

Abstract
Traumatic brain injury often results in acute metabolic crisis. We recently demonstrated that this is associated with chronic brain atrophy, which is most prominent in the frontal and temporal lobes. Interestingly, the neuropsychological profile of traumatic brain injury is often characterized as ‘frontal-temporal’ in nature, suggesting a possible link between acute metabolic crisis related-brain atrophy and neurocognitive impairment in this population. While focal lesions and diffuse axonal injury have a well-established role in the neuropsychological deficits observed following traumatic brain injury, no studies to date have examined the possible contribution of acute metabolic crisis-related atrophy in the neuropsychological sequelae of traumatic brain injury. In the current study we employed positron emission tomography, magnetic resonance imaging, and neuropsychological assessments to ascertain the relationship between acute metabolic crisis related-brain atrophy and neurocognitive outcome in a sample of 14 right-handed traumatic brain injury survivors. We found that acute metabolic crisis related-atrophy in the frontal and temporal lobes was associated with poorer attention, executive functioning, and psychomotor abilities at 12 months post-injury. Furthermore, participants with gross frontal and/or temporal lobe atrophy exhibited numerous clinically significant neuropsychological deficits in contrast to participants with other patterns of brain atrophy. Our findings suggest that interventions that reduce acute metabolic crisis may lead to improved functional outcomes for traumatic brain injury survivors.
Keywords: traumatic brain injury, metabolic crisis, neuropsychology, brain atrophy, neuroimaging
Introduction
While there is great heterogeneity in the etiology and neuropathology of traumatic brain injury (TBI; National Institutes of Health, 1999; Williamson et al., 1996), most cases are associated with acceleration-deceleration forces and result in diffuse axonal injury (DAI; Adams & Jennett, Adams et al., 1989; 2001; Bigler, 2001; Takaoka et al., 2002; Williamson et al., 1996; Adams et al., 1980) in addition to lesions to ventral frontal lobe and ventral anterior medial temporal areas (Adams et al., 1980; Adams et al., 1989; Wilson et al., 1995). Data from our group indicate another mechanism of chronic brain injury following TBI, early metabolic crisis.
Even in the absence of ischemia, early alterations of oxygen (decreased) and glucose (increased) metabolism in addition to increased lactate levels are common in moderate to severe TBI (Glenn et al., 2003; Vespa et al. 2005). Our work indicates that early metabolic dysfunction in TBI is not a spatially or temporally uniform phenomenon, rather altered metabolism varies greatly by brain region and time since injury (Hattori et al., 2003; Marcoux et al., 2008; Vespa et al., 2004; Vespa et al., 2007; Xu et al., 2010). While pericontusional tissue appears to exhibit the greatest disruption, metabolic disturbances extend to normal appearing brain tissue as well (Xu et al., 2010). Early metabolic crises are associated with chronic atrophy in the frontal, temporal, and parietal lobes, with the frontal and temporal lobes generally exhibiting the greatest atrophic changes (Xu et al., 2010). It is important to note that early metabolic crisis-related atrophy is not associated with ischemia and appears to be distinct from DAI (Hattori et al., 2003; Xu et al., 2010).
Moderate to severe TBI routinely results in marked impairments in attention/speeded processing, memory, and executive functioning (Lezak et al., 2004). The deficit profile associated with TBI has often characterized as reflecting ‘frontal’ (Stuss & Gow, 1992: Vakil, 2005) or ‘frontal-temporal’ (Lezak et al., 2004) dysfunction, as attention, memory, and executive abilities require intact function of the frontal and temporal lobes. Indeed, we have found that distinct ‘frontal’ and ‘temporal’ components contribute to deficits in various forms of memory following severe TBI (Schmitter-Edgecombe & Wright, 2003; Schmitter-Edgecombe & Wright, 2004; Schmitter-Edgecombe et al., 2004; Wright & Schmitter-Edgecombe, 2011; Wright et al., 2010). For example, in a study of verbal memory where we employed a novel item analytic technique for discerning process disruptions on list-learning tests (Wright et al., 2009), we found that severe acceleration-deceleration TBI related-memory deficits were driven by strategic encoding deficits (primary) and consolidation problems (secondary) (Wright et al., 2010). Additionally, our data indicated that while these encoding and consolidation deficits interacted, they made significant independent contributions to the memory impairments observed in our TBI sample. This was an important finding, as it suggested optimal targets for cognitive rehabilitation in TBI and because echoed neuroimaging research that has shown that strategic encoding (reliant on prefrontal-hippocampal interactions; see Bor et al., 2004; Habib et al., 2003) and consolidation/storage (dependant on medial temporal lobe structures—namely, the hippocampus; see Squire, 1994, 1980; Squire & Zola, 1998) of verbal material requires seemingly simultaneous independent and integrative processing (Bohlman & Knight, 1994; Gleissner et al., 1997). That said, not all abilities that rely on frontal and temporal lobe function are necessarily disturbed following moderate to severe TBI (e.g., language) (Schmitter-Edgecombe et al., 1993) and the characterization of cognitive deficits following TBI as ‘frontal-temporal’ should be considered a useful heuristic rather than an absolute.
While the presence of DAI and focal lesions (often to the frontal and temporal lobes) predicts cognitive outcome in TBI (Auerbach, 1986; Bigler, 2001; Fork et al. 2005; Kraus et al., 2007; Lehtonen et al., 2005; Levine et al., 2002; Wallesch et al., 2001; Williamson et al., 1996), it is not clear whether or not early metabolic crisis-related brain atrophy (MCBA) is associated with cognitive impairment following TBI. The ‘frontal-temporal’ cognitive profile of TBI and the ‘frontal-temporal’ impact of MCBA suggests a possible link between chronic cognitive abilities in TBI survivors and acute metabolic crisis. However, the ‘frontal-temporal’ cognitive profile of TBI could be due to higher lesion burden in the frontal and temporal lobes, DAI mediated reductions in connectivity between brain regions, and/or the relative vulnerability of the frontal and temporal lobes owing to their later ontogenetic development (Bentourkia et al., 2000; Head et al., 2004; Raz, 2000).
