

Abstract
The absence of Tsa1, a key peroxiredoxin that scavenges H2O2 in Saccharomyces cerevisiae, causes the accumulation of a broad spectrum of mutations. Deletion of TSA1 also causes synthetic lethality in combination with mutations in RAD51 or several key genes involved in DNA double-strand break repair. In the present study, we propose that the accumulation of reactive oxygen species (ROS) is the primary cause of genome instability of tsa1Δ cells. In searching for spontaneous suppressors of synthetic lethality of tsa1Δ rad51Δ double mutants, we identified that the loss of thioredoxin reductase Trr1 rescues their viability. The trr1Δ mutant displayed a CanR mutation rate 5-fold lower than wild-type cells. Additional deletion of TRR1 in tsa1Δ mutant reduced substantially the CanR mutation rate of tsa1Δ strain (33-fold), and to a lesser extent, of rad51Δ strain (4-fold). Loss of Trr1 induced Yap1 nuclear accumulation and over-expression of a set of Yap1-regulated oxido-reductases with antioxidant properties that ultimately re-equilibrate intracellular redox environment, reducing substantially ROS-associated DNA damages. This trr1Δ -induced effect was largely thioredoxin-dependent, probably mediated by oxidized forms of thioredoxins, the primary substrates of Trr1. Thioredoxin Trx1 and Trx2 were constitutively and strongly oxidized in the absence of Trr1. In trx1Δ trx2Δ cells, Yap1 was only moderately activated; consistently, the trx1Δ trx2Δ double deletion failed to efficiently rescue the viability of tsa1Δ rad51Δ. Finally, we showed that modulation of the dNTP pool size also influences the formation of spontaneous mutation in trr1Δ and trx1Δ trx2Δ strains. We present a tentative model that helps to estimate the respective impact of ROS level and dNTP concentration in the generation of spontaneous mutations.
Introduction
Reactive oxygen species (ROS) are formed in all oxygen-consuming organisms. ROS can attack almost all cell components and can induce many types of DNA damage that can cause mutations and genome rearrangements. Yeasts like Saccharomyces cerevisiae, and other aerobic organisms, have acquired a wide array of mechanisms, including pathways that repair the ROS-induced DNA damage, to prevent the deleterious effects of ROS [1]. Because redox communication occurs between the different cellular compartments [2], the cytosol accumulates endogenous oxidizing compounds arising as byproducts of mitochondrial, peroxisomal and endoplasmic reticulum metabolism. However, under normal growth conditions, steady state levels of ROS in the S. cerevisiae cytoplasm are low [3]. Two main redox systems are involved in reducing the level of ROS, the glutathione (GSH) and thioredoxin (Trx) pathways. The GSH pathway involving glutaredoxins is thought to provide a redox buffering function and GSH is the reductant of the glutaredoxins. The thioredoxin redox system comprising the thioredoxins (Trxs) and thioredoxin reductase (Trr) plays a major role in H2O2 metabolism through the peroxiredoxins [4]. The Trxs are the preferred reductants of the ribonucleotide reductase (RNR) [5] and the 3’-phosphoadenylsulfate reductase (PAPS reductase) [6]. When S. cerevisiae cells experience endogenous or exogenous stresses that disturb redox homeostasis, they respond by altering their transcriptional program [7]. Two transcription factors are mainly involved: Yap1 and Skn7, which function in part cooperatively in the peroxide response [8]. Yap1 is the main regulator that controls the expression of S. cerevisiae genes encoding most antioxidants, components of glutathione and carbohydrate metabolism, and components of different metal and drug response pathways [8]–[10].
S. cerevisiae possesses five peroxiredoxins, which have different sub-cellular localizations. Among them, Tsa1 has the most potent ability to scavenge H2O2 [11]. In addition to its role in peroxide reduction, Tsa1 is also known to have chaperone activity [12]. Tsa1 is the only peroxiredoxin that causes an elevated CanR mutation rate when individually removed, indicating that Tsa1 is the most important peroxiredoxin for preventing ROS-induced mutations [13]. DNA lesions resulting from oxygen metabolism can lead to formation of mutations by action of replicative DNA polymerases and translesion DNA polymerases. ROS-induced DNA damage also activates checkpoint pathways which could stimulate dNTP production [14]. This up-regulation of dNTP synthesis facilitates the repair of DNA lesions but is associated with higher mutation rates resulting in part from more efficient translesion DNA synthesis [15]. Tang and collaborators [16] have shown that the accumulation of ROS in tsa1Δ cells accumulate DNA lesions which, by presumably activating the DNA damage checkpoint, stimulate dNTP production. However, it is not understood whether it is the accumulations of ROS and consequently accumulations of mutagenic DNA lesions or the overproduction of dNTPs that is the primary cause of the elevated CanR mutation rate in tsa1 mutants.
