones with RNeasy mini kit (Qiagen, Milano, Italy). For clones derived from the female patient, the exclusive expression of only one CDKL5 allele was confirmed as previously reported.37
Microarray analysis
Agilent Two-Color Microarray-Based gene expression analysis (Quick Amp Labeling, Agilent Technologies, Torino, Italy) protocol was used for global gene expression analysis. For each clone, four technical replicates were performed to control technical bias. Microarray raw data were analysed by Agilent Feature Extraction Software v9.5 for quality assessment. Gene Spring GX software v11.5 (Agilent Technologies) was used for data processing and analysis. Data for each mutant/control pair were analysed independently. Significantly modulated genes were defined as those with absolute fold change (FC) of >1.5. The lists of deregulated genes obtained for the two mutant/control pairs were compared in order to identify common entries. Microarray data are available in the ArrayExpress database (www.ebi.ac.uk/arrayexpress) under accession number E-MTAB-2223.
Real-time qRT-PCR
Quantitation was performed using commercial TaqMan probes from Applied Biosystem (Life Technologies, Monza, Italy) (GRID1 assay id: Hs00324946_m1; LST1 assay id: Hs00938298_g1; RESP18 assay id: Hs01563187_m1; ODF4 assay id: Hs00537806_m1). The Cyclophilin (CYCLO) gene was used as a reference (assay id: Hs01565700_g1). Student's t-test with a significance level of 95% was used for the identification of statistically significant differences in expression levels among different clones.
Immunoblotting
Proteins from neuronal cell pellets were extracted with a 10-fold excess of RIPA buffer, cleared with an ultracentrifugation step, and analysed by western blot.
Immunoprecipitation
Proteins were immunoprecipitated from mouse brain lysate by incubation with Protein A-covered beads and the specific antibody, before being washed three times and being analysed in western blot.40
Chromatin immunoprecipitation (ChIP)
Chromatin was prepared from human SH-SY5Y neuroblastoma cells as described previously.41 ChIP-isolated chromatin was subjected to quantitative PCR with primers targeting the GRID1 promoter region (GRID1_For: 5′-ATGAGCCAGCAAGGTGACTT-3′ GRID1_Rev: 5′-GGGTGGGTCAGGTTTCACTA-3′). PCR was performed in triplicate. Statistical evaluation was performed using a two-tailed t-test, assuming unequal variance.
Results
Microarray analysis of iPS cells
Expression profiling was performed on iPS clones from two patients (one female and one male) with CDKL5 mutations.37 For the female patient (CDKL5 mutation c.1039C>T, p.(Gln347Ter)) we compared one clone expressing the mutant CDKL5 allele (46) to another clone from the same patient that expresses the wild-type allele (20). Maintenance of X-chromosome inactivation and the resulting monoallelic expression of CDKL5 were confirmed by androgen receptor assay and direct sequencing of CDKL5 mRNA. The clone derived from the male patient (58; CDKL5 mutation c.863C>T, p.(Thr288Ile)) was compared with a clone derived from a normal newborn male (BJ).38 For each mutant/control pair, four technical replicates were performed for a total of eight chip hybridizations. Differences in gene expression between wild-type and mutant clones for each patient were calculated using the FC cutoff of 1.5 and P<0.05. From the first comparison (46 versus 20) we obtained 137 dysregulated genes, whereas for the second comparison (58 versus BJ) 190 genes with altered expression were identified (Supplementary Tables 1 and 2). None of these genes passed the Benjamini and Hochberg correction for FDR, probably because of the very low number of genes with altered expression. We decided, however, to compare the two gene lists to check whether common genes were present and we identified six common genes; four genes were consistently dysregulated in the same direction in both cases, one with decreased levels and the others with increased expression (Table 1). To confirm the dysregulation, real-time RT-PCR analysis was performed on an independent preparation of RNA from the same clones. RNA derived from a second clone from the female patient expressing mutant CDKL5 allele (clone 33) and from an MECP2-mutated iPS clone (15; MeCP2 mutation c.916C>T, p.(Arg306Cys)) were also included. A downregulation (∼50%) of GRID1 gene was confirmed in the CDKL5-mutated clones; intriguingly, a similar reduction was also observed in the clone with MECP2 mutation (Figure 1). GRID1 expression in clone 20 (expressing wild-type CDKL5 allele) was comparable to that observed in BJ control (Figure 1), strongly suggesting that the reduction in expression is a specific consequence of CDKL5 or MECP2 mutation.
