

Summary
Type 1 diabetes remains an important health problem, particularly in Western countries where the incidence has been increasing in younger children1. In 1986, Eisenbarth described Type 1 diabetes as a chronic autoimmune disease. Work over the past 3 ½ decades has identified many of the genetic, immunologic, and environmental factors that are involved in the disease and have led to hypotheses concerning its pathogenesis. Based on these findings, clinical trials have been conducted to test these hypotheses but have had mixed results. In this review, we discuss the findings that have led to current concepts of the disease mechanisms, how this understanding has prompted clinical studies, and the results of these studies. The findings from preclinical and clinical studies support the original proposed model for how type 1 diabetes develops, but have also suggested that this disease is more complex than originally thought and will require broader treatment approaches.
A model of the pathogenesis of type 1 diabetes (T1D) was originally proposed by George Eisenbarth in a landmark paper in 19862. It followed earlier observations of a long prediabetic period identified by the presence of islet cell autoantibodies (ICA) in diabetes with polyendocrine deficiencies3 and described a chronic autoimmune process, initiated by unknown factors that proceeded over many years in which insulin-producing β cells were killed by autoreactive lymphocytes. The bases for this highly original concept were observations from clinical studies examining progression of the disease in relatives of patients who were at risk. When patients with TID received a pancreatic isograft from an identical twin, T cell infiltration was found in the isograft at the time of declining graft function4. In addition, data from a number of intervention studies suggested that immunosuppressive therapies, such as anti-thymocyte globulin and cyclosporin A, could have a positive impact on T1D disease progression 5,6. Since then, extensive human and animal studies have strengthened the concept that this progressive disease is accompanied by β cell destruction, but also β cell dysfunction. At the time of onset, most clinical studies suggest that as much as 30% of β cell mass is present and in many cases residual insulin production can increase soon after disease diagnosis as the dysfunction improves with metabolic control7 (Box 1). This level of residual function is by no means insignificant and warrants preservation. More than 90% of patients with new onset disease, including children, have a level of stimulated C-peptide that is at least 0.2 nmol/l, a level found to be associated with improved glucose control, and reduced risk of severe hypoglycemia and secondary end organ complications (for example, retinopathy and renal disease)8,9. However, there is a linear decline in functional β cell mass. Thus, the proportion of subjects who maintain this level is small 5 years after the initial T1D diagnosis.
Box 1. Clinical aspects of T1D.
Type 1 diabetes (T1D) is one of the most common chronic diseases of childhood. The prevalence of T1D ranges from <5 in every 100,000 individuals in eastern countries to as many as 39.9 in every 100,000 individuals in European and other western countries154. A significant proportion (estimated to be approximately 10%) of adults who present with diabetes have T1D rather than the more common T2D which is not autoimmune in nature. There are strong genetic determinants of the disease (Box 3) but >90% of individuals presenting with new onset disease do not have a relative with T1D57. More than 90% of individuals with T1D have at least one positive autoantibody and the presence of autoantibodies identifies relatives of patients who are at high risk for the disease (typical targets of these autoantibodies include GAD65, ICA512, insulin, ZNT8 and ICA)75,155,156. The peak incidence of disease onset is between 6–15 years of age with a second peak occurring later in adolescence. At the time of presentation, most patients have signs and symptoms of hyperglycemia and insulin deficiency (polyuria, polydipsia, visual change, weight loss, and elevated glycosylated hemoglobin A1c levels) or even more severe metabolic decompensation with ketoacidosis. However, some patients are identified on routine urine or blood tests, before β cell destruction and insulin deficiency have resulted in symptoms. After presentation and metabolic stabilization, many patients enter a clinical ‘honeymoon’ when insulin secretion improves and some patients could even discontinue the use of exogenous insulin. This period is invariably followed by loss of insulin production and increasing dependence on exogenous insulin8,157. Due to the absolute deficiency in insulin production, replacement with exogenous insulin and dietary regulation are the mainstays of treatment. Retention of some endogenous insulin production, which is reflected by the level of C-peptide (the byproduct of processing of proinsulin that is synthesized by β cells) of at least 0.2 pmol/ml, has been associated with improved metabolic control and reduced risk of long term complications such as eye and renal disease as well as the acute complication of insulin induced hypoglycemia8,9. Metabolic control is monitored by measurement of glycosylated hemoglobin A1c (HbA1c) that reflects the glucose control over the previous 2–3 months.
Studies in preclinical models have added to our understanding of the antigens, cells and mechanisms involved in T1D development and progression. Furthermore, recent clinical investigations have refined and in some cases changed these concepts. In this review, we will discuss the concepts that led to clinical studies in patients with T1D and the results of testing those hypotheses in clinical trials. Similar to the clinical observations that led to the original hypothesis that T1D is an autoimmune disease, translational research efforts in humans and animal models continue to be a source of new discoveries that shape the T1D field.
Environmental factors leading to T1D
In many countries in the incidence of T1D has been increasing in younger children, faster than can be accounted for by genetic change alone, highlighting a role for environmental factors1,10. There is evidence of temporal changes over the last 20 or so years in those developing T1D, with increased incidences in the under 5 age group as well as in individuals with lower risk HLA haplotypes, such as HLA-DQB1*0602. These disturbing findings have revived searches for environmental factors that may be responsible for triggering T1D, such as changes in exposure to infectious, environmental or nutritional agents.
Infectious agents and commensal organisms
Infectious agents, including parasites, viruses or bacteria, could have pathogenic or protective roles in T1D. Pathogenesis could be elicited through direct infection of β cells, through a more generalized release of pro-inflammatory or cytotoxic cytokines in response to infection — especially at the pancreatic tissue site — or through antigen mimicry11. Alternatively, it has been proposed that some infectious organisms, such as helminths, can shape the immune system in a manner that is of mutual benefit to parasite and host. These interactions might have historically prevented the onset of T1D and the removal of these infections through increased public health measures might be responsible for the increased incidence of T1D that we see today (this idea is known as the ‘hygiene hypothesis’)12–15.
It has been proposed for decades that enteroviruses are linked to T1D11,16. The report of a 10 year-old patient who died with fulminant T1D and showed acute and convalescent titers against a coxsackie B4 virus that was isolated and shown to cause β cell destruction, supported this notion as did a second case study, which showed that sudden onset of T1D in an adult patient was associated with coxsackie B4 virus infection and natural killer cell-composed insulitis17,18;16. More recent studies have shown that in response to infection, human islets secrete pro-inflammatory cytokines, such as interleukin-6 (IL-6), IL-8, and tumour necrosis factor (TNF), and chemokines, such as CXC-chemokine ligand 10 (CXCL10, also known as IP-10), CC-chemokine ligand 3 (CCL3, also known as MIP1α) and CCL4 (also known as MIP1β)19,20. Moreover, phagocytosis of enterovirus-infected β cells triggers innate immune responses in human dendritic cells (DCs)21.
There is increasing evidence that commensal organisms play a role in molding the host immune system and that alterations of the gut microbiome can have immunological, metabolic and pathological consequences (Figure 1) 22,23. The polysaccharide component of the outer membrane vesicles of Bacteroides fragilis, which is a normal component of the human gut microbiota, has been shown to interact with host DCs, inducing anti-inflammatory cytokine production and generation of Foxp3+ regulatory T (Treg) cells that are capable of inhibiting inflammatory bowel disease. An increase in B. ovatus and the firmicute strain CO19 has been found in the microbiota of patients with T1D in a case controlled study although it is not clear if an associated polysaccharide component in these strains influence disease progression. Moreover, a relative decrease in B fragilis was seen in patients vs controls over time24,25. Other commensal organisms have been shown to influence invariant natural killer T (iNKT) cell activity26. As both Treg cells and iNKT cells have been shown to influence diabetes onset in model systems, the ability of exogenous infectious agents, as well as commensal organisms, to influence these regulatory cell types provides mechanisms by which environmental agents might influence the host immune response. The pathogenic effects of some commensal organisms may, however, be disease-specific. For example, there is evidence that some species, such as segmented filamentous bacteria, can accelerate the onset of arthritis or experimental allergic encephalomyelitis by inducing Th17 cells but inhibit the onset of autoimmune diabetes in non-obese diabetic (NOD) mice27,28.
Figure 1. Revision of the Eisenbarth model.
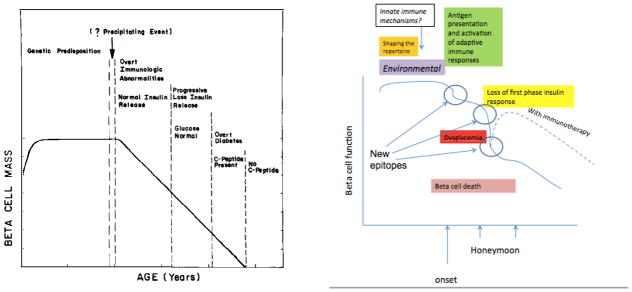
Earlier concepts of the pathogenesis of T1D have been modified with new information. During childhood, there is an increased in β cell function, as a result of which older subjects present with T1D with higher C-peptide responses than younger subjects157,172. The timing of the decline in β cell function may be more acute than previously appreciated173. Nonetheless, the impairement seen at the time of diagnosis may reverse partially but invariably continues with time. Successful immune therapies have altered the decline in C-peptide.
Studies of Myd88−/− NOD mice underscored the importance of the gut microbiota and its interactions with the host innate immune system in modulating diabetes onset29. This signaling pathway is required for autoimmune diabetes development in NOD mice under specific pathogen-free (SPF) conditions. The way in which MYD88 signaling affects disease pathogenesis is through modulation of the gut microbiota, because Myd88−/− NOD germ-free mice develop rampant disease, but disease protection can be transferred with feces from Myd88−/− NOD mice raised under SPF conditions. However, the precise role of the gut microbiota in this setting has not been clarified.
At least three other observations concerning the microbiota are relevant to this discussion. First, the microbiome of healthy children is more diverse and less stable than are the microbiomes from patients with autoimmune diseases. Second, germ-free mice have incomplete immune systems and colonization with microflora is needed for the development of Th17 cell responses as well as Treg cell responses in the gut. Third the gut microbiota regulates intestinal permeability, which may play a role in the initiation of insulitis by altering the transport and subsequent autoimmune triggering antigens to the pancreas25.
Clinical testing targeting environmental factors
There are at least three challenges to identifying organisms that are causative of T1D development in humans. First, infection with the agent may not be closely related in time to presentation with hyperglycemia – these events may be separated by years. Second, the majority (>90%) of patients who develop T1D do not have an affected relative with the disease. Therefore, identification of at risk individuals requires a broad population-based search. Third, it is possible that the pathologic event involves the absence of an immune response to an organism rather than the presence of a protective response and therefore, cannot be identified. For instance, instead of looking for a virus or other environmental antigen that induces disease, the relative risk may increase as a result of failure to develop protective immunity which makes the causal linkage that much more difficult to identify. Two ongoing studies (TEDDY and DAISY) are screening for viral and other pathogenic infections in high risk offspring that are associated with T1D.
Clinical data indirectly supports the notion that early dietary manipulation may affect disease development. Epidemiologic studies have shown that T1D incidence is lower in breast –fed versus bottle-fed offspring of parents with T1D and the timing of exposure to cereal was linked to T1D development 30–32. The TRIGR study is testing whether exposure to cow’s milk is associated with development of autoimmunity and ultimately diabetes33,34.