In the current study, we investigated neuropsychological function at 12 months post-injury in a sample of TBI participants who demonstrated non-ischemic MCBA (see Xu et al., 2010). Our goal was to determine if MCBA following TBI is associated with chronic neuropsychological impairments. Additionally, based on the regional pattern of MCBA and the typical of neurocognitive deficits observed in TBI, we hypothesized that: (1) frontal and temporal lobe MCBA would be associated with neuropsychological impairments to a greater degree than atrophy in other brain regions; (2) TBI participants with gross frontal and/or temporal lobe atrophy would exhibit poorer neuropsychological performances in comparison to TBI participants with other patterns of brain atrophy.
Materials and Methods
Participants
The current study was approved by the institutional review board of the University of California, Los Angeles and all participants consented directly or by proxy to voluntary participation between 09/06/1999 and 03/12/2006. Inclusion/exclusion criteria included (1) a Glasgow Coma Scale (GCS) score of 8 or a GCS between 9 and 15 with evidence of intracranial bleeding on acute computerized tomography brain scans; (2) a level of acute medicophysiologic stability necessary to complete positron emission tomography (PET) and magnetic resonance imaging (MRI) brain studies; (3) follow-up MRI brain study at around 6 months post-injury; (4) acute and chronic brain image quality sufficient for evaluation (e.g., minimal or no motion artifact); and (5) completion of most or all of a brief neuropsychological test battery at around 12 months post-injury. The current study included 14 right hand dominant TBI participants who exhibited non-ischemic MCBA; this sample consists of a subset of participants in a previous imaging study (see Xu et al., 2010). Neuropsychological test completion rates were variable due to injury severity and fatigue; the number of participants completing specific tests ranged from 10–14. All of the TBIs were due to acceleration-deceleration forces and were moderate to severe in nature (Table 1). Seven participants indicated gross frontal/temporal lobe atrophy (FTA) relative to the sample. FTA subgroup membership was defined by bilateral frontal, bilateral temporal, or a least one temporal and frontal lobe with atrophy above the sample median. Mann-Whitney U tests showed that the FTA and other atrophy (OA) groups were not significantly different in terms of injury severity (initial and best in 8 Glasgow Coma Scale (GCS) scores; FTA: Mdn=,8.00, OA: Mdn= 10.00, Z=. −.77, p= .44, r= −.21; FTA: Mdn= 7.00, OA: Mdn= 8.00, Z= −.71, p= .48, r= −.19, respectively), years of age (FTA: Mdn= 22.00, OA: Mdn= 27.00, Z= .00, p= 1.00, r= .00), or years of education (FTA: Mdn= 12.00, OA: Mdn= 16.00, Z= −1.75, p= .08, r= −.47). While the groups were not significantly different in years of education, their achieved educational levels were qualitatively different; the FTA group achieved a high school level education and many of the OA group completed college. Additionally, while the two groups were statistically similar in age, their absolute difference in median age could potentially impact comparisons of their neuropsychological test performances. Differences in age and education during group comparisons were adjusted via normative corrections to neuropsychological test scores (see below for details).
Table 1.
Sample Characteristics
| n | Mean (SD) | Median | Range | % | |
|---|---|---|---|---|---|
|
|
|||||
| Demographics | |||||
| Age (yrs) | 14 | 28.93 (11.71) | 25.50 | 37.00 | - |
| Education (yrs) | 14 | 13.64 (3.48) | 13.50 | 14.00 | - |
| Sex (% male) | 11/14 | - | - | - | 78.60 |
| Ethnicity (%) | |||||
| African American | 3/14 | - | - | - | 21.40 |
| Asian/Pac. Islander | 3/14 | - | - | - | 21.40 |
| Caucasian/White | 7/14 | - | - | - | 50.00 |
| Hispanic/Latino/a | 1/14 | - | - | - | 7.10 |
| Handedness (% Rt.) | 14/14 | - | - | - | 100.00 |
| Injury Characteristics | |||||
| Initial GCS | 14 | 8.57 (3.46) | 9.00 | 10.00 | - |
| Best in 8 GCS | 14 | 7.57 (2.07) | 7.50 | 8.00 | - |
| Injury Type (%) | |||||
| MVA | 6/14 | - | - | - | 42.90 |
| MVA vs. Pedestrian | 2/14 | - | - | - | 14.30 |
| Bicycle Accident | 2/14 | - | - | - | 14.3 |
| Long Fall | 4/14 | - | - | - | 28.6 |
| Closed Head Injury | 7/14 | - | - | - | 50.00 |
Note. Yrs= years; % male= percentage of sample that was male; Initial GCS= Glasgow Coma Scale score obtained in the emergency room; Best in 8 GCS= best of eight Glasgow Coma Scale scores obtained during hospitalization; MVA= motor vehicle accident; Long Fall= any fall of the participants height or greater.
Procedures
Acute care protocol
All participants were admitted to a neurointensive care unit following surgery or initial stabilization in the emergency room. Craniotomies were performed for evacuation of intracranial mass lesions and hematomas. We measured intracranial pressure (ICP), which was maintained below 20mmHg using a standardized treatment protocol that including head of bed up to 301, mild hyperventilation (PaCO2 = 30 to 35mmHg), external ventriculostomy with cerebrospinal fluid drainage, moderate sedation with low doses of propofol, normoglycemia (80 to 120mg/dL), and maintenance of mild hypernatremia (sodium = 140 to 145 mmol/L). Refractory ICP was managed by pentobarbital-induced burst suppression coma. We kept cerebral perfusion pressure above 60mmHg with volume repletion and vasopressors. Continuous jugular venous oxygen saturation was monitored and kept at 60% to 70% via adjustments in cerebral perfusion pressure. Possible seizure activity and barbiturate effects were assessed via continuous electroencephalographic monitoring. Additionally, all participants received acute rehabilitation and 50% received Commission on Accreditation of Rehabilitation Facilities (CARF) accredited rehabilitation. No other data regarding participant therapies was collected for the current study.