Table 1. Common deregulated genes.
| Gene symbol | Gene name | Accession number | Fold change 58/BJ | Fold change 46/20 |
|---|---|---|---|---|
| GRID1 | Glutamate receptor, ionotropic, δ-1 | NM_017551 | −2.1036077 | −1.6889609 |
| LST1 | Leukocyte-specific transcript 1 | NM_007161 | 3.537233 | 1.5218107 |
| SLC23A3 | Solute carrier family 23 (nucleobase transporters), member 3 | AK055730 | 2.0275977 | 3.1108685 |
| RESP18 | Regulated endocrine-specific protein 18 | NM_001007089 | 2.1038992 | 2.5666316 |
| HLA-DQA2a | Major histocompatibility complex, class II, DQ α2 | NM_020056 | −1.5860842 | 1.5345135 |
| GUSBP3a | Glucuronidase, β pseudogene 3 | NR_027386 | −2.231126 | 1.6598059 |
These genes have been excluded from RT-PCR validation because of the opposite values of fold change observed in the two sample pairs.
Figure 1.

Real-time RT-PCR analysis of GRID1 mRNA levels in iPS clones. CDKL5 (33, 46, and 58)- and MECP2 (15)-mutated clones were analysed. BJ is a control clone derived from a healthy newborn male and clone 20 is a clone harbouring the CDKL5 mutation in one X chromosome but expressing the wild-type allele from the other X chromosome because of X-chromosome inactivation. The y axis shows 2−ΔΔCt values (±SD). *P≤0.01; **P≤0.05; ***P≤0.001.
GRID1 expression in NPCs and neurons
GRID1 encodes GluD1, a member of the orphan δ-glutamate ionotropic receptor family. We thus decided to validate our results on NPCs and mature neurons. To reduce the potentially confounding effect of genetic background, for CDKL5 mutations only the clones derived from the female patient were used. The iPSCs were differentiated toward a neuronal fate and NPCs were isolated from differentiating cultures using a MicroBeads-conjugated antibody that recognizes PSA-NCAM42 (Figure 2a). Flow cytometry analysis on purified NPCs confirmed the efficiency of enrichment (Figure 2b). GRID1 expression in NPCs appears more variable with significantly different expression between BJ control and clone 20 (Figure 2c). Surprisingly, a significant overexpression was observed in NPCs from both clone15 (MECP2 mutation) and clones 46 and 33 (CDKL5 mutation c.1039C>T, p.(Gln347Ter)). Neurons were isolated from mature cultures using a MicroBeads-conjugated anti-CD24 antibody39 (Figure 2a). Immunofluorescence staining with β-III-Tubulin confirmed the neuronal identity of the sorted cells (Supplementary Figure 1). Analysis of mature neurons confirmed GRID1 overexpression, although the difference did not reach statistical significance for the MECP2-mutated clone (Figure 2d). To verify whether the increased mRNA levels were accompanied by alterations in protein levels, GluD1 protein was quantitated in extracts from mature neurons differentiated with the same protocol. Because of the difficulty of isolating sufficient amounts of protein from sorted neurons, the analysis was performed on nonsorted cultures. The analysis confirmed a significant increase in protein levels in extracts obtained from both CDKL5-mutated and MECP2-mutated clones (Figure 3).
Figure 2.

(a) Overview of the neuronal differentiation protocol showing the timing of specific passages expressed in days respect to day 0 (d0) defined as the first day of the protocol. The arrows indicate the day at which neuronal precursors (PSA-NCAM+) or neurons (CD24+) were sorted for expression analysis. (b) FACS analysis of differentiating cultures before sorting (left) and after sorting with anti-PSA-NCAM microbeads (right) showing a significant enrichment in positive cells. The x axis shows SSC (side scatter), and the y axis shows fluorescence intensity. (c) GRID1 expression analysis in PSA-NCAM+ NPCs (day 18) mRNA derived from neuronal differentiation of CDKL5 (20, 46, and 33)- and MECP2 ( 15)-mutated clones (for clone identification see Figure 1 legend). (d) Expression analysis in mature neurons at day 70 of differentiation protocol sorted with anti-CD24 microbeads. The y axis in (c) and (d) shows 2−ΔΔCt values (±SD). *P≤0.01; **P≤0.0001; ***P≤0.05.
Figure 3.

(a) Anti-GluD1 antibody characterization. Different amounts of mouse brain lysate as well as immunoprecipitates (with mouse nonspecific IgG as control) were loaded and probed for two different antibody batches (26 and 27) to double-proof the identity of the recognized epitope. (b) GluD1 expression in iPSC-derived neurons. Each lane was loaded with 30 μg of total protein. Experiments were performed in triplicate (n=3). Immunosignals were detected using autoradiographic films, and glutamate-δ-1 receptor levels were quantified using Photoshop CS3 software (Adobe Systems, San Jose, CA, USA) and normalized against α-tubulin levels (**P<0.01, Student's t-test).
Considering the role of MeCP2 as a transcriptional regulator, we wondered whether the alteration in GRID1 expression in the MECP2-mutant clone might indicate that MeCP2 protein directly binds to the GRID1 promoter to regulate its expression. To answer this question, ChIP experiments with an anti-MeCP2 antibody were performed in SHSY-5Y neurons. The analysis indeed demonstrated a modest but statistically significant binding of MeCP2 protein to the GRID1 promoter (Figure 4).