Breaking tolerance to autoantigens
Creating the autoimmune repertoire: autoantigens, T cells and B cells
In the original model, the basis for the breakdown in tolerance to self proteins remained unclear and this question persists today (Box 2). The unresolved issues include the identity of the critical self antigen(s) that drives initiation and perpetuation of disease, the nature of the tolerance defect, and the individual roles of central versus peripheral compartments in propagating diabetes. The discovery of autoantibodies in the serum of patients was direct evidence suggesting that T1D was an autoimmune disease 3. Although autoantibodies identify the ongoing autoimmune response, the main way in which of B cells contribute to T1D pathology appears to be through their antigen presentation35. Antibody-mediated depletion of B cells, even at the time of onset of hyperglycemia, can prevent or reverse disease in NOD mice 36,37
Box 2. Mechanisms of tolerance relevant to T1D.
Tolerance to self proteins is controlled by a number of checkpoints during development of T and B cells, a process referred to as central tolerance, as well as in the peripheral tissues to ensure that potentially autoreactive cells do not respond to the tissues, termed peripheral tolerance 158. Central tolerance purges the mature repertoire of T and B cells expressing autoreactive receptors in the thymus and bone marrow, respectively. This can occur by inducing apoptosis of T or B cells, or in the case of B cells, altering the specificity of the B cell receptor (BCR) in a process termed receptor editing 159,160. These mechanisms are restricted to antigens that are presented or expressed in those compartments and as a result, potentially autoreactive T and B cells may escape into the periphery. Both cell intrinsic and extrinsic mechanisms are involved in controlling the activation of those cells. The former includes modulators of BCR and T cell receptor (TCR) signaling, such as induction of inhibitory receptors (for example, CTLA4, PD-1 and LAG3) or ubiquitin ligases (for example,. Cbl). Extrinsic factors involve restriction of the required costimulatory ligands on APCs (for example, the B7 molecules CD80 and CD86, which are required for CD28-mediated costimulation of T cells), limiting the availability of survival factors (such as BAFF and IL-7), or exposure to inhibitory cytokines (such as IL-10) or regulatory populations (for example, Treg cells). The latter may develop in the thymus or in the periphery 161. Treg cells are characterized by the expression of the X-linked forkhead transcription factor FOXP3, a master regulator required for their maximal development and function. A genetic mutation of FOXP3 abolishes Treg cell function leading to the immunodysregulation polyendocrinopathy enteropathy X-linked (IPEX) syndrome in human, a severe multiorgan autoimmune syndrome caused by uncontrolled immune activation.
Over the course of several years, the targets of these autoantibodies have been identified and include insulin, proinsulin, GAD65, IGRP, IA-2, and most recently the zinc transporter in β cells, ZNT838. A combination of human genome studies (Box 3) and functional studies in animal models of disease have implicated insulin (or proinsulin) 39,40 as the primary autoantigen for disease initiation while other islet-specific molecules, such as IGRP and chromogranin A have been suggested to promote disease progression. Preclinical studies have highlighted the progression of disease through intramolecular and intermolecular spreading41,42. Although many autoantigens have been implicated as targets and drivers of T1D, there is limited direct evidence for a single autoantigen in the development of the disease. Elimination of proinsulin or insulin completely prevents insulitis and diabetes in NOD mice, but the removal of IGRP, another self antigen that is targeted by T cells, did not show this protective effect 39,40. In humans, the primacy of insulin as the major autoantigen for diabetes initiation has not been proven, although in young children with diabetes, autoantibodies against insulin tend to appear before autoantibodies with other specificities43, and cytotoxic T lymphocytes have been isolated from a patient with T1D that kill β cells through a glucose-related preproinsulin epitope44.
Box 3. Genetics of T1D.
There is a strong genetic basis for the disease. Overall the risk of disease for siblings of patients with type 1 diabetes (T1D) is approximately 6%, which is 15-fold higher than in the general population 162,163. The risk for identical twins has been reported to be as low as 30% but more recent data have suggested that with a longer observation period (to age 60) 65% are concordant164. The most important susceptibility alleles are within the major histocompatibility complex (MHC) – the link to the MHC had originally suggested the autoimmune basis for T1D. The MHC complex has an odds ratio for disease of approximately 6.857,165 The HLA DRB1*04-DQA1*0301-DQB1*0302 and DRB1*03-DQA1*0501-DQB1*0201 haplotypes are the strongest T1D risk factors in European populations: heterozygosity for both risk haplotypes confers the greatest known genetic risk. Other alleles have been associated with T1D in non-European populations (for example, DRB1*0405-DQB1*0401 and DRB1*0901-DQB1*0303 in Japanese and Korean populations). In addition, strongly protective alleles (HLA-DQB1*0602) have a dominant effect 166. Recently, the application of genome-wide SNP typing technology to large sample sets and comparisons with results from other immune-mediated diseases have provided convincing support for 19 non-MHC T1D loci, all with allelic odds ratios of less than 1.3 165. These include IL-2RA, with an odds ratio of approximately 1.6, PTPN22, with an odds ratio of 2.0, and CTLA4 with an odds ratio of approximately 1.25. Some of the immune response related loci are shared with other autoimmune diseases, while other susceptibility alleles appear to be disease specific. For example, IDDM2, with an odds ratio of 2.1, is in the insulin promoter and thus may affect insulin expression in the thymus and negative selection.
Several studies continue to point to the potential importance of central tolerance mechanisms in preventing immune reactivity to autoantigens, and failure of these mechanisms promotes the progression of autoimmune diabetes in mice, and potentially humans40,45,46. A significant percentage of patients with autoimmune polyendocrinopathy candidiasis ectodermal dystrophy (APECED) develop autoimmune diabetes47,48. APECED is caused by mutations in the transcription factor AIRE, which is expressed in thymic medullary epithelial cells and promotes their expression of tissue-specific antigens. This process enables the development of central tolerance to peripheral proteins, such as insulin, that are only expressed at restricted tissue sites. In mice, the Ins1 gene is predominantly expressed by β cells in the pancreatic islets, but the Ins2 gene is expressed both in the thymus and by islet β cells. Although Ins1−/− mice exhibited reduced autoimmune diabetes, Ins2−/− mice showed markedly accelerated disease development, presumably owing to a defect in central tolerance 46,49,50. In an analogous manner, the susceptibility alleles of the IDDM2 gene locus, a polymorphism of the insulin (INS) promoter, cause lower thymic INS expression compared with the alleles associated with diabetes resistance (Box 3).
Certain novel epitopes of disease-associated antigens may not be presented in the thymus resulting in escape of autoreactive T cells into the periphery. For instance, an insulin peptide (B12–20), which is distinct from the B13–21 peptide that is presented in the thymus, is presented by antigen-presenting cells in the islets to “type B T cells”. The antigen-presenting cells in the islets accomplish this without interaction with H2-DM, and the responding cells, which can cause T1D do not recognize peptide derived from the intracellular processing of native protein 51–53. Moreover, Kappler and colleagues have shown that insulin peptides can bind to MHC molecules, such as I-Ag7 in distinct registers due to flexibility in the binding grove of MHC class II molecule53–55. The so-called ‘register 3’ binding is of low affinity and leads to the escape of the peptide-specific CD4+ T cells from the thymus53–55. In the periphery, the unique trimolecular complex of T cell receptor (TCR), MHC, and peptide can activate autoreactive CD4+ T cells. The absence of presentation and expression of these tissue-specific antigens in the thymus provides a potential mechanism that might be perturbed in autoimmune diseases like T1D, although this has yet to be established 39–43.
Defects in both central and peripheral B cell tolerance mechanisms have also been identified in patients with T1D56. The frequency of polyclonal and Hep-2 reactive autoantibodies is increased in these patients suggesting failures of central and peripheral tolerance checkpoints, respectively. A similar increase in these types of autoantibodies is seen in patients with rheumatoid arthritis or in otherwise healthy subjects with the R620W variant of the PTPN22 allele. The PTPN22 allele, which is found on a gene locus that is associated with T1D (IDDM3), encodes the lymphoid protein tyrosine phosphatase (LYP). LYP also acts in a complex with C-terminal Src kinase (CSK) to negatively regulate signaling from the TCR and variant PTPN22 has been shown to alter negative selection in the thymus by altering TCR signaling57. Thus these defects in central and peripheral T and B cell tolerance can establish an autoreactive lymphocyte repertoire that may drive T1D development.
Mechanisms of β cell destruction
In preclinical models such as the NOD mouse and BB/W rat, β cell destruction is caused by T cells 58,59. Two ‘checkpoints’ during the pathogenesis of the disease have been described60. The first checkpoint is the recognition of islet antigens and this has been associated with β cell death, which can be developmentally programmed or occur following cellular damage. The second checkpoint is the conversion from a non-destructive to a destructive insulitis, a process that is also enhanced by cellular damage. The latter checkpoint may involve acquisition of new effector functions by T cells, a lack of negative signaling, the enhanced production of pro-inflammatory mediators, the exposure of previously inaccessible β cell antigens that fuel the disease process, or abrogation of regulatory control caused by a defect in the number or function of Treg cells (see below for more detailed discussion on this).
Both diabetes antigen specific CD4+ and CD8+ T cell clones can transfer disease to naïve recipients and their destructive mechanisms involve the generation of cytokines, such as TNF and IFNγ, in addition to their direct cytotoxic effects61,62. While some have suggested that the CD4+ T cells that infiltrate the islets of NOD mice possess a heterogeneous phenotype, in terms of T cell receptor (TCR) usage and antigen specificity63,64, sequencing studies of early infiltrates suggested that TCR usage by islet-infiltrating T cells is fairly limited65. In humans, restricted and preferential TCR usage has been observed in intra-islet T cells from T1D patients 66. Some of this restricted usage may be due to the recognition of insulin peptides by germline-encoded TCR elements 67. A critically important future goal will be to better understand the TCR usage and antigen specificity of the islet-infiltrating T cell repertoire. T cell retrogenic technology is a platform in which this can be accomplished in a reasonable time frame68–71. Studies using this technology have shown that islet antigen expression is a key factor in determining the ability of a given T cell population to accumulate in the islets. Cell extrinsic mechanisms do not result in the accumulation of bystander cells indicating that islet entry and accumulation is a cell-autonomous event63. The network for pancreatic organ donors with diabetes (nPOD) and similar resources will likely prove invaluable for this work 72. Studies with this material have shown multifocal infiltration in addition to widespread expression of MHC class I on pancreatic islets. Using tetramer technology it was furthermore possible to identify the islet antigen specificity of autoreactive CD8+ T cells in insulitic lesions from recent onset and long-term T1D patients73.
Clinical testing of agents that target autoreactive lymphocyte responses
‘Natural history’ studies have shown that there is a strong association between the risk of developing T1D disease and the presence of autoantibodies to known islet antigens74–76. Interestingly, the number of different antigens that were targeted by autoantibodies (including GAD65, IA-2 or ICA512, IGRP, insulin, and islet cell antibody (ICA)) rather than the overall titer of autoantibodies was the most important determinant of risk75. Indeed, among unaffected first degree relatives of T1D patients, with positive islet cell antibody and ‘dysglycemia’ or 4 autoantibodies, over 75% will develop T1D over the following 5–6 years with a median time to onset in those with dysglycemia of 2.81 and 4.24 years for 8–17 and 18–45 years old, respectively.
A number of studies have attempted to induce tolerance to specific antigens by parenteral, or intranasal vaccination, or by oral administration (Figure 2). The rationale for these approaches was to induce tolerance by affecting specific lymphocyte populations, or to promote bystander suppression or even infectious tolerance 77. However, the results from trials of antigen-specific therapies have been disappointing. Trials to induce tolerance to insulin have been largely unsuccessful in altering disease progression once autoimmune destruction had ensued 78,79. High-risk relatives of patients administered parenteral (in the DPT-1) or even intranasal administration of insulin showed no evidence for modification of disease progression80. Although a prevention trial, in which insulin was administered orally to at-risk relatives of patients with diabetes (that is, to induce oral tolerance), failed to show any significant delay in the onset of diabetes, the treatment did induce a significant delay in disease onset in individuals with the highest titer of anti-insulin antibodies 81. Another pilot study suggested a delay in the decline of C-peptide levels when patients with T1D were administered GAD65 in alum adjuvant to generate autoantigen-specific regulatory T cells82. The rationale for this study was that by modifying the immune response to this antigen through vaccination, the disease progression could be curtailed. However, two subsequent trials failed to corroborate these findings, even though the immunization increased the titers of GAD65 autoantibodies and the frequency of T cells producing inhibitory cytokines82–84. These results have been disappointing but highlight that the selection of antigen and patient may be paramount for the success of this strategy.