Neuroimaging protocol and processing
We employed PET and MRI to ascertain metabolic state and atrophy, respectively. Our PET protocol was initiated at a median of 5 days post-injury and included serial O-15 PET scans (C15O, O15O, H215O) using dynamic blood sampling to assess regional cerebral blood flow [CBF; ml per (100 g/min)], oxygen extraction fraction (OEF), and cerebral metabolic rate of oxygen [CMRO2; ml per (100 g/min)]. While in the PET scanner, each participant received continuous physiologic monitoring of ICP, end-tidal CO2, arterial blood gases, arterial blood pressure, electroencephalography, core temperature, and heart rate. These physiologic parameters were maintained to those present in the intensive care unit. OEF images were generated from the initial 5min of the O15O study and were assessed via compartmental model for oxygen that accounts for recirculated H215O (Ohta et al, 1992). The O15O images (1.471×1.471×2.45mm3) were smoothed with a three-dimensional filter (in plane full-width at half-maximum=2.942mm, axial full-width at half-maximum= 2.45mm) before OEF images were generated on a voxel by voxel basis. Subsequently, a 2-Deoxy-2-[18F] fluoro-D-Glucose PET scan was obtained using a quantitative technique for the calculation of the regional cerebral metabolic rate of glucose [mg per (100 g/min)]. Each PET scan contained 63 slices, and each brain lobe contained 20 to 30 slices.
PET coregistration with volumetric MRI was accomplished using a six-parameter rigid-body transformation program and visual evaluation (Lin et al.,, 1994) with realignment and partitioning. Four slices with equal intervals were selected, and the brain lobe regions of interest (ROIs) were drawn manually with the contusion areas excluded. The means of the metabolic parameters for the ROIs over four planes were calculated for each lobe. This image processing was carried via Janus Program version 6.3 (Los Angeles, CA, USA; http://149.142.143.7/BIRC/INDEX.HTML).
Acute (median of 8 days post-injury) and chronic (median of 195 days post-injury) structural imaging studies were conducted using a Siemens Sonata 1.5T MRI scanner (Siemens, Munich, Germany). Both acquisitions included volumetric T1-weighted magnetization-prepared gradient echo [MP-RAGE; TR (repetition time)=1,900ms, echo time (TE)=3.5ms, field of view(FOV)=256×256, 1mm slice thickness], axial fluid-attenuated inversion recovery (FLAIR; TR=9,590ms, TE=70ms, FOV=512×384, slice thickness 3mm), axial diffusion weighted [DWI: TR=6,000ms, TE=105ms, FOV=96×128, 7mm slice, b=0 and 1,000 (3 directions), 1-scan trace] imaging studies. The DWI study was used to generate apparent diffusion coefficient (ADC) images.
An independent radiological expert blinded to the clinical data performed volumetric measurements on the T1-weighted images. Lobar ROIs were drawn using home-written plug-in software for ImageJ (NIH, Bethesda, MD, USA) 1.37v (http://rsb.info.nih.gov) to perform semiautomated lobar segmentation. ImageJ was utilized to determine lobar volume, which accounted for volume elements (voxels) metabolic parameters in each lobar ROI, and multiplied by the voxel volume. Lobar boundaries were defined using a standard approach (Hayman, 1992) and an anatomic atlas (http://www.med.harvard.edu/AANLIB/cases/caseNA/pb9.htm). Additionally, we utilized acute lobar ROIs as a qualitative guide to exclude primary contusions in the chronic scans. To accommodate shift due to reabsorbing blood and resolving macroscopic edema, careful attention was paid to multiple anatomic landmarks. Thus, consistent verification of lobar geographic limits was maintained. Contusions, dura matter, blood vessels, cranial nerves, the superior sagittal sinus, and the choroid plexus were excluded. FLAIR images were used to identify and exclude contusions. The primary contusion was judged to be the region containing a GRE hypointense lesion with a volume >1cm3. Brain edema was controlled for via a computational approach using LONI Pipeline Software (UCLA, Los Angeles, CA, USA) (http://www.loni.ucla.edu/Software/Pipeline; see Dinov et al., 2009). An automated pipeline extracted brain volumes, classified tissue voxels (background, normal gray matter, normal white matter, cerebrospinal fluid), and quantified brain volume (normal gray matter + normal white matter) from T1-weighted MP-RAGE images. Automatically generated volumes were visually inspected for errors. Acute and chronic volumetric studies were compared to derive the percent of lobar atrophy [((acute lobe volume – chronic lobe volume)/acute lobe volume) × 100]. For each participant, coregistered DWI-ADC and GRE images and a structured visual scoring system were used to discern acute ischemic injury. All ADC hypointense lesions >0.1cm in diameter were visually identified. An ischemic lesion was defined as any lesion with >0.1cm diameter that exhibited ADC reduced to a threshold of ≤550μm2/sec without a reduction in GRE signal intensity. Diffuse axonal lesions were defined as lesions >0.1cm in diameter in where ADC was ≤550μm2/sec with GRE hypointensity. Lesions having hyperintense FLAIR signal and ADC increase in the absence of GRE hypointensity were considered ischemic lesions that had progressed beyond the acute stage. Additionally, to address small vessel ischemia that may have been precipitated during the early posttraumatic period we inspected DWI-ADC lesions in the border zone regions of the anterior and middle cerebral arteries. Brainsuite Software (UCLA, Los Angeles, CA, USA) (Shattuck and Leahy, 2002) was used for final three-dimensional visualization. All atrophy values attributed to sources other than acute metabolic crisis were excluded from the present analysis.
Neuropsychological assessment
Neuropsychological assessments were conducted by a clinical neuropsychologist. We utilized a modified version of a test battery designed for TBI clinical trials (Clifton et al., 1992). All tests were administered and scored in accordance with standard instructions. The battery of tests required approximately 40min to complete. The assessments were conducted at approximately 12 months (M=11.50, SD=1.40) post-injury. Test scores were age and education corrected for group comparisons. This was accomplished by calculating T-scores via common normative datasets for the Symbol Digit Modalities Test (SDMT; Smith, 1991; norms: Smith, 1991), Controlled Oral Association Test (COWAT, F-A-S; Benton, Hemsher, Varney, and Spreen, 1983; norms: Mitrushina et al., 2005), 6-trial version of the Selective Reminding Test (SRT-6; Hannay and Levine, 1985; norms: Larrabee et al., 2000), part B of the Trail Making Test (TMT-B; Army Individual Test Battery, 1944; norms: Mitrushina et al., 2005), Rey-Osterrieth Complex Figure Test (ROCFT; Corwin and Bylsma, 1993; norms: Mitrushina et al., 2005); and Grooved Pegboard Test dominant hand performance (GPT; Matthews and Kløve, 1964; norms: Mitrushina et al., 2005). Additionally, Disability Rating Scale (DRS) and Glasgow Coma Scale Extended (GOSE) data were collected from all participants during the assessment visit.