Figure 4.
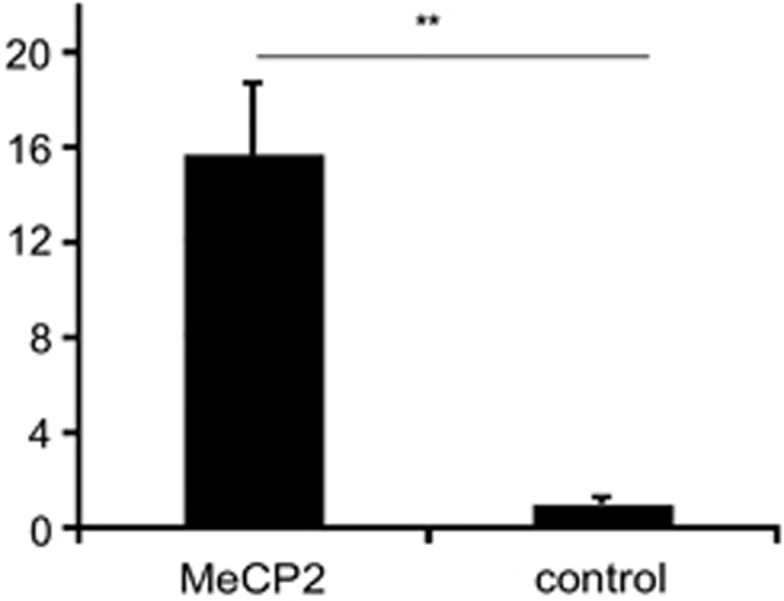
Chromatin immunoprecipitation with anti-MeCP2 and control antibodies shows MeCP2 binding to the GRID1 promoter in SHSY-5Y neuroblastoma cells. The graph shows fold enrichment of target DNA isolated by ChIP using MeCP2 NH2 antibody normalized to a control preimmune antibody. Error bars show SEM. **P=0.0048 by two-tailed t-test, assuming unequal variance.
Discussion
RTT is a monogenic disease due to de novo mutations in either MECP2 (classic and Zappella variant) or CDKL5 (early-onset seizures variant) genes on the X chromosome. The overlapping phenotype observed in patients with mutations in the two genes suggests that they might act, at least in part, on common pathways. MeCP2 is a transcriptional regulator involved in both activation and repression of gene expression.43 CDKL5 encodes for a kinase protein that exerts multiple functions inside the cell.23, 24, 44, 45 It directly interacts with Dnmt1 and mediates its phosphorylation and it is involved in the structural organization and dynamics of nuclear speckles, suggesting that, like MeCP2, it might be implicated in the regulation of gene expression.24, 45 We thus hypothesized that mutations in the two genes may lead to similar phenotypes by dysregulating the expression of common genes. To test this hypothesis, we used iPS cells derived from patients mutated in MECP2 and CDKL5. Expression profiling in CDKL5-mutated iPS cells from two patients with different mutations resulted in six common genes and a larger number of clone-specific alterations. None of these genes is present among those recently reported by Nectoux et al46 in fibroblasts from three patients with frameshift CDKL5 mutations. This is not an unexpected finding, considering the different cell types analysed (iPS versus fibroblasts). Moreover, very limited overlap is usually present among dysregulated genes resulting from different expression profiling analyses, as widely documented from studies on MECP2.11, 14, 47 Validation of the six common genes demonstrated that GRID1 was consistently downregulated in both CDKL5-mutated and MECP2-mutated iPS cells (Figure 1). Analysis of neuronal precursors derived from these clones demonstrated that the absence of a functional MeCP2 or CDKL5 protein resulted in a significant upregulation of GRID1 mRNA in these cells (Figure 2). The upregulation persists in mature neurons.