Figure 2. Results of immune therapy trials.

Clinical trials of rituximab, CTLA4Ig, anti-CD3 mAb have shown decreased rates of decline in C-peptide during the first 1 or 2 years of T1D, whereas others, such as antigens, MMF/DZB, thymoglobulin, or anti-IL-1 reagents have not85,86,91,135,137,138,141,142. A trial of rapamycin and IL-2, thought to enhance Tregs showed transient decline in C-peptide responses. HSP60, believed to bind to TLR2 also showed effecdts on glucagon stimulated C-peptide responses. A trial of sTNF receptor (Etanercept) resulted in lower HgbA1c and increased insulin production in a pilot trial174.
Some surprising results, based on prior experiences with anti-thymocyte globulin and cyclosporin A, was that neither treatment with mycophenylate mofetil and daclizumab (a monoclonal antibody specific for the α subunit of the IL-2 receptor) nor treatment with thymoglobulin (which eliminated T cells and some B cells) was able to affect the decline in C-peptide seen in T1D patients with new onset disease 85(Gitelman SE, Fisher L, Gottlieb P, Gottschalk M, Moore W, Moran A, Rigby M, Willi S, Keyes-Elstein L, Ding L, and Ehlers, M unpublished observations). In contrast, eliminating B cells with a 4 week course of rituximab delayed the decline of C-peptide at 1 year in patients with new onset T1D86. Insulin and GAD65 autoantibodies but not ICA-512 antibodies were reduced by rituximab treatment suggesting a kinetic hierarchy of antigens, but interestingly, the T cell proliferative responses to islet antigens were not reduced87,88. Despite its efficacy at 1 year, the beneficial effects of rituximab treatment were no longer detectable at year 2. Collectively, these studies suggest that induction of immune regulation, rather than cell depletion, may be a more effective strategy for inhibiting T1D disease in the long term (see below).
In addition to modulating specific autoreactive T cells directly, blocking their activation has been more successful. In mice, blockade of CD28–B7 costimulation signals with human CTLA4-Ig at 2–4 wks, but not later, prevented diabetes primarily due to preventing CD86 (also known as B7.2)–CD28 signaling89. However, mouse CTLA4-Ig transgenic or NOD mice treated with CD80 (also known as B7.1)-specific antibody had exacerbated disease90. In humans, treatment with CTLA4-Ig (Abatacept) delayed disease progression (by 9.6 months) in a randomized placebo controlled study in subjects with new onset T1D who received 27 infusions of the drug over a 2-year period.91. This finding suggests either that the timing of priming of diabetogenic cells is later than predicted in the NOD model or that CD80/86-dependent mechanisms may be involved in the function of pathogenic T cells very late in the disease course. It should also be noted that in spite of continued treatment the C-peptide levels declined in parallel with the placebo treated group after the first 6 months, possibly reflecting the action of co-stimulation independent cells in this later stage, or β cell loss that is independent of CD80/86-dependent immune mechanisms (Figure 2).
Role of innate immune cells
Numerous studies have implicated cells of the innate immune system in both the initiation and development of diabetes. Analysis of NOD mice and human diabetic islets has revealed the infiltration of macrophages, DCs and natural killer (NK) cells along with cells of the adaptive immune response92. In addition to MHC class I-restricted killing of β cells by cytolytic T cells and Fas–FasL interactions, pathways mediated by innate immune cells have been implicated in the selective death of β cells. These include interaction of NK cell-expressed NKG2D or NKp46 with β cell-expressed RAE1 or NKp46 ligand, respectively, and cytokine mediated effects, including reactive oxygen species (ROS) induction. Human and mouse β cells express NKp46 ligands as well as ligands for NKG2D, which, together with the presence of CD107+ NK cells in the diabetic mouse pancreas, has lead to the suggestion that NK cells may play a role in β cell death93. However, recent depletion studies of NK1.1+ cells have questioned the significance of NK or NKT cells in NOD mice, as NKG2D is also expressed by activated T cells and NKp46 is expressed by γδ T cells and some αβ T cells as well as by innate lymphoid cells 94.
Blockade of macrophage entry into the pancreas or inhibition of macrophage function in mice prevents diabetes onset suggesting a key role for this population in β cell demise. Cytokines produced by cells of the innate immune system, including macrophages and DCs, have been implicated in β cell dysfunction in the diabetic pancreas in mice and humans. IL-1β can inhibit insulin production and IL-1β, TNF and IFNγ may directly contribute to β cell death95,96. Treatment with IL-1-specific antibody or genetic deficiency in IL-1 receptor expression delays but does not prevent T1D in NOD mice97,98.
Pro-inflammatory cytokines upregulate expression of MHC class I on islets in vitro. Interestingly, in humans with T1D, there is high levels of expression of MHC class I molecules on islet cells with and without cellular infiltrates raising the possibility of a sustained inflammatory response within islets due to viral or other environmental insults, abnormalities in MHC class I peptide processing or the effects of systemic cytokines on the β cell environment73,99. The collective consequences of these immunological insults may make β cells more susceptible to CD8+ T cell-mediated killing. In addition to classical DCs, that play a key role in the initiation of diabetes through activation of autoreactive T cells, plasmacytoid DCs (pDCs) have also been implicated in diabetes development. These cells make large amounts of type 1 IFNs, as well as IL-12 and pro-inflammatory cytokines, and there is evidence from several models that type 1 interferons can enhance diabetes onset. Lastly, non-specific inhibition of inflammation with alpha-1 anti-trypsin (AAT), which inhibits enzymes that are release by innate immune cells such as neutrophils, was shown to reverse new onset NOD diabetes100. As a result of treatment with AAT, increased β cell proliferation and insulin sensitivity were also seen in NOD mice.
Clinical testing based on innate immune cell targets
Preclinical studies had suggested that high doses of nicotinamide affected ADP-ribosylation and other reactions in β cells as well as in immune cells and the endothelium. These reactions are thought to play a role in signaling through certain TLRs and other innate inflammatory responses 101. Cell death pathways and gene-expression patterns were modified, leading to improved β cell survival and an altered immunoregulatory balance. This mechanism also prevented the depletion of nicotinamide adenine dinucleotide (NAD) in β cells. Based on these preclinical studies and the notion that islet damage was an important driver of the disease, a trial of nicotinamide in autoantibody-positive relatives with dysglycemia of patients with T1D was performed102. This study failed, however, to show any reduction in the high rate of progression among the active drug-treated participants compared with the placebo-treated participants.
A clinical trial of the IL-1 receptor antagonist (IL-1Ra) anakinra in patients with type 2 diabetes showed improvement in metabolic parameters and these effects were largely attributed to a direct effect of anakinra on β cells103. A pilot trial of anakinra in patients with T1D showed biologic efficacy104. However, in two recent trials in patients with new onset T1D, canakinumab (an IL-1β-specific antibody) and anakinra failed to affect the decline in C-peptide responses within the first year of the disease. These studies illustrate the many variables in translating results in animal studies to those in humans including differences in the drug itself, timing and dosing of the interventions, and lack of uniformity (i.e. possibility of disease subtypes) of the human disease (Box 4).
Box 4. Pitfalls in translation.
There have been notable failures of effective therapies in preclinical models, most often in NOD mice, to achieve the same success in human trials. Included among these are successful therapies of oral insulin, GAD65 immunization, and IL-1 blockade81,97,98,167, 168(in press). Effects on C-peptide and insulin use were seen in clinical trials of anti-CD3 mAbs but the permanent reversal of disease that was so striking in diabetic NOD mice was not achieved. Moreover, at least one therapy that was relatively unsuccessful in NOD mice (CTLA4Ig) did improve C-peptide responses in patients with new onset disease89. These experiences have led some to question the value of preclinical studies in models and their necessity for the design of clinical studies.
However, a careful analysis of the preclinical studies suggests that their ability to predict outcomes is strong but details concerning a broad number of variables, including dosage of agents (e.g. oral insulin) and timing (e.g. anti-IL-1 reagents), may not have been fully considered in the clinical trial design169. Nonetheless, there clearly are important differences between murine and human autoimmune diabetes that complicate the translation. In fact, there may be difference among subsets of humans with what is lumped together as a single disease. It has been suggested that the ability of murine β cells to regenerate may be more robust than human β cells, but more studies are needed 170. The kinetics of diabetes in NOD mice appears to be more abbreviated based on the timing of metabolic decompensation. There may be differences between the development of T1D in young versus older humans. When patients present with new onset T1D, the majority retain a stimulated C-peptide level of at least 0.2 nmol/l and lose this clinically significant level only over a period of years after onset8. In contrast, β cell function and mass in NOD mice is rapidly lost after the first appearance of hyperglycemia – attributes that may be more similar in very young humans than in young adolescents and adults where the therapies are often tested first. There are clearly differences in the innate and adaptive immune response pathways that are affected by therapeutics and therefore, it is to be expected that there may be differences171. Finally, NOD mice are inbred and live in a protected pathogen-free environment. Patients with T1D live in the real world but even this can vary enormously in different geographical locations and economic states. Thus the primary weakness may not be in the NOD mouse model per se but rather in our interpretation and utilization of the data derived from its use.
Where does this leave us? Clinical experience would suggest that therapies that have dramatic effects on diabetes in NOD mice may not achieve the same degree of therapeutic benefit in humans because of patient heterogeneity, differences in kinetics of disease, the responses of β cells to stress and injury, and even subtle differences in immune responses. However, the animal models have been very effective in elucidating mechanisms of action that are relevant to human disease. The challenge is how to apply the data from preclinical models to the patient population. Endpoints of clinical trials should be carefully chosen to identify biologic proof of efficacy and mechanism of action, with outcomes that are important for the design of a combinatorial approach that will successfully achieve clinical endpoints. Considering the heterogeneity of human subjects, efforts to identify individuals most likely to respond to a particular intervention, based on clinical parameters or immunologic markers may be very valuable.
Immunization with a peptide of heat shock protein 60 (HSP60), DiaPep277, has been postulated to enhance the function of CD4+CD25+ Tregs (see below) via signaling through TLR2 105 was shown to reduce the decline in C-peptide responses in patients with new onset T1D, most recently in a Phase III trial106(presented at EASD 2012). The mechanistic basis may involve activation of Toll-like receptor 2 (TLR2) in regulatory T cells that show activation of PKC, PI3K and p38, and secretion of transforming growth factor β (TGFβ) and IL-10 105. Finally, studies are ongoing to test the effects of AAT in patients with new onset T1D.
Defects and mechanisms in the control of tolerance by immune cells
It has become increasingly evident over the past 10–15 years that the immune system is under tight control mediated by specialized cell subsets that suppress immune reactivity. The most prominent of the suppressive cell subsets are Treg cells. This infrequent T cell population, generally identified as CD4+CD25+CD127lo T cells, is critical for the maintenance of peripheral tolerance in many autoimmune diseases, including autoimmune diabetes107,108. Peripheral tolerance is dependent on the balance of effector and regulatory T cells to maintain immune and tissue homeostasis, and defects in Treg cell populations may play a critical role in disease pathogenesis (Box 2).