Statistical analyses
A threshold of p< .05 for statistical significance was set for all analyses and we corrected for multiple tests via q-values with a predetermined false discovery rate (FDR) cut-off of .05 (Storey, 2002). We did not utilize q-value corrections for analyses of confounding variables; p-values for potential group differences with regard to age, education, and injury severity and correlational analyses between GCS and MCBA were not corrected for so that any indication of confounding could be detected. Nonparametric statistics were utilized in the current study due to our sample size and non-normal distribution of some the neuropsychological and neuroimaging data. Spearman rank correlations were computed to determine associations between neuroimaging, injury severity, and raw 12 month post-injury test values. Mann-Whitney U tests were utilized to determine differences between age, education, injury severity, and neuropsychological performances between participants with FTA and OA. Finally, one-sample Wilcoxon signed rank tests were employed to examine the degree to which each group differed from the norm (T= 50) to assess the clinical significance of their cognitive deficits. For group comparisons and group-norm comparisons, raw neuropsychological test scores were converted to T-scores via common normative datasets (see above).
Results
Brain atrophy-cognitive associations
MCBA values excluded any identified area contusion or lesion. Prior to determining the association between MCBA and neuropsychological test data, we assessed if lobar MCBA values were related to initial GCS (thought to reflect degree of DAI; Hattori et al., 2003). Initial GCS was not significantly related to atrophy in any lobe (ρs= −.09–.21, ps>.05). Additionally, even though participants indicated chronic partial disability on the DRS (M= 2.43, SD= 2.44, Mdn= 3.0, range= 0.0–7.0) and the GOSE (M= 4.5, SD= 0.76, Mdn= 5.0, range= 3.0–5.0) these scores failed to correlate with total MCBA (ρs= −.38–.20, ps>.05).
As can be seen in Table 2, MCBA was associated with neuropsychological function at 12 months post-injury. Total atrophy was associated with attention (SDMT, oral). Atrophy in the left frontal lobe was related to poor executive ability (TMT-B) and psychomotor function (GPT). Temporal lobe atrophy was associated with reduced attention (SDMT, oral-both lobes, written-left lobe), executive skills (TMT-B-both lobes), and psychomotor ability (GPT-both lobes). No other significant lobar MCBA-neuropsychological associations were found.
Table 2.
Associations (Spearman’s rho (ρ)) between Early Metabolic Crisis-Related Brain Atrophy and Neuropsychological Performances 12 months Post-Injury
| Total Atrophy | Rt. Temporal | Lt. Temporal | Rt. Frontal | Lt. Frontal | Rt. Parietal | Lt. Parietal | Rt. Occipital | Lt. Occipital | |
|---|---|---|---|---|---|---|---|---|---|
|
|
|||||||||
| SDMT Oral | .62a* | .60a* | .66a* | .39c | .63d | .21b | .47b | −.06a | .06a |
| SDMT Written | .39a | .56 | .60a* | .18c | .40d | .20b | .31b | .13a | .18a |
| ROCFT Imm. | .48a | .33a | .27a | .35c | .45d | .28b | .46b | .00a | −.26a |
| ROCFT Delay | .42b | .30b | .28b | .34d | .62e | .20b | .49b | −.23b | −.23b |
| SRT-6 Total | .11a | .10a | .18a | −.18c | .07d | .08d | −.08d | .42a | .25a |
| SRT-6 Delay | −.07a | −.16a | −.04a | −.09c | −.17d | −.13c | −.06b | .33a | .27a |
| TMT-B | −.51a | −.65a* | −.71a** | −.33c | −.67d* | −.19b | −.42b | .02a | −.05a |
| COWAT | .36a | .49a | .54a | .38c | .45d | .43b | .42b | −.12a | −.12a |
| GPT Dom. | −.53b | −.62b* | −.66b* | −.42d | −.69e* | −.26c | −.36c | .10b | .06b |
Note. SDMT Oral= Symbol Digit Modalities Test, oral trial (attention); SDMT Written= Symbol Digit Modalities Test, written trial (attention); ROCFT Imm.= Rey-Osterrieth Complex Figure Test, immediate recall trial (nonverbal memory); ROCFT Delay= Rey-Osterrieth Complex Figure Test, delayed recall trial (nonverbal memory); SRT-6 Total= Selective Reminding Test, 6-trial version, total learning trial recall (verbal memory); SRT-6 Delay= Selective Reminding Test, 6-trial version, delayed recall (verbal memory); TMT-B= Trail Making Test, part B (executive ability); COWAT= Controlled Oral Association Test, F-A-S version (executive ability); GPT Dom. = Grooved Pegboard Test, dominant hand (psychomotor ability). Performance on the TMT-B and the GPT are based on time (lower scores are better); the other test scores on based on correct responses (higher scores are better).
n = 14,
n = 13,
n = 12,
n = 11,
n = 10
p < 0.05, FDR < .05
p < 0.01, FDR < .05
Atrophy group differences
Brain atrophy for both groups is listed in Table 3 and the overall brain atrophy in the sample from a representative participant is displayed in Figure 1. As can be seen in Table 3, the groups differed on percent median total atrophy (Z= −3.13, p= .002, q= .002, r= −.84), parietal lobe atrophy (right: Z= −2.71, p= .007, q= .003, r= −.75; left: Z= −2.29, p= .02, q= .006, r= −.63), frontal lobe atrophy (right: Z= −2.43, p= .02, q= .005, r= −.67; left: Z= −2.24, p= .03, q= .006, r= −.65), and temporal lobe atrophy (right: Z= −3.13, p=.002, q= .002, r= −.84; left: Z= −2.75, p= .006, q= .003, r= −.73); there was no group difference in occipital lobe atrophy (right: Z= −.19, p=.85, q= .16, r= −.05; left: Z= −.70, p= .48, q= .10, r= −.19).