GRID1 encodes the orphan δ-glutamate receptor subunit-1 (GluD1) and, together with GRID2, constitutes the δ-family of ionotropic glutamate receptors.48 However, no channel function has been presently demonstrated either alone or in combination with other receptor subunits, and available data point to a role in synapse development rather than functioning.49, 50, 51 In particular, the postsynaptic GluD1 binds to presynaptic neurexin through cerebellin (Cbln) to induce presynaptic differentiation. Depending on the specific brain region, this can result in the formation of glutamatergic (cerebellum and hippocampus) or GABAergic (cortex and hippocampus) synapses, although GABAergic differentiation seems to be the prevalent outcome.51, 52, 53 Our experiments show that GluD1 is overexpressed in mature neurons derived from both MECP2- and CDKL5-mutated iPS cells. ChIP analysis in SHSY-5Y neurons indicate that MeCP2 binds to the GRID1 promoter, suggesting that the increased expression might arise from the absence of a direct repression by MeCP2. In Mecp2-mutant mice, a significant reduction in dendritic spine number has been reported, suggesting decreased excitatory synapse capacity.54, 55 Accordingly, MECP2-null neurons have a significant reduction in VGLUT/PSD-95 puncta and excitatory postsynaptic current (EPSC) amplitude, whereas MeCP2 overexpression results in the opposite effects, suggesting that MeCP2 might regulate the excitation/inhibition balance in the CNS by controlling the number of excitatory synapses.56, 57 A similar impairment has been reported in both CDKL5-silenced mouse hippocampal neurons and patient iPS-derived neurons.23 Considering the ability of GluD1 to induce the maturation of GABAergic synapses in telencephalic neurons, it can be hypothesized that its overexpression in the absence of both MeCP2 and CDKL5 might contribute to the reduction in excitatory synapses number by causing a shift toward differentiation of inhibitory synapses. Although no such alteration has been evidenced in the above-mentioned studies, analyses on Mecp2 mouse models have demonstrated extensive region-specific variation in synaptic defects.51, 58, 59, 60 Additional studies will thus be necessary to verify whether an increased GABAergic differentiation is indeed present in our culture conditions and to possibly characterize the identity of the neuronal population involved.
Although available data point to a role for GRID1 in neuronal maturation, we already detected alteration of GRID1 expression in NPCs. No data are presently available on what could be the function of GluD1 in these cells. However, both glutamate and GABA ionotropic receptor subunits and functional receptor channels are expressed very early during brain development in proliferating neuroepitelial cells and are considered important for events such as precursor proliferation, migration, differentiation, and survival.61, 62, 63 It is thus possible that an alteration of GluD1 levels in precursor cells might influence one of these processes and result in alteration of subsequent brain development.
Apart from the six common dysregulated genes, independent analysis of expression profiling results for the two CDKL5-mutated clones identified more than 300 clone-specific genes. The easiest explanation for this very limited overlap is the presence of different CDKL5 mutations that might have different consequences on protein functionality: the female patient has a truncating mutation (p.Gln347Ter) that leads to a protein lacking the domains responsible for the interaction with MeCP2 and DNMT1 and the nuclear export signal, whereas the male patient has a missense change (p.Thr288Ile) in an highly conserved amino acid inside the catalytic kinase domain that might result in a protein with altered kinase activity.37 This scenario is consistent with the phenotypic differences observed between the two patients. In fact, the female patient has been diagnosed as having an early-onset seizures variant of RTT, whereas the male has X-linked epileptic encephalopathy. Although we are conscious that comparing cells from patients with different phenotypes might reduce the number of common genes identified, we selected this strategy as it might lead to the identification of those genes that represent the basal targets of CDKL5. In support of our hypothesis, a similar mutation-specific effect has also been reported by Nectoux et al46 in CDKL5-mutated fibroblasts. Finally, we cannot exclude that some of the clone-specific alterations might be a nonspecific consequence of the integration site of reprogramming virus rather than a consequence of CDKL5 absence.
The finding that GluD1 expression is altered in the presence of a mutation in MECP2 or CDKL5 appears particularly intriguing as mutations in GluD1 presynaptic partner are associated with Pitt–Hopkins-like syndrome 1 (OMIM #610042) that shares phenotypic features with RTT.64, 65 The other gene mutated in this syndrome is CNTNAP2 (also known as NRXN4), another member of Neurexin gene family. Data recently obtained from our group indicate that CNTNAP2 might be involved in the modulation of the phenotype arising from mutations in MECP2.66 In light of these results, it seems likely that all these genes act on the regulation of common processes and thus the alteration of one or more of them results in overlapping phenotypic outcomes. If this is the case, we would expect that alterations in GRID1 might also cause a neurological phenotype. Indeed, linkage and association studies have established GRID1 as a strong candidate gene for schizophrenia, major depressive disorder, and autism spectrum disorders (ASD).67, 68, 69, 70 Moreover, GRID1-null mice present aberrant social and emotional behaviours, further stressing the importance of this gene for normal cognition.71
In conclusion, our results demonstrate that GRID1 gene expression is downregulated in both MECP2- and CDKL5-mutant iPS cells and upregulated in neuronal precursors and mature neurons. Our data provide the first potential functional link between MECP2 and CDKL5 genes. These data provide novel insights into disease pathophysiology and identify possible new targets for therapeutic treatments of RTT.