Homologous recombination is involved in repair of many types of DNA lesions. We have previously shown that combining a tsa1Δ mutation with a rad51Δ mutation or mutations inactivating other key genes that function in DNA double-strand break (DSB) repair results in cell death [17]. Oxygen metabolism likely takes part in the inviability of tsa1Δ rad51Δ double mutants since anaerobic conditions were found to restore the viability of tsa1Δ rad51Δ double mutants [18]. One explanation for this is that in cells growing in aerobic conditions, the absence of Tsa1 results in a high level of DNA damage that is lethal in the absence of key DNA repair pathways such as DSB repair. To better understand the cause of the elevated mutation rate of the tsa1Δ mutants and the synthetic lethality of tsa1Δ rad51Δ double mutants, we screened for spontaneous mutations that suppress synthetic lethality of tsa1Δ rad51Δ double mutants. We found that mutations in TRR1, which encodes the cytosolic thioredoxin reductase, are able to rescue the growth of tsa1Δ rad51Δ double mutant and suppress the genomic instability phenotype of tsa1Δ mutants. Our results support a model in which the majority of spontaneous mutations that occur in wild-type strains are formed from lesions generated by ROS.
Results
ROS are the primary cause of genome instability in tsa1Δ cells
Tsa1 plays a key role in preventing mutations and genome rearrangements [13]. The mutator phenotype of tsa1Δ cells might be primarily due to their inability to reduce H2O2. The endogenous concentrations of H2O2 and alkyl hydroperoxides in wild-type and tsa1Δ strains have so far not been precisely determined, because available ROS sensors were not selective or sensitive enough [19]. However, several laboratories have detected a significant increased intracellular ROS levels in tsa1Δ strains [16], [20]–[23]. The conversion of this excess of endogenous H2O2 to hydroxyl radical through the Fenton reaction, believed to occur at or near DNA, could induce a variety of types of DNA damage [24]. Yet the increase of intracellular H2O2 in tsa1Δ cells versus wild-type cells was not high enough to change the nuclear and cytoplasmic GSH/GSSG redox state detectable by the genetically encoded redox sensor rxYFP [25] (Figure S1). It is generally accepted that the CanR mutation rate in cells is indicative of the level of DNA damage, which itself follows the variations of ROS concentration. One way to decrease endogenous ROS concentrations is to grow yeast cells under anaerobiosis. The CanR mutation rates of wild-type and tsa1Δ strains, grown in aerobic or anaerobic conditions, were measured by fluctuation analysis [26]. As shown in Table 1, the CanR mutation rates were similar for wild-type cells grown under both conditions. An identical observation was published by Northam et al. [27]. In contrast, the CanR mutation rate of the tsa1Δ strain decreased substantially under anaerobiosis and was the same as the CanR mutation rate of the wild-type strain. This observation supports the view that scavenging intracellular H2O2 is one of the major cellular functions of Tsa1 that protects the nuclear genome from damage by ROS. These results suggest that ROS in tsa1Δ mutant grown under aerobic conditions is a major source of DNA damage underlying the formation of CanR mutations. It may be not so surprising that the anaerobiosis does not reduce the basal level of CanR mutation rate in the wild-type strain. In fact, anaerobiosis was shown to invoke a stress response in yeast cells [28], [29]. In addition, osmotic and DNA replication stress elicit mutagenesis [27], [30], [31]. Although the level of ROS is greatly reduced in anaerobic conditions and consequently the level of DNA damage generated, wild-type cells (as well as tsa1Δ cells) could sense and respond to anoxia. Indeed, hypoxia constitutes a profound cellular stress for mammalian cells that can promote genetic instability [32].
Table 1. CanR mutation rates of wild-type and tsa1Δ strains grown in aerobic and anaerobic conditions.
| Mutation rates (× 10−7) | |||
| Strain | Genotype | Aerobic growth | Anaerobic growth |
| GF4729 | wild-type | 2.13 (1.63–3.17) | 3.03 (2.67–4.27) |
| GF5270 | tsa1Δ | 33.07 (25.16–64.02) | 2.72 (2.43–4.24) |
Mutation rates were determined as described in Material and Methods and were calculated each from 19 parallel cultures, using fluctuation analysis. 95% confidence intervals are indicated.
Yap1 is not activated in tsa1Δ mutant
Yap1 is a redox sensitive transcription factor, considered as a central node in the oxidative stress response network [33]. Yap1 regulates the expression of several hundreds of genes (http://www.yeastgenome.org) among which are genes coding for enzymes with antioxidant properties, numerous oxido-reductases and transcription factors. In non-stressed wild-type cells, Yap1 exists mainly in its reduced form and is distributed equally throughout the cytoplasm and nucleus [34]. In response to oxidative stress, Yap1 rapidly forms two disulfide bonds, resulting in the inhibition of the Yap1 interaction with the nuclear export protein Crm1, and consequently its accumulation within the nucleus [34]. Thus, an excess of H2O2 in tsa1Δ cells may increase endogenous oxidative stress, and Yap1 should be oxidized. Published results have proven the difficulties to distinguish by Western blotting oxidized and reduced forms of Yap1 in W303 derived strains [35], [36]. As an alternative approach, we followed by qRT-PCR analysis the expression levels of several Yap1-target genes [8]–[10] in wild-type cells and in cells harbouring deletions tsa1Δ, yap1Δ or both (Table 2). Yap1 was found not activated in the tsa1Δ strain, as only small variations were observed between wild-type and tsa1Δ strains. These variations are in no way comparable to those produced by exogenous oxidative stress [37].