Table 3.
Percent Median Atrophy for entire sample and by subgroup
| Sample | n | FTA | n | OA | n | |
|---|---|---|---|---|---|---|
|
|
||||||
| Total | .05 | 14 | .12 | 7 | .03 | 7 |
| Rt. Temporal Lobe | 5.54 | 14 | 14.80 | 7 | 2.91 | 7 |
| Lt. Temporal Lobe | 8.98 | 14 | 14.76 | 7 | 4.14 | 7 |
| Rt. Frontal Lobe | 4.78 | 13 | 11.68 | 6 | 2.16 | 7 |
| Lt. Frontal Lobe | 3.06 | 12 | 5.36 | 6 | 1.56 | 6 |
| Rt. Parietal Lobe | 9.72 | 13 | 16.28 | 7 | 4.00 | 6 |
| Lt. Parietal Lobe | 6.60 | 13 | 12.86 | 7 | 2.27 | 6 |
| Rt. Occipital Lobe | 3.39 | 14 | 3.38 | 7 | 3.60 | 7 |
| Lt. Occipital Lobe | 5.08 | 14 | 7.26 | 7 | 4.83 | 7 |
Note. Atrophy values exclude any identified area contusion or lesion.
Percent atrophy = ((acute volume − chronic volume)/acute volume) × 100.
FTA= gross frontal/temporal lobe atrophy group; OA= other atrophy group.
FIG. 1.
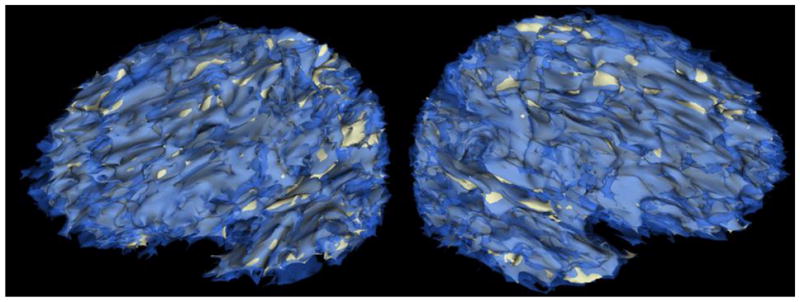
Three-dimensional representation of atrophy in the left and right hemisphere (left and right images, respectively) derived from co-registered acute (transparent blue) and chronic (grey) magnetic resonance images from a study participant. Note that areas of blue over grey indicate atrophy. This participant, like most of our participants, evidenced gross cortical atrophy. Images represent total atrophy, not just that due to early metabolic crisis.
Additional Mann-Whitney U tests for group comparisons on neuropsychological tests failed to reveal any significant differences (Zs = −.29 – −1.98, ps= .05 – .78, qs= .047 – .78, rs= −.08 – −.53), although there was a trend toward a significant difference on a test of attention (SDMT oral; FTA: Mdn= 36.32, OA: Mdn= 44.78, Z= −1.98, p= .047, q= .32, r= −.53).
Median age and education corrected scores were contrasted with normative data (T= 50) for each neuropsychological test to assess the clinical significance of the deficits exhibited by each group. One-sample Wilcoxon signed rank tests revealed that the FTA group exhibited many clinically significant deficits at 12 months post-injury. Specifically, they performed significantly worse than the norm on measures of attention (SDMT written, Z= −2.20, p= .03, q= .04, r= −.83; SDMT oral, Z= −2.37, p= .02, q= .04, r= −.89), executive skill (TMT-B, Z= −2.37, p= .02, q= .04, r= −.89; COWAT, Z= −2.20, p= .03, q= .04, r= −.83), verbal memory (SRT-6 delayed, Z= −2.03, p= .04, q= .048, r= −.77), nonverbal memory (ROFCT immediate, Z= −2.03, p= .04, q= .048, r= −.77; ROFCT delayed, Z= −2.20, p= .03, q= .04, r= −.83), and psychomotor function (GPT, Z= −2.20, p= .03, q= .04, r= −.90). While the FTA group’s verbal learning performances were not significantly different from the norm, we found a trend toward a significant difference (SRT-6 total, Z= −1.86, p= .06, q= .06, r= −.70).
One-sample Wilcoxon signed rank tests revealed that the OA group did not significantly differed from the norm at 12 months post-injury (Zs = −.51 – −2.37, ps= .02 –.61, qs= .16 – .61, rs= −.19 – −.89), although a trend toward a significant difference in verbal learning was detected (SRT-6 total, Z= −2.37, p= .02, q= .16, r= −.89).
Discussion
Our primary objective was to determine if MCBA following TBI is associated with neuropsychological impairments at 12 months post-injury. We found that nonischemic MCBA was strongly correlated with attention, executive functioning, and psychomotor abilities at 12 months post-injury. It is important to note that we accounted for all identified contusions and lesions in our quantification of MCBA. We did not find significant cognitive differences between participants with early metabolic crisis-related FTA and OA as we had hypothesized. However, participants with FTA demonstrated clinically significant deficits (−2SDs or more from the norm) in attention, memory (verbal and nonverbal), executive ability, and psychomotor function, while those with OA did not indicate a pattern of clinically significant neuropsychological deficits. That said, the participants with FTA also demonstrated greater nonischemic early metabolic crisis-related parietal lobe atrophy in contrast to the OA participants. Nevertheless, our data suggest that cognitive impairments associated with MCBA are regionally driven, as chronic neuropsychological impairments were only related to frontal (executive and psychomotor performances) and temporal (attention, executive, and psychomotor performances) lobe atrophy, although the study was underpowered to detect smaller brain-behavior relationships.
Previous work from our group has indicted that acute metabolic disturbances are related to chronic atrophy in the frontal, temporal, and parietal lobes, with the frontal and temporal lobes generally exhibiting the greatest atrophy (Xu et al., 2010). Interestingly, neuropsychological data have often characterized TBI-related deficits in attention/speeded processing, memory, and executive functioning as ‘frontal-temporal’ in nature (Lezak et al., 2004; Stuss & Gow, 1992; Vakil, 2005). The current study suggests that MCBA may significantly contribute to the ‘frontal-temporal’ profile of TBI.