Treg cells develop in the thymus with a unique antigen-recognition repertoire skewed toward self-antigens109. FOXP3 expression remains critical throughout life to maintain the Treg cell population and prevent autoimmunity. In fact, FOXP3 can be turned on during activation of human conventional T cells and, under the right inflammatory conditions and cytokine milieu, the expression of FOXP3 is stabilized by demethylation of the conserved non-coding sequence 2 (CNS2) that is found within the FOXP3 promoter. This results in the development of a peripheral Treg cell compartment that has a repertoire overlapping that of conventional T cells109. In NOD mice, depletion of CD4+CD25+ Treg cells greatly accelerates the development of diabetes110,111. Similarly, removing critical co-stimulatory or proliferative signals necessary for Treg cell development or survival, such as IL-2 or CD28, exacerbates diabetes in NOD mice111. Thus, it is clear that Treg cells function as the major peripheral cells controlling tolerance and immune homeostasis. However, it should be noted that there are multiple types of suppressor cells that have been identified and may contribute to modulating autoimmune diabetes onset and/or progression. Examples include IL-10-producing regulatory B cells, suppressor macrophages, tolerogenic DCs and additional FOXP3− TGFβ-dependent Th3 cells, IL-10-dependent T regulatory 1 (Tr1) cells and CD8+ regulatory cells112–117. Treg cells can be subdivided into multiple subsets and tissue-specific subpopulations108. In addition, it has been postulated that the Treg cell transcriptional programs, and conceivably their suppression mechanisms, can be tailored to the nature of the effector response they regulate118. Thus, the collective magnitude of suppressive activities may reflect the functions of individual Treg cell subsets in different tissues with distinct dynamics and unique immunological effects.
The basis of Treg cell functional suppression is quite complex and includes a number of cell surface and soluble factors that control immune activation directly and via bystander suppression. Some of their most prominent activities include production of IL-10, TGFβ and IL-35, cytokines that shut down antigen-presenting DCs and activated T cells. Cell surface molecules including CTLA4, PD1 and LAG3 are also important for Treg cell-mediated suppression113,119–122. For instance, CTLA4 can function by competing with CD28 for binding to CD80 and CD86, and in some studies CTLA4 was found to strip the molecules off the cell surface of the APCs or to deliver a negative signal to APCs through those ligands. In addition, factors produced directly and indirectly as a result of Treg cell function (such as IL-10, IL-35, indoleamine 2,3-dioxygenase 1 (IDO) and TGFβ) can promote the development of other regulatory cells in their vicinity, leading to so-called infectious tolerance and a robust local regulation114,121,123,124.
Several genetic loci important for Treg cell biology have been linked to increased susceptibility to T1D including: CD25 and IL-2, the critical growth factor receptor/ligand for Treg cell growth and survival, CTLA4, a major functional receptor on Treg cells and the HLA locus, which can alter Treg cell repertoires57,125,126 (Box 2). In addition, PTPN22 alters TCR signaling leading to less IL-2 production by effector T cells. This suboptimal IL-2 production by effector T cells in the islets could locally compromise Treg cell homeostasis. Furthermore, phosphorylation of the critical IL-2-induced intracellular signaling molecule STAT5 is reduced in patients with T1D, which may be the reason for reduced Treg cell numbers in some patients with the disease127. Reduced IL-2 receptor signaling has been linked to the IL-2RA susceptibility allele (rs12722495)125. In fact, treatment of diabetic mice with IL-2 reverses diabetes 128.
Treg cells have been shown to be unstable in a variety of autoimmune settings, including in mouse and human studies of T1D. Specifically, in NOD mice, the lack of IL-2 expression in the islets can lead to a loss of CD25 expression, reduction of FOXP3 expression and increased numbers of ‘exFOXP3+’ cells; these exFOXP3+ T cells are potentially pathogenic as they can recognize islet antigens in a specific manner and produce IFNγ (Figure 3). Adoptive transfer of autoreactive exFoxp3 cells led to the rapid onset of diabetes109. In patients, the frequency of Treg cells producing pro-inflammatory cytokines, such IFNγ or IL-17, is elevated in T1D patients129. A final point to consider is that, rather than a primary defect existing in Treg cell functions, it has been suggested that conventional effector T cells in patients with T1D are resistant to regulation mediated by Treg cell127.
Figure 3. Seeding the periphery with ex-Tregs with pathogenic potential.

In normal individuals, developing T cells that are not highly self reactive mature and leave the thymus whereas highly autoreactive T cells are deleted during development as part of negative selection. Regulatory T cells, which also develop in the thymus, are selected on self-antigens and demethylate the FOXP3 and express the protein (green dots). In patients with T1D, as well as other autoimmune diseases, cells that are autoreactive but retain the ability to make pathogenic cytokines and do not completely demethylate the FOXP3 gene. These cells are postulated to participate in pathologic immune responses to self antigens.
Clinical testing based on targeting Treg cells
Studies in NOD mice have shown that immunotherapies ranging from anti-CD3, anti-thymocyte globulin and rapamycin may stabilize and expand Treg cell populations130–132. More recent studies in conventional and humanized mice identified a mechanism whereby anti-CD3 monoclonal antibody induces migration of T cells to the gut, where they acquire a regulatory phenotype and produce TGFβ (in conventional mice) or IL-10 (in conventional and humanized mice)133,134. Thus, a number of recent therapeutic opportunities have focused on altering the effector T cell/regulatory T cell balance in patients with T1D (Figure 2).
Initial trials of two Fc receptor (FcR) non-binding anti-CD3 monoclonal antibodies (teplizumab and otelixizumab) showed that the decline in C-peptide was reduced for up to 3 years after a single course of drug treatment in new onset patients. Furthermore, these antibody therapies also showed efficacy in subgroups of patients with longer duration disease, importantly, without the need for continuous immune suppression 135–138. Samples from the drug-treated subjects suggested that treatment with the CD3-specific antibodies induced CD8+ T cells with regulatory function 139,140. However, two Phase III studies with these drugs failed to meet their primary endpoints141. In the study with otelixizumab, the drug administered was approximately 1/10th the dose used in the previous successful Phase II trial. In the other trial with teplizumab, the primary endpoint (number of subjects with hemoglobin A1c <6.5% and utilizing < 0.5U/kg/d of insulin) was not met but an effect on preservation of C-peptide secretion was still seen. Moreover, 2 recent trials have shown that teplizumab treatment can preserve C-peptide levels in new onset patients and even in younger patients with longer duration of disease142(Herold KC, Gitelman SE, Ehlers MR, Gottlieb PA, Greenbaum CJ, Hagopian W, Boyle KD, Keyes-Elstein L, Aggarwal S, Phippard D, Sayre PH, McNamara J, Bluestone JA, unpublished observations)
A pilot trial of IL-2 with rapamycin was performed after preclinical studies and suggested that this combination therapy would increase the number of regulatory T cells and lead to disease reversal130,143. However, the treatment transiently worsened C-peptide responses. This outcome has been attributed to the potentiating effects of IL-2 on pathogenic cells, such as NK cells. Further studies with lower doses of IL-2 without rapamycin are in progress.
How can we improve treatments for T1D based on these findings and experiences?
T1D is a complex disease influenced by genetic and environmental factors and involving innate and adaptive arms of the immune system. It is therefore likely that a multifaceted solution will be required for prevention, treatment and a durable cure. A key component of this process is to continue to develop a complete understanding of the disease process, as well as better biomarkers to identify patients who will develop T1D as early as possible to maximize the success of intervention (Box 5). This will require better animal disease models as well as direct human experimentation.
Box 5. Unresolved areas of translational investigation.
Although there has been much learned about the pathogenesis of T1D as a result of preclinical and clinical studies, several key questions have arisen and remain unanswered. Among these include:
What are the initiating factors?
Are viruses involved?
Are these unique or common? Are any of these factors intrinic to β cells in T1D patients?
Which antigens are presented and does this change over time or in different patients?
How does the microbiome affect the induction or progression of autoimmunity?
How are innate responses involved?
What is the role of epigenetic changes in the penetrance of disease?
How does the immune repertoire differ in patients who will develop T1D?
What is the antigen specificity of pathogenic T cells and how can these cells be identified?
How much of disease heterogeneity stems from stochastic variation in immune development versus exposure to natureal pathogens versus normal responses to one’s environment?
Why does it take so long to destroy all of the β cells?
Are there unusual features of autoreactive T cell development pathways?
How do immune response and other genes affect disease in general, or the daibetogenic potential of T cells specifically?
What is the role of cell instrisic regulatory mechanisms?
What are the roles of natural and adaptive Tregs?
What are the mechanisms of β cell death?
Which cells are involved?
Do human β cells regenerate, differentiate or divide, and does this differ in very young children versus adults?
Can β cell regeneration, transdifferentiation or division be induced?
Why is β cell death segmental and in a lobular distribution?
How can treatment be improved?
What are the appropriate and realistic parameters for determining success in a clinical trial with a given modality (eg. should restoration of β cell function be an expected outcome with a therapy that targets the autoimmune response if β cell regeneration, transdifferentiation or division does not occur)?
What are the mechanisms of long term failure?
Does it reflect recurrence of the autoimmune response or failure of β cells independent of immune attack?
How does metabolic control affect responses to immune therapies?
How can responders and non-responders be identified? When should interventions be initiated?
Can any of the interventions prevent T1D?
What combinations are optimal and how can the regulatory path for the development of these combinations be optimized?
There are some essential components that will need to be included in any therapy. First, it is likely that treatment will be given for short periods of time, or at best intermittently, to avoid long term off-target effects on fundamental protective immune functions, which are likely to be seen with all except the safest therapeutics. Second, the primary rationale for modality selection should be approaches that are distinct but complementary, with data that supports efficacy. Third, this should include therapies that engage or enhance regulatory mechanisms without the need for chronic immune suppression, which is often associated with long-term risks of infection and tumors. These tolerogenic therapies will be needed to reinstate robust central and peripheral tolerance. Examples might include targeting antigen-specific or other regulatory cells either through drugs or cell-based therapeutics. These efforts could build on current approaches, such as treatment with CD3-specific monoclonal antibodies, cytokines like IL-2, drugs like rapamycin and/or administration of ex vivo expanded Treg cell populations. Additional informed therapies directed at genetic pathways associated with T1D, such as the polymorphisms of IL-2RA and STAT5 signaling or phosphatases such as PTPN22 or PTPN2, may suggest specific targets to induce immune tolerance.
In many cases, these pro-tolerogenic therapies may need to enhance regulatory mechanisms rather than simply inducing unresponsiveness in, or deleting, pathogenic T cells. Combination therapies that can control multiple cells, through mechanisms such as infectious tolerance144, and ensure the durability and stability of these regulatory populations are optimal. In addition, short-term treatment with drugs that silence pro-inflammatory responses and drugs that eliminate the effector/memory T cells, which are resistant to standard regulatory processes, could be used in order to stop the aggressive ongoing destruction and rapid deterioration of glucose tolerance that occurs in the first several months of the disease145, 144,145.
It is important to recognize that with all these therapies, not all patients will respond and therefore, a number of strategies are needed. Data from clinical studies suggest that there are ‘responders’ and ‘non-responders’87. Therefore, in addition to biomarkers that can identify the biologic efficacy of molecules in the short term, identification of the genetic, metabolic and immunologic features that differentiate responders and non-responders may help to select therapies for subjects in order to improve efficacy and safety, and to guide how combinations might be constructed.
The role of the β cell in the progression of the disease is also an area that is not well understood. In response to immunologic stress, β cells secrete a number of factors including cytokines (e.g. IL-1β) and chemokines such as CXCL10 (IP-10) which attracts pathologic cells 146,147. A β cell dysfunction likely plays a role in the acute decline of insulin secretion seen at onset and the rapid recovery after metabolic stabilization148,149. The reversal of the latter most likely explains the ‘honeymoon’ that is seen soon after T1D diagnosis and metabolic stabilization. The rate of β cell death is greater in patients with new onset disease but analysis of immune therapies might consider the impact on cell death as well as function in order to identify those which are most likely to have lasting impact on the disease 150. Moreover, β cell dedifferentiation has been proposed as a mechanism that may explain loss of β cell function under conditions of metabolic or other stress151.