Table 2. Expression of Yap1-regulated oxido-reductase genes in tsa1Δ cells.
| Strains | Genotype | TSA2 | TRX2 | GRX2 | GLR1 | GSH1 | CCP1 |
| GF4729 | wild-type | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 |
| GF5265 | tsa1Δ | 1.7 | 1.2 | 0.7 | 0.6 | 0.9 | 1.0 |
| GF5384 | yap1Δ | 0.2 | 0.5 | 0.3 | 0.4 | 0.5 | 0.4 |
| GF5381 | tsa1Δ yap1Δ | 1.2 | 0.5 | 0.4 | 0.4 | 0.25 | 0.9 |
The expression of six Yap1-regulated oxido-reductase genes was followed by quantitative RT-PCR. mRNA levels were calculated as ratios relative to that of the wild-type strain GF4729.
What could be the cause of the non- or weak activation of Yap1 in tsa1Δ cells? It has been proposed that the Gpx3 sensor regulates the cellular H2O2 homeostasis in S. cerevisiae and is necessary for Yap1 activation. Gpx3 senses H2O2 and converts this signal into a cysteine-based redox cascade that culminates in oxidation of Yap1 [38], [39]. A third component, Ybp1 seems to be crucial for H2O2-induced Yap1 activation. Ybp1 forms an H2O2-induced complex with Yap1 and the transient Yap1-Gpx3 intermediate cannot be formed in the absence of Ybp1 [40], [41], although this mechanism is not fully understood. Strains harboring the W303 genetic background contain a mutated YBP1 gene. In this context, Yap1 activation appears to be Gpx3-independent but requires Tsa1 [35]. The yeast strains used in this study are derived in part from W303 strain. We sequenced YBP1 gene of GF4729 strain and identified all the mutations previously described in allele ybp1–2 [35] (data not shown). Thus the absence of a functional Ybp1 protein might be responsible for the absence of Yap1 activation in the tsa1Δ strain. To test this hypothesis, we replaced the ybp1–2 allele by the YBP1 gene isolated from an S288c strain that is free of any mutation. We monitored by qRT-PCR analysis the expression level of six Yap1-target genes in GF4729 (wild-type, ybp1–2), GF5652 (wild-type, YBP1), GF5270 (tsa1Δ ybp1–2) and GF5606 (tsa1Δ, YBP1) strains. As shown in Figure 1, the expressions of these target genes were very similar between GF4729 (wild-type, ybp1–2) and GF5270 (tsa1Δ ybp1–2) strains. The expression of target genes changed within 2-fold between GF4729 (wild-type, ybp1–2) and GF5652 (wild-type, YBP1) strains, and between GF5270 (tsa1Δ ybp1–2) and GF5606 (tsa1Δ, YBP1) strains. Yet, these small variations did not affect significantly the CanR mutations rates (Table 3). However, as shown by Okazaki and coworkers [35], it remains possible that a strain like GF5606 (tsa1Δ YBP1) can use preferentially the Ybp1/Gpx3 pathway to activate Yap1 in response to a strong exogenous oxidative stress.
Figure 1. Quantitative RT-PCR analysis for indicated genes in GF4729 (wild-type ybp1–2), GF5652 (wild-type YBP1), GF5270 (tsa1 .

Δ ybp1–2 ) and GF5606 ( tsa1 YBP1 ). Quantitative RT-PCR analysis was performed as described in Material and Methods. mRNA levels of each gene were calculated as ratios relative to that of the wild-type ybp1–2 strain (set as 1). The reported values are the mean of three independent experiments and the error bars represent the standard error of the mean.
Table 3. CanR mutation rates of different mutant strains.
| Strain | Genotype | Mutation rate × 10−7 | 95% confidence interval |
| GF4729 | wild-type | 4.20 | 3.98–4.82 |
| GF5652 | wild-type YBP1 | 3.76 | 3.44–4.40 |
| GF5270 | tsa1Δ | 70.7 | 61.60–101.50 |
| GF5606 | tsa1ΔYBP1 | 65.57 | 52.76–77.23 |
| GF5968 | tsa2Δ | 3.60 | 3.03–4.41 |
| GF5965 | tsa1Δ tsa2Δ | 70.61 | 57.13–96.93 |
| GF5505 | trr1Δ | 0.84 | 0.55–1.05 |
| GF5499 | trr1Δ tsa1Δ | 2.15 | 1.88–2.78 |
| GF6054 | trr1Δ tsa2Δ | 1.05 | 0.62–1.45 |
| GF5967 | trr1Δ tsa1Δ tsa2Δ | 3.28 | 2.71–3.58 |
| GF5668 | trx1Δ trx2Δ | 2.11 | 1.74–2.42 |
| GF5898 | trr1Δ trx1Δ trx2Δ | 2.71 | 2.26–2.98 |
| GF5674 | trx1Δ trx2Δ tsa1Δ | 5.75 | 5.21–7.05 |
| GF6067 | trx1Δ trx2Δ tsa1Δ tsa2Δ | 5.87 | 4.94–7.25 |
| GF5675 | rad51Δ | 38.26 | 30.50–46.42 |
| GF5506 | trr1Δ rad51Δ | 9.41 | 7.30–12.35 |
| GF6020 | trx1Δ trx2Δ rad51Δ | 80.54 | 65.35–97.57 |
| GF5494 | trr1Δ tsa1Δ rad51Δ | 25.53 | 20.89–35.09 |
| GF5959 | trr1Δ tsa1Δ tsa2Δ rad51Δ | 73.43 | 67.41–77.85 |
Each mutation rate was calculated from 19 to 57 independent cultures.