Acknowledgments
We are grateful to A Bartolini for help with culture maintenance and differentiation. The work was partially funded by Telethon grant (GGP11110A), by Italian Health Ministry ‘Ricerca finalizzata 2008' (RF-TOS-2008_1225570) and ‘Ricerca finalizzata 2010' (RF-2010-2317597) grants, and by AIRETT (Associazione Italiana Rett) 2011 grant to AR. The ‘Cell lines and DNA bank of Rett Syndrome, X-linked mental retardation and other genetic diseases', member of the Telethon Network of Genetic Biobanks (project no. GTB12001), funded by Telethon Italy, and of the EuroBioBank network, provided us with specimens.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies this paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Supplementary Material
References
- Rett A. Ueber ein eigenartiges hirnatrophisches Syndrom bei Hyperammonaemie im Kindesalter. Wien Med Wochenschrift. 1966;116:723–726. [PubMed] [Google Scholar]
- Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007;56:422–437. doi: 10.1016/j.neuron.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Hagberg B, Aicardi J, Dias K, Ramos O. A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett's syndrome: report of 35 cases. Ann Neurol. 1983;14:471–479. doi: 10.1002/ana.410140412. [DOI] [PubMed] [Google Scholar]
- Williamson SL, Christodoulou J. Rett syndrome: new clinical and molecular insights. Eur J Hum Genet. 2006;14:896–903. doi: 10.1038/sj.ejhg.5201580. [DOI] [PubMed] [Google Scholar]
- Artuso R. Early-onset seizure variant of Rett syndrome: definition of the clinical diagnostic criteria. Brain Dev. 2010;32:17–24. doi: 10.1016/j.braindev.2009.02.004. [DOI] [PubMed] [Google Scholar]
- Neul J, Kaufmann W, Glaze D, et al. Rett syndrome: revised diagnostic criteria and nomenclature. Ann Neurol. 2010;68:944–950. doi: 10.1002/ana.22124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir RE, Zoghbi HY. Rett syndrome: methyl-CpG-binding protein 2 mutations and phenotype-genotype correlations. Am J Med Genet. 2000;97:147–152. doi: 10.1002/1096-8628(200022)97:2<147::aid-ajmg6>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- LaSalle JM, Goldstine J, Balmer D, Greco CM. Quantitative localization of heterogeneous methyl-CpG-binding protein 2 (MeCP2) expression phenotypes in normal and Rett syndrome brain by laser scanning cytometry. Hum Mol Genet. 2001;10:1729–1740. doi: 10.1093/hmg/10.17.1729. [DOI] [PubMed] [Google Scholar]
- Shahbazian MD, Antalffy B, Armstrong DL, Zoghbi HY. Insight into Rett syndrome: MeCP2 levels display tissue- and cell-specific differences and correlate with neuronal maturation. Hum Mol Genet. 2002;11:115–124. doi: 10.1093/hmg/11.2.115. [DOI] [PubMed] [Google Scholar]
- Jordan C, Li HH, Kwan HC, Francke U. Cerebellar gene expression profiles of mouse models for Rett syndrome reveal novel MeCP2 targets. BMC Med Genet. 2007;8:36. doi: 10.1186/1471-2350-8-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tudor M, Akbarian S, Chen RZ, Jaenisch R. Transcriptional profiling of a mouse model for Rett syndrome reveals subtle transcriptional changes in the brain. Proc Natl Acad Sci USA. 2002;99:15536–15541. doi: 10.1073/pnas.242566899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux JC, Zala D, Panayotis N, Borges-Correia A, Saudou F, Villard L. Modification of Mecp2 dosage alters axonal transport through the Huntingtin/Hap1 pathway. Neurobiol Dis. 2012;45:786–795. doi: 10.1016/j.nbd.2011.11.002. [DOI] [PubMed] [Google Scholar]
- Belichenko PV, Oldfors A, Hagberg B, Dahlstrom A. Rett syndrome: 3-D confocal microscopy of cortical pyramidal dendrites and afferents. Neuroreport. 1994;5:1509–1513. [PubMed] [Google Scholar]
- Colantuoni C, Jeon OH, Hyder K, et al. Gene expression profiling in postmortem Rett Syndrome brain: differential gene expression and patient classification. Neurobiol Dis. 2001;8:847–865. doi: 10.1006/nbdi.2001.0428. [DOI] [PubMed] [Google Scholar]
- Blue ME, Naidu S, Johnston MV. Altered development of glutamate and GABA receptors in the basal ganglia of girls with Rett syndrome. Exp Neurol. 1999;156:345–352. doi: 10.1006/exnr.1999.7030. [DOI] [PubMed] [Google Scholar]
- Johnston MV, Blue ME, Naidu S. Rett syndrome and neuronal development. J Child Neurol. 2005;20:759–763. doi: 10.1177/08830738050200091101. [DOI] [PubMed] [Google Scholar]
- Evans J, Archer H, Colley J, et al. Early onset seizures and Rett-like features associated with mutations in CDKL5. Eur J Hum Genet. 2005;13:1113–1120. doi: 10.1038/sj.ejhg.5201451. [DOI] [PubMed] [Google Scholar]