Finally, it is important to note that for hundreds of thousands of patients without any insulin-producing β cells, it is essential that any immune-based therapy will also incorporate a β cell replacement strategy. This may even be an issue in new onset disease as there is increasing evidence that the ongoing assault of the islets by the immune response may initiate necrotic and apoptotic death pathways that may be irreversible following expression of proapoptotic genes 152. Thus, efforts in the embryonic stem cell and induced pluripotent stem cell fields will be an essential and complimentary effort to ensure that once the immune problem is ‘solved’ there will be an effective and ample supply of β cells to replace the damaged tissue153.
Acknowledgments
KCH is supported by the National Institutes of Health (U19 AI082713, DK045735, DK057846, UL1RR024139) and the JDRF (2011-248, 2007-1059). D.A.A.V. is supported by the National Institutes of Health (DK089125, AI039480, AI091977, AI052199), the St Jude National Cancer Institute Comprehensive Cancer Center (CA21765) and the American Lebanese Syrian Associated Charities (ALSAC). J.A.B is supported by the National Institutes of Health (AI50834, AI046643, JDRF 4-2011-248, U19 AI056388). AC is supported by the MRC, Wellcome Trust and EU.
Glossary Terms
- CD28/B7 costimulation
A critical receptor–ligand interaction that is required for maximal T cell activation and survival
- Insulitis
Inflammation of the islets of Langerhans in the pancreas that comprises a complex cellular infiltrate that invades and destroys the islets of Langerhans. The cellular composition includes T cells (CD4+, CD8+ and Treg cells), B cells, DCs, NK cells, and macrophages
- FOXP3
A forkhead/winged-helix family transcription factor (forkhead box P3) that is a critical master regulator of Treg development and function
- Gut microbiome
This is the collective community of bacteria in the small and large intestines
- iNKT cell
Invariant natural killer T cells that share properties of T cells and NK cells and recognize the non-polymorphic CD1d molecule, an antigen-presenting molecule that binds self- and foreign lipids and glycolipids. They recognize GalCer and have restricted TCR usage
- Regulatory T (Treg) cells
A rare subpopulation of CD4+ T cells that are endowed with potent suppressive capacity. They typically express the transcription factor Foxp3+.. Both naturally occurring (which develop in the thymus) and adaptive (which acquire their regulatory activity in the periphery) have been described
- Specific pathogen free (SPF) mice
SPF mice are guaranteed to be free of a defined list of mouse pathogens
- Germ-free mice
Germ-free (also known as gnotobiotic) mice do not harbor any bacteria, viruses or parasites
- T helper 17 (Th17) cell
A CD4+ T cell subset that is characterized by its expression of the transcription factors RORγ, RORα and STAT3. They are involved in inflammatory responses, and normally play an important protective role at epithelial and mucosal surfaces. Their development involves a combination of TGFβ, IL-21, IL-23 and IL-1β, and they secrete IL-17, IL-22, IL-22 and in some circumstances GM-CSF and/or IFNγ
- anti-thymocyte globulin
Polyclonal antibodies against human T cells that are produced by immunizing rabbits or horses
- cyclosporin A
An immunosuppressive drug that inhibits calcineurin, a Ca2+-dependent serine/threonine phosphatase necessary for the nuclear translocation of the transcription factor NFAT (nuclear factor of activated T cells)
- C-peptide
The connecting peptide that joins the A-chain and B-chain of insulin in the proinsulin molecule
- Hygiene hypothesis
The theory that the lack of early childhood exposure to infectious agents, symbiotic microorganisms (for example, changes in gut microflora) and parasites increases susceptibility to allergic and autoimmune diseases by modulating immune system development
- Hep-2
A human epithelial cell line that is commonly used as a target for immunofluorescent detection of a wide range of nuclear- and cytoplasmic-staining antibodies. Distinct staining patterns are associated with particular antibody specificities. For example, a homogeneous nuclear-staining pattern is indicative of antibodies that react with double-stranded DNA or chromatin, whereas a speckled nuclear-staining pattern is indicative of antibodies that react with small nuclear ribonucleoproteins
- Rapamycin
An immunosuppressive drug that, in contrast to calcineurin inhibitors (such as cyclosporin A and FK506), does not prevent T-cell activation but blocks interleukin-2-mediated clonal expansion by blocking mTOR (mammalian target of rapamycin). It does not interfere with the function and expansion of naturally occurring regulatory T cells
Footnotes
Dedication: The authors would like to dedicate this manuscript to the remarkable contributions and memory of George Eisenbarth who inspired all of us.
References
- 1.Ziegler AG, et al. Accelerated progression from islet autoimmunity to diabetes is causing the escalating incidence of type 1 diabetes in young children. J Autoimmun. 2011;37:3–7. doi: 10.1016/j.jaut.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eisenbarth GS. Type I diabetes mellitus. A chronic autoimmune disease. N Engl J Med. 1986;314:1360–1368. doi: 10.1056/NEJM198605223142106. [DOI] [PubMed] [Google Scholar]
- 3.Bottazzo GF, Florin-Christensen A, Doniach D. Islet-cell antibodies in diabetes mellitus with autoimmune polyendocrine deficiencies. Lancet. 1974;2:1279–1283. doi: 10.1016/s0140-6736(74)90140-8. [DOI] [PubMed] [Google Scholar]
- 4.Sibley RK, Sutherland DE, Goetz F, Michael AF. Recurrent diabetes mellitus in the pancreas iso- and allograft. A light and electron microscopic and immunohistochemical analysis of four cases. Lab Invest. 1985;53:132–144. [PubMed] [Google Scholar]
- 5.Eisenbarth GS, et al. Anti-thymocyte globulin and prednisone immunotherapy of recent onset type 1 diabetes mellitus. Diabetes Res. 1985;2:271–276. [PubMed] [Google Scholar]
- 6.Stiller CR, et al. Effects of cyclosporine immunosuppression in insulin-dependent diabetes mellitus of recent onset. Science. 1984;223:1362–1367. doi: 10.1126/science.6367043. [DOI] [PubMed] [Google Scholar]
- 7.Faber OK, Binder C. B-cell function and blood glucose control in insulin dependent diabetics within the first month of insulin treatment. Diabetologia. 1977;13:263–268. doi: 10.1007/BF01219710. [DOI] [PubMed] [Google Scholar]
- 8.Palmer JP, et al. C-peptide is the appropriate outcome measure for type 1 diabetes clinical trials to preserve beta-cell function: report of an ADA workshop, 21–22 October 2001. Diabetes. 2004;53:250–264. doi: 10.2337/diabetes.53.1.250. [DOI] [PubMed] [Google Scholar]
- 9.Steffes MW, Sibley S, Jackson M, Thomas W. beta-Cell function and the development of diabetes-related complications in the diabetes control and complications trial. Diabetes Care. 2003;26:832–836. doi: 10.2337/diacare.26.3.832. [DOI] [PubMed] [Google Scholar]
- 10.Dunne DW, Cooke A. A worm’s eye view of the immune system: consequences for evolution of human autoimmune disease. Nat Rev Immunol. 2005;5:420–426. doi: 10.1038/nri1601. [DOI] [PubMed] [Google Scholar]
- 11.Atkinson MA, et al. Cellular immunity to a determinant common to glutamate decarboxylase and coxsackie virus in insulin-dependent diabetes. J Clin Invest. 1994;94:2125–2129. doi: 10.1172/JCI117567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Strachan DP. Hay fever, hygiene, and household size. BMJ. 1989;299:1259–1260. doi: 10.1136/bmj.299.6710.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bach JF, Chatenoud L. The hygiene hypothesis: an explanation for the increased frequency of insulin-dependent diabetes. Cold Spring Harb Perspect Med. 2012;2:a007799. doi: 10.1101/cshperspect.a007799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cooke A, Zaccone P, Raine T, Phillips JM, Dunne DW. Infection and autoimmunity: are we winning the war, only to lose the peace? Trends Parasitol. 2004;20:316–321. doi: 10.1016/j.pt.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 15.David T, Thomas C, Zaccone P, Dunne DW, Cooke A. The impact of infection on the incidence of autoimmune disease. Curr Top Med Chem. 2004;4:521–529. doi: 10.2174/1568026043451258. [DOI] [PubMed] [Google Scholar]
- 16.Dotta F, et al. Coxsackie B4 virus infection of beta cells and natural killer cell insulitis in recent-onset type 1 diabetic patients. Proc Natl Acad Sci U S A. 2007;104:5115–5120. doi: 10.1073/pnas.0700442104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yoon JW, Austin M, Onodera T, Notkins AL. Isolation of a virus from the pancreas of a child with diabetic ketoacidosis. N Engl J Med. 1979;300:1173–1179. doi: 10.1056/NEJM197905243002102. [DOI] [PubMed] [Google Scholar]
- 18.Akatsuka H, et al. A case of fulminant type 1 diabetes with coxsackie B4 virus infection diagnosed by elevated serum levels of neutralizing antibody. Diabetes Res Clin Pract. 2009;84:e50–52. doi: 10.1016/j.diabres.2009.03.009. [DOI] [PubMed] [Google Scholar]
- 19.Berg AK, Korsgren O, Frisk G. Induction of the chemokine interferon-gamma-inducible protein-10 in human pancreatic islets during enterovirus infection. Diabetologia. 2006;49:2697–2703. doi: 10.1007/s00125-006-0429-7. [DOI] [PubMed] [Google Scholar]
- 20.Christen U, McGavern DB, Luster AD, von Herrath MG, Oldstone MB. Among CXCR3 chemokines, IFN-gamma-inducible protein of 10 kDa (CXC chemokine ligand (CXCL) 10) but not monokine induced by IFN-gamma (CXCL9) imprints a pattern for the subsequent development of autoimmune disease. J Immunol. 2003;171:6838–6845. doi: 10.4049/jimmunol.171.12.6838. [DOI] [PubMed] [Google Scholar]
- 21.Schulte BM, et al. Phagocytosis of enterovirus-infected pancreatic beta-cells triggers innate immune responses in human dendritic cells. Diabetes. 2010;59:1182–1191. doi: 10.2337/db09-1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chung H, et al. Gut immune maturation depends on colonization with a host-specific microbiota. Cell. 2012;149:1578–1593. doi: 10.1016/j.cell.2012.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mathis D, Benoist C. The influence of the microbiota on type-1 diabetes: on the threshold of a leap forward in our understanding. Immunol Rev. 2012;245:239–249. doi: 10.1111/j.1600-065X.2011.01084.x. [DOI] [PubMed] [Google Scholar]