Limitations
While our study was the first to demonstrate a link between MCBA and chronic neuropsychological deficits in TBI, our results are based on data from a small sample of participants. While this reinforces the magnitude of the relationship between MCBA and cognition, it also means that we had relatively low statistical power and may have missed significant associations between cognitive functioning and MCBA in the parietal and occipital lobes, as well as additional associations in the frontal and temporal lobes. Also, sample size precluded contrasting neuropsychological performances in multiple atrophy groups to determine the most meaningful patterns and levels of MCBA as they relate to neuropsychological outcome. Additionally, although we went to great lengths to control for contusions, ischemic lesions, and axonal injuries and found no correlation between MCBA and an indirect indicator of DAI (initial GCS score), we cannot completely rule out the possible impact of one or more of these factors on our results, although our atrophy data is most clearly related to acute metabolic crisis in contrast to any other obvious causes. That said, additional studies are needed to determine how lesion burden, DAI mediated reductions connectivity between brain regions, and acute metabolic crisis interact to contribute to TBI-related cognitive impairment.
Conclusions
This study was the first to demonstrate an association between acute metabolic crisis and neuropsychological outcome in TBI. Specifically, our data showed that MCBA in the frontal and temporal lobes was very strongly associated with deficits in attention, executive functioning, and psychomotor abilities at 12 months post-injury. These findings provide a meaningful link between our work showing that MCBA is most prominent in the frontal and temporal lobes (Xu et al., 2010) and neuropsychological studies characterizing TBI-related cognitive impairments as ‘frontal-temporal’ in nature. Moreover, we found that participants with gross frontal and/or temporal lobe pathology exhibited more clinically significant neuropsychological deficits relative to clinical norms. While it is currently unclear how various neuropathological processes interact and impact neuropsychological functioning in TBI, it is clear that MCBA plays a prominent role in the neuropsychological sequelae of TBI. That said, additional studies are needed to determine if various markers of acute brain metabolism may be salient predictors of outcome in TBI survivors and if interventions focused on reducing acute metabolic crisis might result in greater functional outcomes for TBI survivors. Moreover, future work should also investigate the interaction between MCBA and other TBI-related neuropathology.
Acknowledgments
This research was supported by NS049471, NS02089, P01-NS058489 and the California State Neurotrauma Initiative.
References
- Adams JH, Doyle D, Ford I, Gennarelli TA, Graham DI, McLellan DR. Diffuse axonal injury in head injury: definition, diagnosis, and grading. Histopathology. 1989;15:49–59. doi: 10.1111/j.1365-2559.1989.tb03040.x. [DOI] [PubMed] [Google Scholar]
- Adams JH, Doyle D, Graham DI, Parker L, Scott G. Brain damage in fatal non-missile head injury. Journal of Clinical Pathology. 1980;33:1132–1145. doi: 10.1136/jcp.33.12.1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams JH, Jennett DIG. The structural basis of moderate disability after traumatic brain injury. Journal of Neurology, Neurosurgery, Psychiatry. 2001;71:521–524. doi: 10.1136/jnnp.71.4.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Army Individual Test Battery. Manual of directions and scoring. Washington, D.C: War Department, Adjutant General’s Office; 1944. [Google Scholar]
- Auerbach SH. Neuroanatomical correlates of attention and memory in traumatic brain injury: an application of neurobehavioral subtypes. Journal of Head Trauma Rehabilitation. 1986;1:1–12. doi: 10.1097/00001199-198609000-00004. [DOI] [Google Scholar]
- Benton AL, Hamsher K, Varney NR, Spreen O. Contributions to neuropsychological assessment. New York: Oxford; 1983. [Google Scholar]
- Bentourkia M, Bol A, Ivanoiu A, Labar D, Sibomana M, Coppens A, De Volder AG. Comparison of regional cerebral blood flow and glucose metabolism in the normal brain: effect of aging. Journal of Neurological Science. 2000;181:19–28. doi: 10.1016/S0022-510X(00)00396-8. [DOI] [PubMed] [Google Scholar]
- Bigler ED. Quantitative magnetic resonance imaging in traumatic brain injury. Journal of Head Trauma Rehabilitation. 2001;16:117–134. doi: 10.1097/00001199-200104000-00003. [DOI] [PubMed] [Google Scholar]
- Bohlman L, Knight RT. Electrophysiological dissociation of rapid memory mechanisms in humans. NeuroReport. 1994;5:1517–1521. doi: 10.1097/00001756-199407000-00027. [DOI] [PubMed] [Google Scholar]
- Bor D, Cumming N, Scott CEL, Owen AM. Prefrontal cortical involvement in verbal encoding strategies. European Journal of Neuroscience. 2004;19:3365–3370. doi: 10.1111/j.1460-9568.2004.03438.x. [DOI] [PubMed] [Google Scholar]
- Clifton GL, Hayes RL, Levin HS, Michel ME, Choi SC. Outcome measures for clinical trials involving traumatically brain-injured patients: report of a conference. Neurosurgery. 1992;31:975–978. doi: 10.1227/00006123-199211000-00028. [DOI] [PubMed] [Google Scholar]
- Corwin J, Bylsma FW. “Psychological Examination of Traumatic Encephalopathy” by A. Rey and “The Complex Figure Copy Test” by P. A. Osterrieth. The Clinical Neuropsychologist. 1993;7:3–21. [Google Scholar]