- Scala E, Ariani F, Mari F, et al. CDKL5/STK9 is mutated in Rett syndrome variant with infantile spasms. J Med Genet. 2005;42:103–107. doi: 10.1136/jmg.2004.026237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montini E, Andolfi G, Caruso A, et al. Identification and characterization of a novel serine-threonine kinase gene from the Xp22 region. Genomics. 1998;51:427–433. doi: 10.1006/geno.1998.5391. [DOI] [PubMed] [Google Scholar]
- Lin C, Franco B, Rosner MR. CDKL5/Stk9 kinase inactivation is associated with neuronal developmental disorders. Hum Mol Genet. 2005;14:3775–3786. doi: 10.1093/hmg/ddi391. [DOI] [PubMed] [Google Scholar]
- Rusconi L, Salvatoni L, Giudici L, et al. CDKL5 expression is modulated during neuronal development and its subcellular distribution is tightly regulated by the C-terminal tail. J Biol Chem. 2008;283:30101–30111. doi: 10.1074/jbc.M804613200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusconi L, Kilstrup-Nielsen C, Landsberger N. Extrasynaptic N-methyl-D-aspartate (NMDA) receptor stimulation induces cytoplasmic translocation of the CDKL5 kinase and its proteasomal degradation. J Biol Chem. 2011;286:36550–36558. doi: 10.1074/jbc.M111.235630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricciardi S, Ungaro F, Hambrock M, et al. CDKL5 ensures excitatory synapse stability by reinforcing NGL-1-PSD95 interaction in the postsynaptic compartment and is impaired in patient iPSC-derived neurons. Nat Cell Biol. 2012;14:911–923. doi: 10.1038/ncb2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kameshita I, Sekiguchi M, Hamasaki D, et al. Cyclin-dependent kinase-like 5 binds and phosphorylates DNA methyltransferase 1. Biochem Biophys Res Commun. 2008;377:1162–1167. doi: 10.1016/j.bbrc.2008.10.113. [DOI] [PubMed] [Google Scholar]
- Nectoux J, Fichou Y, Rosas-Vargas H, et al. Cell cloning-based transcriptome analysis in Rett patients: relevance to the pathogenesis of Rett syndrome of new human MeCP2 target genes. J Cell Mol Med. 2010;14:1962–1974. doi: 10.1111/j.1582-4934.2010.01107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mari F, Azimonti S, Bertani I, et al. CDKL5 belongs to the same molecular pathway of MeCP2 and it is responsible for the early-onset seizure variant of Rett syndrome. Hum Mol Genet. 2005;14:1935–1946. doi: 10.1093/hmg/ddi198. [DOI] [PubMed] [Google Scholar]
- Bertani I, Rusconi L, Bolognese F, et al. Functional consequences of mutations in CDKL5, an X-linked gene involved in infantile spasms and mental retardation. J Biol Chem. 2006;281:32048–32056. doi: 10.1074/jbc.M606325200. [DOI] [PubMed] [Google Scholar]
- Carouge D, Host L, Aunis D, Zwiller J, Anglard P. CDKL5 is a brain MeCP2 target gene regulated by DNA methylation. Neurobiol Dis. 2010;38:414–424. doi: 10.1016/j.nbd.2010.02.014. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Lowry WE, Richter L. Signaling in adult stem cells. Front Biosci. 2007;12:3911–3927. doi: 10.2741/2360. [DOI] [PubMed] [Google Scholar]
- Marchetto MC, Brennand KJ, Boyer LF, Gage FH. Induced pluripotent stem cells (iPSCs) and neurological disease modeling: progress and promises. Hum Mol Genet. 2011;20:R109–R115. doi: 10.1093/hmg/ddr336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbach A, Bar-Nur O, Daley GQ, Benvenisty N. Differential modeling of fragile X syndrome by human embryonic stem cells and induced pluripotent stem cells. Cell Stem Cell. 2010;6:407–411. doi: 10.1016/j.stem.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain SJ, Chen PF, Ng KY, et al. Induced pluripotent stem cell models of the genomic imprinting disorders Angelman and Prader-Willi syndromes. Proc Natl Acad Sci U S A. 2010;107:17668–17673. doi: 10.1073/pnas.1004487107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetto MC, Carromeu C, Acab A, et al. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell. 2010;143:527–539. doi: 10.1016/j.cell.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung AY, Horvath LM, Grafodatskaya D, et al. Isolation of MECP2-null Rett Syndrome patient hiPS cells and isogenic controls through X-chromosome inactivation. Hum Mol Genet. 2011;20:2103–2115. doi: 10.1093/hmg/ddr093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ananiev G, Williams EC, Li H, Chang Q. Isogenic pairs of wild type and mutant induced pluripotent stem cell (iPSC) lines from Rett syndrome patients as in vitro disease model. PLoS One. 2011;6:e25255. doi: 10.1371/journal.pone.0025255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amenduni M, De Filippis R, Cheung AY, et al. iPS cells to model CDKL5-related disorders. Eur J Hum Genet. 2011;19:1246–1255. doi: 10.1038/ejhg.2011.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotta A, Cheung AY, Farra N, et al. Isolation of human iPS cells using EOS lentiviral vectors to select for pluripotency. Nat Methods. 2009;6:370–376. doi: 10.1038/nmeth.1325. [DOI] [PubMed] [Google Scholar]