- 24.Round JL, Mazmanian SK. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci U S A. 2010;107:12204–12209. doi: 10.1073/pnas.0909122107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Giongo A, et al. Toward defining the autoimmune microbiome for type 1 diabetes. ISME J. 2011;5:82–91. doi: 10.1038/ismej.2010.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perez-Cano FJ, Dong H, Yaqoob P. In vitro immunomodulatory activity of Lactobacillus fermentum CECT5716 and Lactobacillus salivarius CECT5713: two probiotic strains isolated from human breast milk. Immunobiology. 2010;215:996–1004. doi: 10.1016/j.imbio.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 27.Lee YK, Menezes JS, Umesaki Y, Mazmanian SK. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2011;108 (Suppl 1):4615–4622. doi: 10.1073/pnas.1000082107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kriegel MA, et al. Naturally transmitted segmented filamentous bacteria segregate with diabetes protection in nonobese diabetic mice. Proc Natl Acad Sci U S A. 2011;108:11548–11553. doi: 10.1073/pnas.1108924108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wen L, et al. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature. 2008;455:1109–1113. doi: 10.1038/nature07336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knip M, Virtanen SM, Akerblom HK. Infant feeding and the risk of type 1 diabetes. Am J Clin Nutr. 2010;91:1506S–1513S. doi: 10.3945/ajcn.2010.28701C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ziegler AG, Schmid S, Huber D, Hummel M, Bonifacio E. Early infant feeding and risk of developing type 1 diabetes-associated autoantibodies. Jama. 2003;290:1721–1728. doi: 10.1001/jama.290.13.1721. [DOI] [PubMed] [Google Scholar]
- 32.Norris JM, et al. Timing of initial cereal exposure in infancy and risk of islet autoimmunity. Jama. 2003;290:1713–1720. doi: 10.1001/jama.290.13.1713. [DOI] [PubMed] [Google Scholar]
- 33.Akerblom HK, et al. Dietary manipulation of beta cell autoimmunity in infants at increased risk of type 1 diabetes: a pilot study. Diabetologia. 2005;48:829–837. doi: 10.1007/s00125-005-1733-3. [DOI] [PubMed] [Google Scholar]
- 34.Knip M, et al. Dietary intervention in infancy and later signs of beta-cell autoimmunity. N Engl J Med. 2010;363:1900–1908. doi: 10.1056/NEJMoa1004809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wong FS, et al. Investigation of the role of B-cells in type 1 diabetes in the NOD mouse. Diabetes. 2004;53:2581–2587. doi: 10.2337/diabetes.53.10.2581. [DOI] [PubMed] [Google Scholar]
- 36.Fiorina P, et al. Targeting CD22 reprograms B-cells and reverses autoimmune diabetes. Diabetes. 2008;57:3013–3024. doi: 10.2337/db08-0420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hu CY, et al. Treatment with CD20-specific antibody prevents and reverses autoimmune diabetes in mice. J Clin Invest. 2007;117:3857–3867. doi: 10.1172/JCI32405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wenzlau JM, et al. The cation efflux transporter ZnT8 (Slc30A8) is a major autoantigen in human type 1 diabetes. Proc Natl Acad Sci U S A. 2007;104:17040–17045. doi: 10.1073/pnas.0705894104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krishnamurthy B, et al. Responses against islet antigens in NOD mice are prevented by tolerance to proinsulin but not IGRP. J Clin Invest. 2006;116:3258–3265. doi: 10.1172/JCI29602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nakayama M, et al. Prime role for an insulin epitope in the development of type 1 diabetes in NOD mice. Nature. 2005;435:220–223. doi: 10.1038/nature03523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bot A, et al. Plasmid vaccination with insulin B chain prevents autoimmune diabetes in nonobese diabetic mice. J Immunol. 2001;167:2950–2955. doi: 10.4049/jimmunol.167.5.2950. [DOI] [PubMed] [Google Scholar]
- 42.von Herrath M, Sanda S, Herold K. Type 1 diabetes as a relapsing-remitting disease? Nat Rev Immunol. 2007;7:988–994. doi: 10.1038/nri2192. [DOI] [PubMed] [Google Scholar]
- 43.Yu L, et al. Early expression of antiinsulin autoantibodies of humans and the NOD mouse: evidence for early determination of subsequent diabetes. Proc Natl Acad Sci U S A. 2000;97:1701–1706. doi: 10.1073/pnas.040556697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Skowera A, et al. CTLs are targeted to kill beta cells in patients with type 1 diabetes through recognition of a glucose-regulated preproinsulin epitope. J Clin Invest. 2008;118:3390–3402. doi: 10.1172/JCI35449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jaeckel E, Lipes MA, von Boehmer H. Recessive tolerance to preproinsulin 2 reduces but does not abolish type 1 diabetes. Nat Immunol. 2004;5:1028–1035. doi: 10.1038/ni1120. [DOI] [PubMed] [Google Scholar]
- 46.Thebault-Baumont K, et al. Acceleration of type 1 diabetes mellitus in proinsulin 2-deficient NOD mice. J Clin Invest. 2003;111:851–857. doi: 10.1172/JCI16584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mathis D, Benoist C. Back to central tolerance. Immunity. 2004;20:509–516. doi: 10.1016/s1074-7613(04)00111-6. [DOI] [PubMed] [Google Scholar]
- 48.Anderson MS, et al. The cellular mechanism of Aire control of T cell tolerance. Immunity. 2005;23:227–239. doi: 10.1016/j.immuni.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 49.Moriyama H, et al. Evidence for a primary islet autoantigen (preproinsulin 1) for insulitis and diabetes in the nonobese diabetic mouse. Proc Natl Acad Sci U S A. 2003;100:10376–10381. doi: 10.1073/pnas.1834450100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chentoufi AA, Polychronakos C. Insulin expression levels in the thymus modulate insulin-specific autoreactive T-cell tolerance: the mechanism by which the IDDM2 locus may predispose to diabetes. Diabetes. 2002;51:1383–1390. doi: 10.2337/diabetes.51.5.1383. [DOI] [PubMed] [Google Scholar]
- 51.Mohan JF, et al. Unique autoreactive T cells recognize insulin peptides generated within the islets of Langerhans in autoimmune diabetes. Nat Immunol. 2010;11:350–354. doi: 10.1038/ni.1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mohan JF, Petzold SJ, Unanue ER. Register shifting of an insulin peptide-MHC complex allows diabetogenic T cells to escape thymic deletion. J Exp Med. 2011;208:2375–2383. doi: 10.1084/jem.20111502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stadinski B, Kappler J, Eisenbarth GS. Molecular targeting of islet autoantigens. Immunity. 2010;32:446–456. doi: 10.1016/j.immuni.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 54.Crawford F, et al. Specificity and detection of insulin-reactive CD4+ T cells in type 1 diabetes in the nonobese diabetic (NOD) mouse. Proc Natl Acad Sci U S A. 2011;108:16729–16734. doi: 10.1073/pnas.1113954108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stadinski BD, et al. Diabetogenic T cells recognize insulin bound to IAg7 in an unexpected, weakly binding register. Proc Natl Acad Sci U S A. 2010;107:10978–10983. doi: 10.1073/pnas.1006545107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Menard L, et al. The PTPN22 allele encoding an R620W variant interferes with the removal of developing autoreactive B cells in humans. J Clin Invest. 2011;121:3635–3644. doi: 10.1172/JCI45790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Concannon P, Rich SS, Nepom GT. Genetics of type 1A diabetes. N Engl J Med. 2009;360:1646–1654. doi: 10.1056/NEJMra0808284. [DOI] [PubMed] [Google Scholar]
- 58.Christianson SW, Shultz LD, Leiter EH. Adoptive transfer of diabetes into immunodeficient NOD-scid/scid mice. Relative contributions of CD4+ and CD8+ T-cells from diabetic versus prediabetic NOD.NON-Thy-1a donors. Diabetes. 1993;42:44–55. doi: 10.2337/diab.42.1.44. [DOI] [PubMed] [Google Scholar]
- 59.Miller BJ, Appel MC, O’Neil JJ, Wicker LS. Both the Lyt-2+ and L3T4+ T cell subsets are required for the transfer of diabetes in nonobese diabetic mice. J Immunol. 1988;140:52–58. [PubMed] [Google Scholar]
- 60.Andre I, et al. Checkpoints in the progression of autoimmune disease: lessons from diabetes models. Proc Natl Acad Sci U S A. 1996;93:2260–2263. doi: 10.1073/pnas.93.6.2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Katz JD, Wang B, Haskins K, Benoist C, Mathis D. Following a diabetogenic T cell from genesis through pathogenesis. Cell. 1993;74:1089–1100. doi: 10.1016/0092-8674(93)90730-e. [DOI] [PubMed] [Google Scholar]
- 62.Verdaguer J, et al. Acceleration of spontaneous diabetes in TCR-beta-transgenic nonobese diabetic mice by beta-cell cytotoxic CD8+ T cells expressing identical endogenous TCR-alpha chains. J Immunol. 1996;157:4726–4735. [PubMed] [Google Scholar]
- 63.Lennon GP, et al. T cell islet accumulation in type 1 diabetes is a tightly regulated, cell-autonomous event. Immunity. 2009;31:643–653. doi: 10.1016/j.immuni.2009.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.van den Elzen P, et al. Limited clonality in autoimmunity: drivers and regulators. Autoimmun Rev. 2004;3:524–529. doi: 10.1016/j.autrev.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 65.Baker FJ, Lee M, Chien YH, Davis MM. Restricted islet-cell reactive T cell repertoire of early pancreatic islet infiltrates in NOD mice. Proc Natl Acad Sci U S A. 2002;99:9374–9379. doi: 10.1073/pnas.142284899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Codina-Busqueta E, et al. TCR bias of in vivo expanded T cells in pancreatic islets and spleen at the onset in human type 1 diabetes. J Immunol. 2011;186:3787–3797. doi: 10.4049/jimmunol.1002423. [DOI] [PubMed] [Google Scholar]
- 67.Nakayama M, et al. Germline TRAV5D-4 T-cell receptor sequence targets a primary insulin peptide of NOD mice. Diabetes. 2012;61:857–865. doi: 10.2337/db11-1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Arnold PY, Burton AR, Vignali DA. Diabetes incidence is unaltered in glutamate decarboxylase 65-specific TCR retrogenic nonobese diabetic mice: generation by retroviral-mediated stem cell gene transfer. J Immunol. 2004;173:3103–3111. doi: 10.4049/jimmunol.173.5.3103. [DOI] [PubMed] [Google Scholar]
- 69.Holst J, et al. Generation of T-cell receptor retrogenic mice. Nat Protoc. 2006;1:406–417. doi: 10.1038/nprot.2006.61. [DOI] [PubMed] [Google Scholar]
- 70.Holst J, Vignali KM, Burton AR, Vignali DA. Rapid analysis of T-cell selection in vivo using T cell-receptor retrogenic mice. Nat Methods. 2006;3:191–197. doi: 10.1038/nmeth858. [DOI] [PubMed] [Google Scholar]
- 71.Bettini ML, Bettini M, Vignali DA. T-cell receptor retrogenic mice: a rapid, flexible alternative to T-cell receptor transgenic mice. Immunology. 2012;136:265–272. doi: 10.1111/j.1365-2567.2012.03574.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Campbell-Thompson M, et al. Network for Pancreatic Organ Donors with Diabetes (nPOD): developing a tissue biobank for type 1 diabetes. Diabetes Metab Res Rev. 2012;28:608–617. doi: 10.1002/dmrr.2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Coppieters KT, et al. Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J Exp Med. 2012;209:51–60. doi: 10.1084/jem.20111187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sherr J, Sosenko J, Skyler JS, Herold KC. Prevention of type 1 diabetes: the time has come. Nat Clin Pract Endocrinol Metab. 2008;4:334–343. doi: 10.1038/ncpendmet0832. [DOI] [PubMed] [Google Scholar]
- 75.Orban T, et al. Pancreatic islet autoantibodies as predictors of type 1 diabetes in the Diabetes Prevention Trial-Type 1. Diabetes Care. 2009;32:2269–2274. doi: 10.2337/dc09-0934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Krischer JP, et al. Screening strategies for the identification of multiple antibody-positive relatives of individuals with type 1 diabetes. J Clin Endocrinol Metab. 2003;88:103–108. doi: 10.1210/jc.2002-020760. [DOI] [PubMed] [Google Scholar]