- Dinov ID, Van Horn JD, Lozev KM, Magsipoc R, Petrosyan P, Liu Z, MacKenzie-Graham A, Toga AW. Efficient, distributed and interactive neuroimaging data analysis using the LONI pipeline. Frontiers in Neuroinformation. 2009;3:1–10. doi: 10.3389/neuro.11.022.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fork M, Bartels C, Ebert AD, Grubich C, Synowitz H, Wallesch C. Neuropsychological sequelae of diffuse traumatic brain injury. Brain Injury. 2005;19:101–108. doi: 10.1080/02699050410001726086. [DOI] [PubMed] [Google Scholar]
- Gleissner U, Helmstaedter C, Kurthen M, Elger CE. Evidence of very fast memory consolidation: an intracarotid amytal study. Neuroreporter. 1997;8:2893–2896. doi: 10.1097/00001756-199709080-00018. [DOI] [PubMed] [Google Scholar]
- Glenn TC, Kelly DF, Boscardin WJ, McArthur DL, Vespa P, Oertel M, Martin NA. Energy dysfunction as a predictor of outcome after moderate or severe head injury: indices of oxygen, glucose, and lactate metabolism. Journal of Cerebral Blood Flow Metabolism. 2003;23:1239–1250. doi: 10.1097/01.WCB.0000089833.23606.7F. http://0dx.doi.org.torofind.csudh.edu/10.1097/01.WCB.0000089833.23606.7. [DOI] [PubMed] [Google Scholar]
- Habib R, Nyberg L, Tulving E. Hemispheric asymmetries of memory: the HERA model revisited. Trends in Cognitive Science. 2003;7:241–245. doi: 10.1016/S1364-6613(03)00110-4. [DOI] [PubMed] [Google Scholar]
- Hannay HJ, Levin HS. Selective reminding test: an examination of the equivalence of four forms. Journal Clinical Experimental Neuropsychology. 1985;7:251–263. doi: 10.1080/01688638508401258. [DOI] [PubMed] [Google Scholar]
- Hattori N, Hung SC, Wu HM, Yeh E, Glenn TC, Vespa PM, Bergsneider M. Correlation of regional metabolic rates of glucose with Glasgow Coma Scale after traumatic brain injury. Journal of Nuclear Medicine. 2003;44:1709–1716. [PubMed] [Google Scholar]
- Hayman LA. Adult cerebrum. In: Hayman ALA, Hinck V, editors. Clinical brain imaging: normal structure and functional anatomy. St Louis: Mosby Year book; 1992. pp. 130–137. [Google Scholar]
- Head D, Buckner RL, Shimony JS, Williams LE, Akbudak E, Conturo TE, Snyder AZ. Differential vulnerability of anterior white matter in nondemented aging with minimal acceleration in dementia of the Alzheimer type: evidence from diffusion tensor imaging. Cerebral Cortex. 2004;14:410–423. doi: 10.1093/cercor/bhh003. doi:0.1093/cercor/bhh003. [DOI] [PubMed] [Google Scholar]
- Kraus MF, Susmaras T, Caughlin BP, Walker CJ, Sweeney JA, Little DM. White matter integrity and cognition in chronic traumatic brain injury: a diffusion tensor imaging study. Brain. 2007;130:2508–2519. doi: 10.1093/brain/awm216. [DOI] [PubMed] [Google Scholar]
- Larrabee GJ, Trahan DE, Levin HS. Normative data for a six-trial administration of the verbal selective reminding test. Clinical Neuropsycholgy. 2000;14:110–118. doi: 10.1076/1385-4046(200002)14:1;1-8;FT110. [DOI] [PubMed] [Google Scholar]
- Lehtonen S, Stringer AY, Millis S, Boake C, Englander J, Hart &, Whyte J. Neuropsychological outcome and community re-integration following traumatic brain injury: the impact of frontal and non-frontal lesions. Brain Injury. 2005;19:239–256. doi: 10.1080/0269905040004310. [DOI] [PubMed] [Google Scholar]
- Levine B, Cabeza R, McIntosh AR, Black SE, Grady CL, Stuss DT. Functional reorganization of memory after traumatic brain injury: a study with H215O positron emission topography. Journal of Neurology, Neurosurgery, & Psychiatry. 2002;73:173–181. doi: 10.1136/jnnp.73.2.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lezak MD, Howieson DB, Loring DW, Hannay HJ, Fischer JS. Neuropsychological assessment. 4. New York, NY, US: Oxford University Press; 2004. pp. 158–194. [Google Scholar]
- Lin K, Huang S, Baxter L, Phelps M. A general technique for inter-study registration of multi-function and multimodality images. IEEE Transactions on Nuclear Science. 1994;41:2850–2855. [Google Scholar]
- Marcoux J, McArthur DA, Miller C, Glenn TC, Villablanca P, Martin …, Vespa PM. Persistent metabolic crisis as measured by elevated cerebral microdialysis lactate-pyruvate ratio predicts chronic frontal lobe brain atrophy after traumatic brain injury. Critical Care Medicine. 2008;36:2871–2877. doi: 10.1097/CCM.0b013e318186a4a0. [DOI] [PubMed] [Google Scholar]
- Matthews CG, Klǿve K. [Instruction manual] for the Adult Neuropsychology Test Battery. Madison, WI: University of Wisconsin Medical School; 1964. [Google Scholar]
- Mitrushina M, Boone KB, Razani J, D’Elia LF. Handbook of normative data for neuropsychological assessment. 2. New York, NY, US: Oxford University Press; 2005. pp. 648–760.pp. 782pp. 969 [Google Scholar]
- National Institutes of Health. NIH consensus development panel on rehabilitation of persons with traumatic brain injury. JAMA. 1999;282:974–983. [PubMed] [Google Scholar]
- Ohta S, Meyer E, Thompson CJ, Gjedde A. Oxygen consumption of the living human brain measured after a single inhalation of positron emitting oxygen. Journal of Cerebral Blood Flow Metabolism. 1992;12:179–192. doi: 10.1038/jcbfm.1992.28. [DOI] [PubMed] [Google Scholar]
- Raz N. Aging of the brain and its impact on cognitive performance: integration of structural and functional findings. In: Craik FIM, Salthouse TA, editors. The handbook of aging and cognition. 2. Mahwah, NJ, US: Lawrence Erlbaum Associates Publishers; 2000. pp. 1–90. [Google Scholar]
- Schmitter-Edgecombe M, Wright MJ. Event-based prospective memory following severe closed-head injury. Neuropsychology. 2004;18:353–361. doi: 10.1037/0894-4105.18.2.353. [DOI] [PubMed] [Google Scholar]