Thioredoxins are thought to be the physiological reducing agents and are responsible for the negative regulation of the Yap1 transcriptional activity [42]–[44]. In non-stressed wild-type cells, Trxs are kept almost exclusively in their reduced forms [42], [45], [46]. An increase of endogenous H2O2 concentration leads to the oxidation of Tsa1 that will be reduced in turn at the expense of reduced Trxs [4]. We estimated the redox state of Trxs in tsa1Δ cells using rabbit anti-Trx1 and Trx2 antibodies and found that both Trxs were in a reduced state in tsa1Δ cells as in wild type cells (Figure 2A, lanes 1, 2; Figure 2B, lanes 8, 9). Taken together, we propose that an increased endogenous concentration of H2O2 in tsa1Δ cells could result from the combined effect of the absence of Tsa1 itself and the inhibition of Yap1 activation by the reduced Trxs.
Figure 2. Oxidation state of Trx1 and Trx2 in different strains defective in maintaining redox homeostasis.

Wild-type and mutated cells were processed for western blotting as described in Material and Methods. Proteins were detected with rabbit polyclonal anti-Trx2 (A, C) and anti-Trx1 (B) antibodies. Extracts from wild-type cells, treated with 0.5 mM H2O2 for 5 min in YPG, was loaded as a control (B, lane 7).
Identification of a suppressor of the lethality of tsa1Δ rad51Δ double mutants
We previously observed that tsa1Δ rad51Δ double mutants were inviable under aerobic conditions but viable under anaerobic conditions [17], [18]. To screen for spontaneous mutations that rescue the viability of tsa1Δ rad51Δ double mutants under aerobic conditions, tsa1Δ rad51Δ cells were grown under anaerobic conditions, harvested and about 4×106 cells were plated under aerobic conditions. Incubation under aerobic conditions resulted in massive cell death and the appearance of a small number of viable colonies. One viable colony from each of 20 independent experiments was selected and further characterized. These 20 strains each containing a potential suppressor mutation were then crossed with a tsa1Δ strain. Analysis of the meiotic segregants showed that the putative suppressor mutations (named sup1 to sup20) were all single mutations, recessive and mapped at loci that were not linked to the starting rad51Δ mutation.
We used a cloning scheme employing the red/white sectored colony method described by Zhao and co-workers [47] to identify the suppressor genes. Briefly, ADE2 and ADE3 were disrupted in the tsa1Δ rad51Δ sup1 strain and the plasmid p1591 (CEN, URA3, TSA1, ADE3) was introduced, yielding the strain GF5377. The presence of this plasmid, which is not needed for cell viability, confers a red color to the colonies on selective medium (SC-uracil). Cells growing in the absence of selection for this plasmid can spontaneously lose it and form red/white sectored or white colonies. As the sup1 mutation was recessive, we expected that the introduction of a plasmid harboring the SUP1 wild-type gene into strain GF5377 would result in red colonies since the resident plasmid p1591 (CEN, URA3, TSA1, ADE3) would now be required for growth. After transformation of strain GF5377 with a CEN-LEU2 plasmid-based genomic library, candidate SUP1 transformants were identified as solid red colonies on plates lacking leucine. After retesting 137 red colonies and then screening the resulting 21 strains for lack of growth on media containing 5-FOA, 7 plasmid-containing strains were obtained. Plasmids were rescued from these strains and sequenced which revealed that they each contained an ∼10 kb-long insert with an overlapping region of chromosome IV containing two full-length ORFs, YDR352w and TRR1. Subcloning of these two ORFs and further analysis showed that only TRR1 could complement the sup1 mutation in the GF5377 strain. We then determined the DNA sequence of the TRR1 gene from each of the 20 independently isolated suppressor strains as well as that of the wild-type strain GF4729 revealing that each of the suppressor strains contained a mutation in TRR1 (Table S4). To further confirm that inactivation of TRR1 suppresses the inviability of the tsa1Δ rad51Δ double mutant, we deleted TRR1 in a tsa1Δ/TSA1 rad51Δ/RAD51 diploid strain and showed by tetrad analysis that the TRR1 deletion restored the viability of double-mutant tsa1Δ rad51Δ in aerobic conditions (Table 4).
Table 4. trr1Δ deletion suppresses the lethality of tsa1Δ rad51Δ.
| Segregant genotype | Number observed | Colony size |
| tsa1Δ rad51Δ trr1Δ | 17 | m |
| tsa1Δ rad51ΔTRR1 | 0 | |
| tsa1Δ RAD51 trr1Δ | 22 | m |
| tsa1ΔRAD51 TRR1 | 18 | G |
| TSA1 rad51Δ trr1Δ | 11 | p |
| TSA1 rad51Δ TRR1 | 23 | G |
| TSA1 RAD51 trr1Δ | 15 | p |
| TSA1 RAD51 TRR1 | 17 | G |
Tetrad analysis of meiotic products of diploid strain tsa1Δ/TSA1 rad51Δ/RAD51 trr1Δ/TRR1. These three loci are unlinked. 40 tetrads were dissected. 20 segregants of each type were expected. Colony size: p <m <G.
TRR1 encodes the cytosolic thioredoxin reductase, whose known major function is to reduce the oxidized forms of Trx1 and Trx2. Thioredoxin reductase and thioredoxins act as a disulfide reductase system and protect cells against both oxidative and reductive stress [4], [48]. Our results therefore suggest that deregulation of the thioredoxin redox system resulting from the trr1 mutation may promote the mechanism suppressing lethality of tsa1Δ rad51Δ double mutants.