- Yuan SH, Martin J, Elia J, et al. Cell-surface marker signatures for the isolation of neural stem cells, glia and neurons derived from human pluripotent stem cells. PLoS One. 2011;6:e17540. doi: 10.1371/journal.pone.0017540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safieddine S, Wenthold RJ. The glutamate receptor subunit delta1 is highly expressed in hair cells of the auditory and vestibular systems. J Neurosci. 1997;17:7523–7531. doi: 10.1523/JNEUROSCI.17-19-07523.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasui DH, Peddada S, Bieda MC, et al. Integrated epigenomic analyses of neuronal MeCP2 reveal a role for long-range interaction with active genes. Proc Natl Acad Sci USA. 2007;104:19416–19421. doi: 10.1073/pnas.0707442104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennartz S, Belvindrah R, Tomiuk S, et al. Purification of neuronal precursors from the adult mouse brain: comprehensive gene expression analysis provides new insights into the control of cell migration, differentiation, and homeostasis. Mol Cell Neurosci. 2004;25:692–706. doi: 10.1016/j.mcn.2003.12.011. [DOI] [PubMed] [Google Scholar]
- Chahrour M, Jung SY, Shaw C, et al. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Zhu YC, Yu J, et al. CDKL5, a protein associated with Rett syndrome, regulates neuronal morphogenesis via Rac1 signaling. J Neurosci. 2010;30:12777–12786. doi: 10.1523/JNEUROSCI.1102-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricciardi S, Kilstrup-Nielsen C, Bienvenu T, Jacquette A, Landsberger N, Broccoli V. CDKL5 influences RNA splicing activity by its association to the nuclear speckle molecular machinery. Hum Mol Genet. 2009;18:4590–4602. doi: 10.1093/hmg/ddp426. [DOI] [PubMed] [Google Scholar]
- Nectoux J, Fichou Y, Cagnard N, et al. Cell cloning-based transcriptome analysis in cyclin-dependent kinase-like 5 mutation patients with severe epileptic encephalopathy. J Mol Med (Berl) 2011;89:193–202. doi: 10.1007/s00109-010-0699-x. [DOI] [PubMed] [Google Scholar]
- Traynor J, Agarwal P, Lazzeroni L, Francke U. Gene expression patterns vary in clonal cell cultures from Rett syndrome females with eight different MECP2 mutations. BMC Med Genet. 2002;3:12. doi: 10.1186/1471-2350-3-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki M, Araki K, Shibata A, Mishina M. Molecular cloning of a cDNA encoding a novel member of the mouse glutamate receptor channel family. Biochem Biophys Res Commun. 1992;183:886–892. doi: 10.1016/0006-291x(92)90566-4. [DOI] [PubMed] [Google Scholar]
- Lomeli H, Sprengel R, Laurie DJ, et al. The rat delta-1 and delta-2 subunits extend the excitatory amino acid receptor family. FEBS Lett. 1993;315:318–322. doi: 10.1016/0014-5793(93)81186-4. [DOI] [PubMed] [Google Scholar]
- Mayat E, Petralia RS, Wang YX, Wenthold RJ. Immunoprecipitation, immunoblotting, and immunocytochemistry studies suggest that glutamate receptor delta subunits form novel postsynaptic receptor complexes. J Neurosci. 1995;15:2533–2546. doi: 10.1523/JNEUROSCI.15-03-02533.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasumura M, Yoshida T, Lee SJ, Uemura T, Joo JY, Mishina M. Glutamate receptor delta1 induces preferentially inhibitory presynaptic differentiation of cortical neurons by interacting with neurexins through cerebellin precursor protein subtypes. J Neurochem. 2012;121:705–716. doi: 10.1111/j.1471-4159.2011.07631.x. [DOI] [PubMed] [Google Scholar]
- Kuroyanagi T, Yokoyama M, Hirano T. Postsynaptic glutamate receptor delta family contributes to presynaptic terminal differentiation and establishment of synaptic transmission. Proc Natl Acad Sci USA. 2009;106:4912–4916. doi: 10.1073/pnas.0900892106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu K, Yokoyama M, Yamashita M, Hirano T. Induction of excitatory and inhibitory presynaptic differentiation by GluD1. Biochem Biophys Res Commun. 2012;417:157–161. doi: 10.1016/j.bbrc.2011.11.075. [DOI] [PubMed] [Google Scholar]