- Schmitter-Edgecombe M, Marks W, Wright MJ, Ventura M. Retrieval inhibition in directed forgetting following severe closed-head injury. Neuropsychology. 2004;18:104–114. doi: 10.1037/0894-4105.18.1.104. [DOI] [PubMed] [Google Scholar]
- Schmitter-Edgecombe M, Wright MJ. Content memory and temporal order memory for performed activities after severe closed-head injury. Journal of Clinical and Experimental Neuropsychology. 2003;25:933–948. doi: 10.1076/jcen.25.7.933.16493. doi:0.1076/jcen.25.7.933.16493. [DOI] [PubMed] [Google Scholar]
- Schmitter-Edgecombe M, Marks W, Fahy JF. Semantic priming after severe closed head trauma: automatic and attentional processes. Neuropsychology. 1993;7:136–148. doi: 10.1037/0894-4105.7.2.136. [DOI] [Google Scholar]
- Shattuck DW, Leahy RM. BrainSuite: an automated cortical surface identification tool. Medical Image Analysis. 2002;6:129–142. doi: 10.1007/978-3-540-40889-4_6. [DOI] [PubMed] [Google Scholar]
- Squire LR. Specifying the defect in human amnesia: storage, retrieval, and semantics. Neuropsychology. 1980;18:369–372. doi: 10.1016/0028-3932(80)90134-7. [DOI] [PubMed] [Google Scholar]
- Squire LR. Memory and forgetting: long-term and gradual changes in memory storage. In: Sporns AO, Tononi G, editors. Selectionism and the brain. San Diego, CA, US: Academic Press; 1994. pp. 243–269. [PubMed] [Google Scholar]
- Squire LR, Zola SM. Episodic memory, semantic memory, and amnesia. Hippocampus. 1998;8:205–211. doi: 10.1002/(SICI)1098-1063(1998)8:3<205::AID-HIPO3>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Storey JD. A direct approach to false discovery rates. Journal of the Royal Statistical Society. 2002;64:479–498. [Google Scholar]
- Stuss DT, Gow CA. “Frontal dysfunction” after traumatic brain injury. Neuropsychiatry, Neuropsychology, Behavior & Neurology. 1992;5:272–282. [Google Scholar]
- Takaoka M, Tabuse H, Kumura E, Nakajima S, Tsuzuki T, Nakamura K, Okada A, Sugimoto H. Semi-quantitative analysis of corpus callosum injury using magnetic resonance imaging indicates clinical severity in patients with diffuse axonal injury. Journal of Neurology, Neurosurgery, & Psychiatry. 2002;73:289–293. doi: 10.1136/jnnp.73.3.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vakil E. The effect of moderate to severe traumatic brain injury (TBI) on different aspects of memory: a selective review. Journal of Clinical Experimental Neuropsychology. 2005;27:977–1021. doi: 10.1080/13803390490919245. [DOI] [PubMed] [Google Scholar]
- Vespa P, Bergsneider M, Hattori N, Wu HM, Huang SC, Martin NA, Hovda DA. Metabolic crisis without brain ischemia is common after traumatic brain injury: a combined microdialysis and positron emission tomography study. Journal of Cerebral Blood Flow & Metabolism. 2005;25:763–774. doi: 10.1038/sj.jcbfm.9600073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vespa PM, Miller C, McArthur D, Eliseo M, Etchepare M, Hirt D, Hovda DA. Non-convulsive electrographic seizures after traumatic brain injury result in a delayed, prolonged increase in intracranial pressure and metabolic crisis. Critical Care Medicine. 2007;35:2830–2836. doi: 10.1097/01.CCM.0000295667.66853.BC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vespa P, McArthur D, Alger J, O’Phelan K, Glenn T, Bergsneider B, Hovda DA. Regional heterogeneity of brain metabolism using cerebral microdialysis: concordance with magnetic resonance spectroscopy and positron emission tomography. Brain Pathology. 2004;14:210–214. doi: 10.1111/j.1750-3639.2004.tb00055.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallesch C, Curio N, Kutz S, Jost S, Bartels C, Synowitz H. Outcome after mild-to-moderate blunt head injury: effects of focal lesions and diffuse axonal injury. Brain Injury. 2001;15:401–412. doi: 10.1080/02699050116886. [DOI] [PubMed] [Google Scholar]
- Williamson DJG, Scott JG, Adams RL. Traumatic brain injury. In: Adams ARL, Parsons OA, Culbertson JL, Nixon SJ, editors. Neuropsychology for Clinical Practice: Etiology, Assessment, and Treatment of Common Neurological Disorders. Washington, DC: American Psychological Association; 1996. pp. 9–64. [Google Scholar]
- Wilson JTL, Hadley DM, Wiedmann KD, Teasdale GM. Neuropsychological consequences of two patterns of brain damage shown by MRI in survivors of severe head injury. Journal of Neurology, Neurosurgery, Psychiatry. 1995;59:328–331. doi: 10.1136/jnnp.59.3.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright MJ, Schmitter-Edgecombe M, Woo E. Verbal memory impairment in severe closed-head injury: the role of encoding and consolidation. Journal of Clinical Experimental Neuropsychology. 2010;32:728–736. doi: 10.1080/13803390903512652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright MJ, Schmitter-Edgecombe M. The impact of verbal memory encoding and consolidation deficits during recovery from moderate-to-severe traumatic brain injury. Journal of Head Trauma Rehabilitation. 2011;26:182–191. doi: 10.1097/HTR.0b013e318218dcf9. [DOI] [PubMed] [Google Scholar]
- Wright MJ, Woo E, Schmitter-Edgecombe M, Hinkin CH, Miller EN, Gooding AL. The Item-Specific Deficit Approach (ISDA) to evaluating verbal memory dysfunction: rationale, psychometrics, and application. Journal Clinical Experimental Neuropsychology. 2009;31:790–802. doi: 10.1080/13803390802508918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, McArthur DL, Alger JR, Etchepare M, Hovda DA, Glenn TC, Vespa PM. Early nonischemic oxidative metabolic dysfunction leads to chronic brain atrophy in traumatic brain injury. Journal Cerebral Blood Flow & Metabolism. 2010;30:883–894. doi: 10.1038/jcbfm.2009.263. [DOI] [PMC free article] [PubMed] [Google Scholar]