The oxidized thioredoxins in trr1Δ mutants contribute to Yap1 activation
It was previously reported that Yap1 is constitutively and partially oxidized and that the basal expression of Yap1 targets is elevated in trr1Δ mutants in absence of external stress [42], [49]. Besides, Trr1 inactivation enhances Msn2 response to H2O2 [50]. In fact, the transcriptional response to oxidative stress is known to depend on several transcription factors, including Yap1, Skn7, Msn2 and Msn4. Yap1 and Skn7 control independent but overlapping responses [8]; Msn2 and Msn4 mediate a transcriptional response which is common to many stress including oxidative stress [51]. We investigated whether a specific set of genes is transcriptionally regulated by Yap1/Skn7 and/or Msn2/Msn4 in trr1Δ mutants. The expressions of 13 genes encoding oxido-reductases were monitored by quantitative RT-PCR (Table 5). These genes include TSA2 and AHP1 whose expression is regulated by Yap1/Skn7 and Msn2/Msn4; GLR1, GSH1, CCP1, YKL071w, TSA1, TRR1 and TRX2 regulated by Yap1 or Yap1/Skn7; CTT1, GRX1 and GRX2 regulated by Msn2/Msn4, and finally TRX1 which is not regulated by any of these transcription factors (http://www.yeastgenome.org). YKL071w is a gene of unknown function encoding a member of the dehydrogenase/reductase family [52]. Remarkably, the genes containing one or more Yap1 response elements (YRE) in their promoters were highly induced in the trr1Δ strain compared to the wild-type strain, especially TSA2 (>600-fold), TRX2 (30-fold), CCP1 (30-fold) and YKL071w (>70-fold). As expected, TRX1 was not up-regulated in trr1Δ strains. Interestingly, CTT1 which is a typical target of Msn2/Msn4 transcription factor [50] was not induced in trr1Δ strains. Taken together, these results suggest that the over-expression of the oxido-reductases tested is mainly due to the activation of Yap1. Consistently, we observed that Yap1 accumulated in the nucleus of trr1Δ cells (Figure 3).
Table 5. Expression of Yap1-regulated or -independent oxido-reductases in different strains.
| Strain | Genotype | Set | TSA2 | AHP1 | CTT1 | GRX1 | GRX2 | GLR1 | GSH1 | CCP1 | YKL071w | TSA1 | TRR1 | TRX1 | TRX2 |
| GF4729 | wild-type | A | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 |
| GF5630 | trx1 | A | 1.0 | 1.7 | 1.4 | 0.9 | 1.0 | 1.1 | 1.3 | 1.2 | 0.7 | 1.5 | 1.3 | NA | 1.3 |
| GF5643 | trx2 | A | 1.2 | 1.6 | 1.4 | 1.1 | 0.9 | 1.1 | 1.2 | 1.2 | 1.0 | 1.3 | 1.3 | 1.2 | NA |
| GF5668 | trx1 trx2 | B | 10.4 | 3.9 | 1.2 | 3.9 | 4.2 | 3.3 | 3.9 | 8.0 | 8.8 | 3.0 | 4.5 | NA | NA |
| GF5719 | trx1 trx2 tsa1 | B | 10.3 | 3.6 | 1.5 | 3.2 | 2.5 | 1.5 | 2.1 | 4.2 | 7.2 | NA | 3.2 | NA | NA |
| GF5898 | trr1 trx1 trx2 | B | 15.8 | 5.6 | 0.6 | 3.3 | 2.6 | 2.2 | 3.4 | 11.7 | 11.2 | 4.2 | NA | NA | NA |
| GF5911 | trr1 trx1 trx2 tsa1 | B | 10.9 | 4.7 | 0.5 | 3.3 | 2.5 | 1.7 | 2.8 | 5.8 | 10.9 | NA | NA | NA | NA |
| GF5505 | trr1 | C | 657.4 | 6.1 | 1.4 | 2.5 | 7.3 | 2.5 | 4.2 | 29.1 | 71.7 | 7.0 | NA | 1.0 | 30.8 |
| GF5888 | trr1 trx1 | C | 1017.0 | 11.0 | 0.9 | 3.3 | 8.7 | 2.9 | 5.1 | 45.3 | 113.1 | 12.6 | NA | NA | 105.9 |
| GF5899 | trr1 trx2 | C | 1916.8 | 21.1 | 1.9 | 3.1 | 18.0 | 10.5 | 10.0 | 130.2 | 406.3 | 23.8 | NA | 3.2 | NA |
| GF5909 | trr1 trx1 tsa1 | C | 886.0 | 8.0 | 0.6 | 2.5 | 6.5 | 2.2 | 3.2 | 31.0 | 124.2 | NA | NA | NA | 39.0 |
| GF5501 | trr1 tsa1 | C | 370.0 | 4.2 | 1.3 | 1.9 | 4.4 | 1.7 | 2.4 | 12.2 | 38.0 | NA | NA | 0.9 | 20.1 |
The expression of 13 oxido-reductase genes in 12 strains was measured by quantitative RT-PCR. mRNA levels were calculated as ratios relative to that of the wild-type strain GF4729. Yeast strains were ordered according to their membership to the three sets (A, B and C) highlighted by correspondence analysis (Figure 4). Genes CTT1 and TRX1 did not appear to be regulated by Yap1. NA, not applicable.