- 77.Waldmann H, Cobbold S. How do monoclonal antibodies induce tolerance? A role for infectious tolerance? Annu Rev Immunol. 1998;16:619–644. doi: 10.1146/annurev.immunol.16.1.619. [DOI] [PubMed] [Google Scholar]
- 78.Fourlanos S, et al. Evidence that nasal insulin induces immune tolerance to insulin in adults with autoimmune diabetes. Diabetes. 2011;60:1237–1245. doi: 10.2337/db10-1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chaillous L, et al. Oral insulin administration and residual beta-cell function in recent-onset type 1 diabetes: a multicentre randomised controlled trial. Diabete Insuline Orale group. Lancet. 2000;356:545–549. doi: 10.1016/s0140-6736(00)02579-4. [DOI] [PubMed] [Google Scholar]
- 80.Effects of insulin in relatives of patients with type 1 diabetes mellitus. N Engl J Med. 2002;346:1685–1691. doi: 10.1056/NEJMoa012350. [DOI] [PubMed] [Google Scholar]
- 81.Skyler JS, et al. Effects of oral insulin in relatives of patients with type 1 diabetes: The Diabetes Prevention Trial--Type 1. Diabetes Care. 2005;28:1068–1076. doi: 10.2337/diacare.28.5.1068. [DOI] [PubMed] [Google Scholar]
- 82.Ludvigsson J, et al. GAD treatment and insulin secretion in recent-onset type 1 diabetes. N Engl J Med. 2008;359:1909–1920. doi: 10.1056/NEJMoa0804328. [DOI] [PubMed] [Google Scholar]
- 83.Ludvigsson J, et al. GAD65 antigen therapy in recently diagnosed type 1 diabetes mellitus. N Engl J Med. 2012;366:433–442. doi: 10.1056/NEJMoa1107096. [DOI] [PubMed] [Google Scholar]
- 84.Wherrett DK, et al. Antigen-based therapy with glutamic acid decarboxylase (GAD) vaccine in patients with recent-onset type 1 diabetes: a randomised double-blind trial. Lancet. 2011 doi: 10.1016/S0140-6736(11)60895-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gottlieb PA, et al. Failure to preserve beta-cell function with mycophenolate mofetil and daclizumab combined therapy in patients with new- onset type 1 diabetes. Diabetes Care. 2010;33:826–832. doi: 10.2337/dc09-1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pescovitz MD, et al. Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N Engl J Med. 2009;361:2143–2152. doi: 10.1056/NEJMoa0904452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Herold KC, et al. Increased T Cell Proliferative Responses to Islet Antigens Identify Clinical Responders to Anti-CD20 Monoclonal Antibody (Rituximab) Therapy in Type 1 Diabetes. J Immunol. 2011;187:1998–2005. doi: 10.4049/jimmunol.1100539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yu L, et al. Rituximab selectively suppresses specific islet antibodies. Diabetes. 2011;60:2560–2565. doi: 10.2337/db11-0674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lenschow DJ, et al. Differential effects of anti-B7-1 and anti-B7-2 monoclonal antibody treatment on the development of diabetes in the nonobese diabetic mouse. J Exp Med. 1995;181:1145–1155. doi: 10.1084/jem.181.3.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lenschow DJ, et al. CD28/B7 regulation of Th1 and Th2 subsets in the development of autoimmune diabetes. Immunity. 1996;5:285–293. doi: 10.1016/s1074-7613(00)80323-4. [DOI] [PubMed] [Google Scholar]
- 91.Orban T, et al. Co-stimulation modulation with abatacept in patients with recent-onset type 1 diabetes: a randomised, double-blind, placebo-controlled trial. Lancet. 2011 doi: 10.1016/S0140-6736(11)60886-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Itoh N, et al. Mononuclear cell infiltration and its relation to the expression of major histocompatibility complex antigens and adhesion molecules in pancreas biopsy specimens from newly diagnosed insulin-dependent diabetes mellitus patients. J Clin Invest. 1993;92:2313–2322. doi: 10.1172/JCI116835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ogasawara K, et al. NKG2D blockade prevents autoimmune diabetes in NOD mice. Immunity. 2004;20:757–767. doi: 10.1016/j.immuni.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 94.Beilke JN, et al. NK cells are not required for spontaneous autoimmune diabetes in NOD mice. PLoS One. 2012;7:e36011. doi: 10.1371/journal.pone.0036011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Amrani A, et al. Interleukin-1 effect on glycemia in the non-obese diabetic mouse at the pre-diabetic stage. J Endocrinol. 1996;148:139–148. doi: 10.1677/joe.0.1480139. [DOI] [PubMed] [Google Scholar]
- 96.Rabinovitch A, Baquerizo H, Sumoski W. Cytotoxic effects of cytokines on islet beta-cells: evidence for involvement of eicosanoids. Endocrinology. 1990;126:67–71. doi: 10.1210/endo-126-1-67. [DOI] [PubMed] [Google Scholar]
- 97.Thomas HE, et al. IL-1 Receptor Deficiency Slows Progression to Diabetes in the NOD Mouse. Diabetes. 2004;53:113–121. doi: 10.2337/diabetes.53.1.113. [DOI] [PubMed] [Google Scholar]
- 98.Cailleau C, Diu-Hercend A, Ruuth E, Westwood R, Carnaud C. Treatment with neutralizing antibodies specific for IL-1beta prevents cyclophosphamide-induced diabetes in nonobese diabetic mice. Diabetes. 1997;46:937–940. doi: 10.2337/diab.46.6.937. [DOI] [PubMed] [Google Scholar]
- 99.Foulis AK, McGill M, Farquharson MA. Insulitis in type 1 (insulin-dependent) diabetes mellitus in man--macrophages, lymphocytes, and interferon-gamma containing cells. J Pathol. 1991;165:97–103. doi: 10.1002/path.1711650203. [DOI] [PubMed] [Google Scholar]
- 100.Koulmanda M, et al. Curative and beta cell regenerative effects of alpha1-antitrypsin treatment in autoimmune diabetic NOD mice. Proc Natl Acad Sci U S A. 2008;105:16242–16247. doi: 10.1073/pnas.0808031105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Schilling E, et al. Inhibition of nicotinamide phosphoribosyltransferase modifies LPS-induced inflammatory responses of human monocytes. Innate Immun. 2012;18:518–530. doi: 10.1177/1753425911423853. [DOI] [PubMed] [Google Scholar]
- 102.Gale EA, Bingley PJ, Emmett CL, Collier T. European Nicotinamide Diabetes Intervention Trial (ENDIT): a randomised controlled trial of intervention before the onset of type 1 diabetes. Lancet. 2004;363:925–931. doi: 10.1016/S0140-6736(04)15786-3. [DOI] [PubMed] [Google Scholar]
- 103.Larsen CM, et al. A Randomized, Placebo-controlled Trial of Interleukin-1 receptor Antagonist in type 2 Diabetes Mellitus. New England Journal of Medicine. 2007 doi: 10.1056/NEJMoa065213. in press. [DOI] [PubMed] [Google Scholar]
- 104.Sanda S, et al. Short-term IL-1beta blockade reduces monocyte CD11b integrin expression in an IL-8 dependent fashion in patients with type 1 diabetes. Clin Immunol. 2010;136:170–173. doi: 10.1016/j.clim.2010.04.009. [DOI] [PubMed] [Google Scholar]
- 105.Zanin-Zhorov A, et al. Heat shock protein 60 enhances CD4+ CD25+ regulatory T cell function via innate TLR2 signaling. J Clin Invest. 2006;116:2022–2032. doi: 10.1172/JCI28423. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 106.Raz I, et al. Beta-cell function in new-onset type 1 diabetes and immunomodulation with a heat-shock protein peptide (DiaPep277): a randomised, double-blind, phase II trial. Lancet. 2001;358:1749–1753. doi: 10.1016/S0140-6736(01)06801-5. [DOI] [PubMed] [Google Scholar]
- 107.Liu W, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med. 2006;203:1701–1711. doi: 10.1084/jem.20060772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Brusko TM, Putnam AL, Bluestone JA. Human regulatory T cells: role in autoimmune disease and therapeutic opportunities. Immunol Rev. 2008;223:371–390. doi: 10.1111/j.1600-065X.2008.00637.x. [DOI] [PubMed] [Google Scholar]
- 109.Zhou X, et al. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat Immunol. 2009;10:1000–1007. doi: 10.1038/ni.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Salomon B, Bluestone JA. Complexities of CD28/B7: CTLA-4 costimulatory pathways in autoimmunity and transplantation. Annu Rev Immunol. 2001;19:225–252. doi: 10.1146/annurev.immunol.19.1.225. [DOI] [PubMed] [Google Scholar]
- 111.Salomon B, et al. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 2000;12:431–440. doi: 10.1016/s1074-7613(00)80195-8. [DOI] [PubMed] [Google Scholar]
- 112.Ablamunits V, Henegariu O, Preston-Hurlburt P, Herold KC. NKG2A is a marker for acquisition of regulatory function by human CD8+ T cells activated with anti-CD3 antibody. Eur J Immunol. 2011;41:1832–1842. doi: 10.1002/eji.201041258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Green EA, Gorelik L, McGregor CM, Tran EH, Flavell RA. CD4+CD25+ T regulatory cells control anti-islet CD8+ T cells through TGF-beta-TGF-beta receptor interactions in type 1 diabetes. Proc Natl Acad Sci U S A. 2003;100:10878–10883. doi: 10.1073/pnas.1834400100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.You S, et al. Adaptive TGF-beta-dependent regulatory T cells control autoimmune diabetes and are a privileged target of anti-CD3 antibody treatment. Proc Natl Acad Sci U S A. 2007;104:6335–6340. doi: 10.1073/pnas.0701171104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Levings MK, Sangregorio R, Roncarolo MG. Human cd25(+)cd4(+) t regulatory cells suppress naive and memory T cell proliferation and can be expanded in vitro without loss of function. J Exp Med. 2001;193:1295–1302. doi: 10.1084/jem.193.11.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Levings MK, et al. Human CD25(+)CD4(+) T Suppressor Cell Clones Produce Transforming Growth Factor beta, but not Interleukin 10, and Are Distinct from Type 1 T Regulatory Cells. J Exp Med. 2002;196:1335–1346. doi: 10.1084/jem.20021139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Roncarolo MG, Bacchetta R, Bordignon C, Narula S, Levings MK. Type 1 T regulatory cells. Immunol Rev. 2001;182:68–79. doi: 10.1034/j.1600-065x.2001.1820105.x. [DOI] [PubMed] [Google Scholar]
- 118.Heiber JF, Geiger TL. Context and location dependence of adaptive Foxp3(+) regulatory T cell formation during immunopathological conditions. Cell Immunol. 2012;279:60–65. doi: 10.1016/j.cellimm.2012.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bettini M, et al. Cutting edge: accelerated autoimmune diabetes in the absence of LAG- 3. J Immunol. 2011;187:3493–3498. doi: 10.4049/jimmunol.1100714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tivol EA, et al. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3:541–547. doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 121.Tang Q, et al. Distinct roles of CTLA-4 and TGF-beta in CD4+CD25+ regulatory T cell function. Eur J Immunol. 2004;34:2996–3005. doi: 10.1002/eji.200425143. [DOI] [PubMed] [Google Scholar]
- 122.Alyanakian MA, et al. Transforming growth factor-beta and natural killer T-cells are involved in the protective effect of a bacterial extract on type 1 diabetes. Diabetes. 2006;55:179–185. [PubMed] [Google Scholar]
- 123.Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol. 2004;4:762–774. doi: 10.1038/nri1457. [DOI] [PubMed] [Google Scholar]
- 124.Collison LW, et al. IL-35-mediated induction of a potent regulatory T cell population. Nat Immunol. 2010;11:1093–1101. doi: 10.1038/ni.1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Maier LM, et al. IL2RA genetic heterogeneity in multiple sclerosis and type 1 diabetes susceptibility and soluble interleukin-2 receptor production. PLoS Genet. 2009;5:e1000322. doi: 10.1371/journal.pgen.1000322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ueda H, et al. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature. 2003;423:506–511. doi: 10.1038/nature01621. [DOI] [PubMed] [Google Scholar]