- Belichenko PV, Wright EE, Belichenko NP, et al. Widespread changes in dendritic and axonal morphology in Mecp2-mutant mouse models of Rett syndrome: evidence for disruption of neuronal networks. J Comp Neurol. 2009;514:240–258. doi: 10.1002/cne.22009. [DOI] [PubMed] [Google Scholar]
- Chapleau CA, Calfa GD, Lane MC, et al. Dendritic spine pathologies in hippocampal pyramidal neurons from Rett syndrome brain and after expression of Rett-associated MECP2 mutations. Neurobiol Dis. 2009;35:219–233. doi: 10.1016/j.nbd.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao HT, Zoghbi HY, Rosenmund C. MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron. 2007;56:58–65. doi: 10.1016/j.neuron.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatto CL, Broadie K. Genetic controls balancing excitatory and inhibitory synaptogenesis in neurodevelopmental disorder models. Front Synaptic Neurosci. 2010;2:4. doi: 10.3389/fnsyn.2010.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani VS, Chang Q, Maffei A, Turrigiano GG, Jaenisch R, Nelson SB. Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc Natl Acad Sci USA. 2005;102:12560–12565. doi: 10.1073/pnas.0506071102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medrihan L, Tantalaki E, Aramuni G, et al. Early defects of GABAergic synapses in the brain stem of a MeCP2 mouse model of Rett syndrome. J Neurophysiol. 2008;99:112–121. doi: 10.1152/jn.00826.2007. [DOI] [PubMed] [Google Scholar]
- Zhang ZW, Zak JD, Liu H. MeCP2 is required for normal development of GABAergic circuits in the thalamus. J Neurophysiol. 2010;103:2470–2481. doi: 10.1152/jn.00601.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L, Rigo JM, Rocher V, et al. Neurotransmitters as early signals for central nervous system development. Cell Tissue Res. 2001;305:187–202. doi: 10.1007/s004410000343. [DOI] [PubMed] [Google Scholar]
- Manent JB, Represa A. Neurotransmitters and brain maturation: early paracrine actions of GABA and glutamate modulate neuronal migration. Neuroscientist. 2007;13:268–279. doi: 10.1177/1073858406298918. [DOI] [PubMed] [Google Scholar]
- Aronica E, Crino PB. Inflammation in epilepsy: clinical observations. Epilepsia. 2011;52 (Suppl 3:26–32. doi: 10.1111/j.1528-1167.2011.03033.x. [DOI] [PubMed] [Google Scholar]
- Zweier C, de Jong EK, Zweier M, et al. CNTNAP2 and NRXN1 are mutated in autosomal-recessive Pitt-Hopkins-like mental retardation and determine the level of a common synaptic protein in Drosophila. Am J Hum Genet. 2009;85:655–666. doi: 10.1016/j.ajhg.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marangi G, Ricciardi S, Orteschi D, et al. Proposal of a clinical score for the molecular test for Pitt-Hopkins syndrome. Am J Med Genet A. 2012;158A:1604–1611. doi: 10.1002/ajmg.a.35419. [DOI] [PubMed] [Google Scholar]
- Grillo E, Lo Rizzo C, Bianciardi L, et al. Revealing the complexity of a monogenic disease: Rett syndrome exome sequencing. PLoS One. 2013;8:e56599. doi: 10.1371/journal.pone.0056599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallin MD, Lasseter VK, Avramopoulos D, et al. Bipolar I disorder and schizophrenia: a 440-single-nucleotide polymorphism screen of 64 candidate genes among Ashkenazi Jewish case-parent trios. Am J Hum Genet. 2005;77:918–936. doi: 10.1086/497703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glessner JT, Hakonarson H. Common variants in polygenic schizophrenia. Genome Biol. 2009;10:236. doi: 10.1186/gb-2009-10-9-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo SZ, Huang K, Shi YY, et al. A case-control association study between the GRID1 gene and schizophrenia in the Chinese Northern Han population. Schizophr Res. 2007;93:385–390. doi: 10.1016/j.schres.2007.03.007. [DOI] [PubMed] [Google Scholar]
- Treutlein J, Muhleisen TW, Frank J, et al. Dissection of phenotype reveals possible association between schizophrenia and Glutamate Receptor Delta 1 (GRID1) gene promoter. Schizophr Res. 2009;111:123–130. doi: 10.1016/j.schres.2009.03.011. [DOI] [PubMed] [Google Scholar]
- Yadav R, Gupta SC, Hillman BG, Bhatt JM, Stairs DJ, Dravid SM. Deletion of glutamate delta-1 receptor in mouse leads to aberrant emotional and social behaviors. PLoS One. 2012;7:e32969. doi: 10.1371/journal.pone.0032969. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.