Figure 3. Localization of Yap1-GFP in TRR1 and trr1Δ cells.

Living cells in exponential phase expressing the Yap1-GFP fusion protein under the control of native Yap1 promoter and stained by DAPI were analyzed by fluorescence microscopy. Overlay image of DAPI and GFP signal (merge) are also shown.
To address the question of how trr1 mutations trigger the activation of Yap1, we investigated the involvement of the thioredoxins Trx1 and Trx2 in this process. Thioredoxins are the primary substrates of Trr1 and are thought to be the physiological reducing agents responsible for negatively regulating Yap1 activity [42]–[44]. In unstressed wild-type cells, Trxs are found almost exclusively in their reduced form [42], [45], [46]. We therefore used quantitative RT-PCR to monitor the expression level of the above defined 13 genes coding for oxido-reductases in a set of strains containing different combinations of trr1Δ, trx1Δ and trx2Δ deletion mutations (Table 5, Figure 4). Globally, the tested genes were not over-expressed in strains harboring either trx1Δ or trx2Δ individual deletion, they were moderately over-expressed in strains harboring the trx1Δ trx2Δ double deletion with or without trr1Δ, and they were highly over-expressed in strains harboring the trr1Δ deletion with either trx1Δ or trx2Δ. The double deletion trr1Δ trx1Δ or trr1Δ trx2Δ induced higher expression than trr1Δ with the highest effect in trr1Δ trx2Δ (Table 5, Figure 4). Different extents of Yap1 activation were most visible when comparing expression levels of genes TSA2, AHP1, CCP1 and YKL071w among these mutants. The fact that the tested genes were significantly more activated in a trr1Δ, trr1Δ trx1Δ, or trr1Δ trx2Δ strain than in trr1Δ trx1Δ trx2Δ strain suggest that the presence of Trx1 or Trx2 largely contributes to the strong over-expression of the tested genes in trr1Δ cells. On the other hand, the moderate Yap1 activation in trx1Δ trx2Δ suggests the existence of some Trx1/Trx2-independent mechanisms.
Figure 4. Correspondence analysis of qRT-PCR data.

The correspondence analysis was performed using the program StatEL (Ad Science) and the quantitative RT-PCR data for genes TSA2, AHP1, CTT1, GRX1, GRX2, GLR1, GSH1, CCP1 and YKL071w in 12 strains (Table 5). The percentage of inertia on the different axes is as follows: axis 1: 84.8%, axis 2: 9.7%, axis 3: 3.6%, axis 4: 0.8%, axis 5: 0.8%. Only the positions of the 12 yeast strains analysed are presented. They can be grouped in three distinct sets named A, B and C.
We then analyzed the oxidation state of Trx1 and Trx2 in the wild-type, trx1Δ, trx2Δ, trr1Δ, trr1Δ trx1Δ and trr1Δ trx2Δ contexts using anti-Trx1 and anti-Trx2 antibodies. The anti-Trx1 antibody (kind gift from Dr. Grant) was prepared against Trx1 while slightly cross-reacting with Trx2 [45], [53]. The anti-Trx2 antibody was prepared against Trx2 (see Material and Methods). Western blots using anti-Trx1 antibody revealed that Trx1 was>90% reduced in wild-type and trx2Δ strains whereas no visible signal was observed with cell extracts from trx1Δ strains (Figure 2B, lanes 1, 4 and 3) (NB: The expression levels of TRX2 and TRX1 in trx1Δ and trx2Δ strains respectively, were similar to that in wild-type (Table 5)). The same anti-Trx1 antibody revealed that Trx1 is>90% oxidized in trr1Δ trx2Δ cell extracts (Figure 2B, lane 6) whereas oxidized (70–75%) and reduced (25–30%) forms were detected in trr1Δ and trr1Δ trx1Δ extracts (Figure 2B, lanes 2 and 5), which indicates that anti-Trx1 antibody cross-reacts with Trx2 when Trx2 is over-expressed (Table 5). Using anti-Trx2 antibody, western blots of trr1Δ trx2Δ cell extracts yielded no signal, (Figure 2A, lane 4) whereas in trr1Δ and trr1Δ trx1Δ cell extracts, Trx2 was at least 10-times more expressed than in the wild type strain and 40–50% Trx2 was in its oxidized form (Figure 2A, lanes 3 and 5). The fact that Trx2 is not completely oxidized in trr1Δ cells implies existence of an alternative electron donor. Indeed, recent data suggest that GSH, directly or indirectly, is able to reduce oxidized Trxs [54], [55].
These results raise the question of whether the over-expression of the Yap1-targeted genes in trr1Δ cells is due to a high concentration of oxidized Trxs or conversely the decreased level of reduced Trxs, since the latter form may impede Yap1 activation. If thioredoxins regulate Yap1 activity by the inhibitory effect of their reduced forms, then Yap1 activity in trx1Δ trx2Δ mutant should be at least similar, if not higher, than in trr1Δ mutants, which was not the case (Table 5). Hence we conclude that, very probably, the oxidized thioredoxins intervene in Yap1 activation in trr1Δ trr1Δ trx1Δ or trr1Δ trx2Δ cells, directly or indirectly. Consistently, Yap1 activation is the strongest in GF5899 strain (trr1Δ trx2Δ) (Table 5) in which Trx1 is more than 90% oxidized (Figure 2). In trr1Δ tsa1Δ cells, Yap1 is about 40% less activated than in trr1Δ cells (Table 5) suggesting that Tsa1 might play a role in Yap1 oxidation [36].