- 127.Long SA, et al. Defects in IL-2R signaling contribute to diminished maintenance of FOXP3 expression in CD4(+)CD25(+) regulatory T-cells of type 1 diabetic subjects. Diabetes. 2010;59:407–415. doi: 10.2337/db09-0694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Grinberg-Bleyer Y, et al. IL-2 reverses established type 1 diabetes in NOD mice by a local effect on pancreatic regulatory T cells. J Exp Med. 2010;207:1871–1878. doi: 10.1084/jem.20100209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.McClymont SA, et al. Plasticity of human regulatory T cells in healthy subjects and patients with type 1 diabetes. J Immunol. 2011;186:3918–3926. doi: 10.4049/jimmunol.1003099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Rabinovitch A, Suarez-Pinzon WL, Shapiro AM, Rajotte RV, Power R. Combination therapy with sirolimus and interleukin-2 prevents spontaneous and recurrent autoimmune diabetes in NOD mice. Diabetes. 2002;51:638–645. doi: 10.2337/diabetes.51.3.638. [DOI] [PubMed] [Google Scholar]
- 131.Simon G, et al. Murine antithymocyte globulin therapy alters disease progression in NOD mice by a time-dependent induction of immunoregulation. Diabetes. 2008;57:405–414. doi: 10.2337/db06-1384. [DOI] [PubMed] [Google Scholar]
- 132.Belghith M, et al. TGF-beta-dependent mechanisms mediate restoration of self-tolerance induced by antibodies to CD3 in overt autoimmune diabetes. Nat Med. 2003;9:1202–1208. doi: 10.1038/nm924. [DOI] [PubMed] [Google Scholar]
- 133.Esplugues E, et al. Control of TH17 cells occurs in the small intestine. Nature. 2011;475:514–518. doi: 10.1038/nature10228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Waldron-Lynch F, et al. Teplizumab induces human gut-tropic regulatory cells in humanized mice and patients. Sci Transl Med. 2012;4:118ra112. doi: 10.1126/scitranslmed.3003401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Herold KC, et al. A Single Course of Anti-CD3 Monoclonal Antibody hOKT3{gamma}1(Ala-Ala) Results in Improvement in C-Peptide Responses and Clinical Parameters for at Least 2 Years after Onset of Type 1 Diabetes. Diabetes. 2005;54:1763–1769. doi: 10.2337/diabetes.54.6.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Herold KC, et al. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med. 2002;346:1692–1698. doi: 10.1056/NEJMoa012864. [DOI] [PubMed] [Google Scholar]
- 137.Keymeulen B, et al. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med. 2005;352:2598–2608. doi: 10.1056/NEJMoa043980. [DOI] [PubMed] [Google Scholar]
- 138.Keymeulen B, et al. Four-year metabolic outcome of a randomised controlled CD3-antibody trial in recent-onset type 1 diabetic patients depends on their age and baseline residual beta cell mass. Diabetologia. 2010;53:614–623. doi: 10.1007/s00125-009-1644-9. [DOI] [PubMed] [Google Scholar]
- 139.Bisikirska B, Colgan J, Luban J, Bluestone JA, Herold KC. TCR stimulation with modified anti-CD3 mAb expands CD8 T cell population and induces CD8CD25 Tregs. J Clin Invest. 2005;115:2904–2913. doi: 10.1172/JCI23961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Herold KC, et al. Activation of human T cells by FcR nonbinding anti-CD3 mAb, hOKT3gamma1(Ala-Ala) J Clin Invest. 2003;111:409–418. doi: 10.1172/JCI16090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sherry N, et al. Teplizumab for treatment of type 1 diabetes (Protege study): 1-year results from a randomised, placebo-controlled trial. Lancet. 2011 doi: 10.1016/S0140-6736(11)60931-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Herold KC, et al. Teplizumab treatment may improve C-peptide responses in participants with type 1 diabetes after the new-onset period: a randomised controlled trial. Diabetologia. 2012 doi: 10.1007/s00125-012-2753-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Long SA, et al. Rapamycin/IL-2 Combination Therapy in Patients With Type 1 Diabetes Augments Tregs yet Transiently Impairs beta-Cell Function. Diabetes. 2012;61:2340–2348. doi: 10.2337/db12-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Qin S, et al. “Infectious” transplantation tolerance. Science. 1993;259:974–977. doi: 10.1126/science.8094901. [DOI] [PubMed] [Google Scholar]
- 145.Ablamunits V, et al. Synergistic reversal of type 1 diabetes in NOD mice with anti-CD3 and interleukin-1 blockade: evidence of improved immune regulation. Diabetes. 2012;61:145–154. doi: 10.2337/db11-1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kwon G, Corbett JA, Rodi CP, Sullivan P, McDaniel ML. Interleukin-1 beta-induced nitric oxide synthase expression by rat pancreatic beta-cells: evidence for the involvement of nuclear factor kappa B in the signaling mechanism. Endocrinology. 1995;136:4790–4795. doi: 10.1210/endo.136.11.7588208. [DOI] [PubMed] [Google Scholar]
- 147.Gysemans C, et al. Interferon regulatory factor-1 is a key transcription factor in murine beta cells under immune attack. Diabetologia. 2009;52:2374–2384. doi: 10.1007/s00125-009-1514-5. [DOI] [PubMed] [Google Scholar]
- 148.Akirav E, Kushner JA, Herold KC. Beta-cell mass and type 1 diabetes: going, going, gone? Diabetes. 2008;57:2883–2888. doi: 10.2337/db07-1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sherry NA, et al. Effects of autoimmunity and immune therapy on beta-cell turnover in type 1 diabetes. Diabetes. 2006;55:3238–3245. doi: 10.2337/db05-1034. [DOI] [PubMed] [Google Scholar]
- 150.Akirav EM, et al. Detection of beta cell death in diabetes using differentially methylated circulating DNA. Proc Natl Acad Sci U S A. 2011;108:19018–19023. doi: 10.1073/pnas.1111008108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Talchai C, Xuan S, Lin HV, Sussel L, Accili D. Pancreatic beta Cell Dedifferentiation as a Mechanism of Diabetic beta Cell Failure. Cell. 2012;150:1223–1234. doi: 10.1016/j.cell.2012.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Eizirik DL, Darville MI. beta-cell apoptosis and defense mechanisms: lessons from type 1 diabetes. Diabetes. 2001;50 (Suppl 1):S64–69. doi: 10.2337/diabetes.50.2007.s64. [DOI] [PubMed] [Google Scholar]
- 153.Melton DA. Using stem cells to study and possibly treat type 1 diabetes. Philos Trans R Soc Lond B Biol Sci. 2011;366:2307–2311. doi: 10.1098/rstb.2011.0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Patterson CC, Dahlquist GG, Gyurus E, Green A, Soltesz G. Incidence trends for childhood type 1 diabetes in Europe during 1989–2003 and predicted new cases 2005–20: a multicentre prospective registration study. Lancet. 2009;373:2027–2033. doi: 10.1016/S0140-6736(09)60568-7. [DOI] [PubMed] [Google Scholar]
- 155.Riley WJ, et al. A prospective study of the development of diabetes in relatives of patients with insulin-dependent diabetes. N Engl J Med. 1990;323:1167–1172. doi: 10.1056/NEJM199010253231704. [DOI] [PubMed] [Google Scholar]
- 156.Sabbah E, et al. Diabetes-associated autoantibodies in relation to clinical characteristics and natural course in children with newly diagnosed type 1 diabetes. The Childhood Diabetes In Finland Study Group. J Clin Endocrinol Metab. 1999;84:1534–1539. doi: 10.1210/jcem.84.5.5669. [DOI] [PubMed] [Google Scholar]
- 157.Greenbaum CJ, et al. Fall in C-peptide During First 2 Years From Diagnosis: Evidence of at Least Two Distinct Phases From Composite TrialNet Data. Diabetes. 2012 doi: 10.2337/db11-1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Goodnow CC, Sprent J, Fazekas de St Groth B, Vinuesa CG. Cellular and genetic mechanisms of self tolerance and autoimmunity. Nature. 2005;435:590–597. doi: 10.1038/nature03724. [DOI] [PubMed] [Google Scholar]
- 159.Halverson R, Torres RM, Pelanda R. Receptor editing is the main mechanism of B cell tolerance toward membrane antigens. Nat Immunol. 2004;5:645–650. doi: 10.1038/ni1076. [DOI] [PubMed] [Google Scholar]
- 160.Hertz M, Nemazee D. BCR ligation induces receptor editing in IgM+IgD- bone marrow B cells in vitro. Immunity. 1997;6:429–436. doi: 10.1016/s1074-7613(00)80286-1. [DOI] [PubMed] [Google Scholar]
- 161.Bluestone JA, Abbas AK. Natural versus adaptive regulatory T cells. Nat Rev Immunol. 2003;3:253–257. doi: 10.1038/nri1032. [DOI] [PubMed] [Google Scholar]
- 162.Harjutsalo V, Podar T, Tuomilehto J. Cumulative incidence of type 1 diabetes in 10,168 siblings of Finnish young-onset type 1 diabetic patients. Diabetes. 2005;54:563–569. doi: 10.2337/diabetes.54.2.563. [DOI] [PubMed] [Google Scholar]
- 163.Steck AK, Rewers MJ. Genetics of type 1 diabetes. Clin Chem. 2011;57:176–185. doi: 10.1373/clinchem.2010.148221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Redondo MJ, Jeffrey J, Fain PR, Eisenbarth GS, Orban T. Concordance for islet autoimmunity among monozygotic twins. N Engl J Med. 2008;359:2849–2850. doi: 10.1056/NEJMc0805398. [DOI] [PubMed] [Google Scholar]
- 165.Barrett JC, et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet. 2009;41:703–707. doi: 10.1038/ng.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Fernando MM, et al. Defining the role of the MHC in autoimmunity: a review and pooled analysis. PLoS Genet. 2008;4:e1000024. doi: 10.1371/journal.pgen.1000024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Zhang ZJ, Davidson L, Eisenbarth G, Weiner HL. Suppression of diabetes in nonobese diabetic mice by oral administration of porcine insulin. Proc Natl Acad Sci U S A. 1991;88:10252–10256. doi: 10.1073/pnas.88.22.10252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Tian J, et al. Modulating autoimmune responses to GAD inhibits disease progression and prolongs islet graft survival in diabetes-prone mice. Nat Med. 1996;2:1348–1353. doi: 10.1038/nm1296-1348. [DOI] [PubMed] [Google Scholar]
- 169.Shoda LK, et al. A Comprehensive Review of Interventions in the NOD Mouse and Implications for Translation. Immunity. 2005;23:115–126. doi: 10.1016/j.immuni.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 170.Stoffers DA, Desai BM, DeLeon DD, Simmons RA. Neonatal exendin-4 prevents the development of diabetes in the intrauterine growth retarded rat. Diabetes. 2003;52:734–740. doi: 10.2337/diabetes.52.3.734. [DOI] [PubMed] [Google Scholar]
- 171.Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol. 2004;172:2731–2738. doi: 10.4049/jimmunol.172.5.2731. [DOI] [PubMed] [Google Scholar]
- 172.Steele C, et al. Insulin Secretion in Type 1 Diabetes. Diabetes. 2004;53:426–433. doi: 10.2337/diabetes.53.2.426. [DOI] [PubMed] [Google Scholar]
- 173.Sosenko JM, et al. Patterns of metabolic progression to type 1 diabetes in the Diabetes Prevention Trial-Type 1. Diabetes Care. 2006;29:643–649. doi: 10.2337/diacare.29.03.06.dc05-1006. [DOI] [PubMed] [Google Scholar]
- 174.Mastrandrea L, et al. Etanercept treatment in children with new-onset type 1 diabetes: pilot randomized, placebo-controlled, double-blind study. Diabetes Care. 2009;32:1244–1249. doi: 10.2337/dc09-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]