The absence of Trr1 suppresses mutagenesis in tsa1Δ and rad51Δ strains
Loss of Trr1 can fully rescue the viability of tsa1Δ rad51Δ double mutants and is associated with over-expression of Yap1-controlled oxido-reductases. We reasoned that over-expression of oxido-reductases may reduce significantly intracellular ROS level and therefore suppress genomic instability of tsa1Δ and/or rad51Δ strains. As previously reported, TSA1 deletion resulted in significant increase in CanR mutation rate, 70.70×10−7, compared to the wild-type strain that was 4.20×10−7 (Table 3). CanR mutation rate for a rad51Δ was 38.26×10−7. The synthetic lethality between tsa1Δ and rad51Δ may mean that cells cannot cope with toxic levels of DNA damage in a tsa1Δ rad51Δ haploid strain grown in aerobic conditions. Furthermore, the CanR rate in a trr1Δ tsa1Δ strain was 2.15×10−7 that is a 33-fold reduction compared to a tsa1Δ single mutant and is lower than the CanR mutation rate of the wild-type strain. Similarly but to a lesser extent, CanR mutation rate for a rad51Δ trr1Δ mutant was 9.41×10−7 that is a 4-fold reduction compared to rad51Δ single mutant. These different effects of trr1Δ on tsa1Δ and rad51Δ may suggest that tsa1Δ -induced increased mutagenesis results from increased ROS accumulation and can be efficiently attenuated by over-expression of oxido-reductases while rad51Δ -induced mutagenesis may be only partially linked to ROS accumulation. The CanR mutation rate in tsa1Δ rad51Δ trr1Δ strain was 25.53×10−7, corresponding to a multiplicative effect of mutations of tsa1Δ trr1Δ and rad51Δ trr1Δ (2.15×9.41). Taken together, our results suggest that trr1Δ rescues the viability of tsa1Δ rad51Δ double mutants through suppression of tsa1Δ and rad51Δ -induced mutagenesis that is achieved by redox regulated over-expression of oxido-reductases.
Interestingly, the CanR mutation rate in a trr1Δ strain was 0.84×10−7, 5-fold lower than in a wild-type strain. Due to the over-expression of oxido-reductases, the ROS level in trr1Δ cells should be very low. We can estimate the respective contribution of Tsa1 and Tsa2 in ROS reduction in the trr1Δ context by comparing the CanR mutations rates. The mutation rates of trr1Δ tsa1Δ, trr1Δ tsa2Δ and trr1Δ tsa1Δ tsa2Δ strains were 2.15×10−7, 1.05×10−7 and 3.28×10−7, respectively. These data suggest that the over-expression of Tsa1 and Tsa2 in trr1Δ cells plays a role in reducing ROS level, but others oxido-reductases are also involved.
We analyzed the meiotic products of diploid strain trr1Δ/TRR1 tsa1Δ/TSA1 rad51Δ/RAD51 tsa2Δ/TSA2. Tsa2 was not essential for the viability of a mutant carrying the triple deletion trr1Δ tsa1Δ rad51Δ Table 6. Nevertheless, Tsa1 and Tsa2 contributed to suppress the CanR mutation formation of trr1Δ rad51Δ (9.41×10−7) since CanR mutation rate was 25.53×10−7 and 74.45×10−7 in trr1Δ tsa1Δ rad51Δ and trr1Δ tsa1Δ tsa2Δ rad51Δ strains respectively. Viability of trr1Δ tsa1Δ tsa2Δ rad51Δ could be attributed to the over-expression of other oxido-reductases. To further clarify the relationship between Tsa1/Tsa2 and thioredoxins, we analyzed the oxidation state of Trx2 in trr1Δ, trr1Δ tsa1Δ, trr1Δ tsa2Δ and trr1Δ tsa1Δ tsa2Δ strains using anti-Trx2 antibody. Constant ratios of the reduced and oxidized forms of Trx2 were found in trr1Δ, trr1Δ tsa1Δ and trr1Δ tsa2Δ cells (Figure 2C, lanes 2, 4, 5), whereas the ratio was slightly increased in the trr1Δ tsa1Δ tsa2Δ strain (Figure 2C, lane 6), suggesting that the reduced form of Trx2 is not consumed more by Tsa1 or Tsa2 in trr1Δ strains than in wild-type strains.
Table 6. Effect of tsa2Δ deletion on the lethality of tsa1Δ rad51Δ trr1Δ.
| Segregant genotype | Number observed |
| tsa1Δ tsa2Δ rad51Δ trr1Δ | 47 |
| tsa1Δ tsa2Δ rad51Δ TRR1 | 1 |
| tsa1Δ tsa2Δ RAD51 trr1Δ | 54 |
| tsa1Δ tsa2Δ RAD51 TRR1 | 53 |
| tsa1Δ TSA2 rad51Δ trr1Δ | 42 |
| tsa1Δ TSA2 rad51Δ TRR1 | 0 |
| tsa1Δ TSA2 RAD51 trr1Δ | 44 |
| tsa1Δ TSA2 RAD51 TRR1 | 48 |
| TSA1 tsa2Δ rad51Δ trr1Δ | 37 |
| TSA1 tsa2Δ rad51Δ TRR1 | 48 |
| TSA1 tsa2Δ RAD51 trr1Δ | 34 |
| TSA1 tsa2Δ RAD51 TRR1 | 46 |
| TSA1 RAD51 rad51Δ trr1Δ | 34 |
| TSA1 RAD51 rad51Δ TRR1 | 57 |
| TSA1 RAD51 RAD51 trr1Δ | 43 |
| TSA1 RAD51 RAD51 TRR1 | 55 |
| expected | 55 |
Tetrad analysis of meiotic products of diploid strain tsa1Δ/TSA1 tsa2Δ/TSA2 rad51Δ/RAD51 trr1Δ/TRR1. These four loci are unlinked. 220 tetrads were dissected. 55 segregants of each genotype were expected.
As trx1Δ trx2Δ double deletion exhibits only a moderate effect in inducing overexpression of oxido-reductases, we determined whether this double deletion has a significant effect on the mutation formation in tsa1Δ and rad51Δ strains and is capable of rescuing viability of tsa1Δ rad51Δ mutants. The CanR mutation rate of a trx1Δ trx2Δ strain was 2.11×10−7 (Table 3), that is 2-fold less than in a wild-type strain. The CanR rate in a trx1Δ trx2Δ tsa1Δ strain was 5.75×10−7 that is a 12-fold reduction compared to a tsa1Δ strain but is higher than that in trr1Δ tsa1Δ strain (2.15×10−7). Surprisingly, the CanR mutation rate of the rad51Δ trx1Δ trx2Δ strain, 80.54×10−7, is 2-fold higher compared to rad51Δ strain. Therefore, the inactivation of TRR1 attenuates deleterious mutagenic consequences of both tsa1Δ and rad51Δ deletions, while the trx1Δ trx2Δ double deletion only reduces the mutagenic effect linked to tsa1Δ but increases DNA damage in rad51Δ. Consequently, the tsa1Δ rad51Δ trx1Δ trx2Δ cells may not be able to cope with the high level of DNA damage still formed. Indeed, we determined whether trx1Δ trx2Δ might rescue the synthetic lethality of a strain bearing tsa1Δ rad51Δ We analyzed the meiotic products of diploid strain tsa1Δ/TSA1 rad51Δ/RAD51 trx1Δ/TRX1 trx2Δ/TRX2 and obtained only 10 tsa1Δ rad51Δ trx1Δ trx2Δ segregants instead of the 35 expected (Table 7). The severe slow growth of these segregants prevented us to assess their CanR mutation rates. Therefore, the different effects resulting from TRR1 deletion or TRX1 and TRX2 double deletion seem related to the expression levels of oxido-reductases in these different strains.
Table 7. Effect of trx1Δ trx2Δ double deletion on the lethality of tsa1Δ rad51Δ.
| Segregant genotype | Number observed | Colony size |
| tsa1Δ rad51Δ trx1Δ trx2Δ | 10 | pp |
| tsa1Δ rad51Δ trx1Δ TRX2 | 1 | |
| tsa1Δ rad51Δ TRX1 trx2Δ | 0 | |
| tsa1Δ rad51Δ TRX1 TRX2 | 0 | |
| tsa1Δ RAD51 trx1Δ trx2Δ | 28 | G |
| tsa1Δ RAD51 trx1Δ TRX2 | 29 | G |
| tsa1Δ RAD51 TRX1 trx2Δ | 39 | G |
| tsa1Δ RAD51 TRX1 TRX2 | 38 | G |
| TSA1 rad51Δ trx1Δ trx2Δ | 28 | p |
| TSA1 rad51Δ trx1Δ TRX2 | 32 | m |
| TSA1 rad51Δ TRX1 trx2Δ | 34 | G– |
| TSA1 rad51Δ TRX1 TRX2 | 38 | m |
| TSA1 RAD51 trx1Δ trx2Δ | 45 | m |
| TSA1 RAD51 trx1Δ TRX2 | 28 | m+ |
| TSA1 RAD51 TRX1 trx2Δ | 31 | G |
| TSA1 RAD51 TRX1 TRX2 | 31 | G |
Tetrad analysis of meiotic products of diploid strain tsa1Δ/TSA1 rad51Δ/RAD51 trx1Δ/TRX1 trx2Δ/TRX2. These four loci are unlinked. 140 tetrads were dissected. 35 segregants of each genotype were expected. Colony size: pp <p <m <m+ <G– <G.
Impact of dNTP levels on the CanR mutation rates in trr1Δ and trx1Δ trx2Δ cells
Thioredoxins are physiologically relevant electron donors for ribonucleotide reductase (Rnr) during DNA precursor synthesis. Yeast cells lacking both cytoplasmic thioredoxins (trx1Δ trx2Δ) accumulate oxidized Rnr1, have a low dNTPs pool and a 3-fold longer S phase than wild-type cells [5]. Trx1 and Trx2 was constitutively strongly oxidized in the absence of Trr1. Furthermore, Trx2 expression was at least 10-times higher in the trr1Δ mutants than in wild-type cells and about 40–50% of Trxs was in oxidized form (Figure 2). These changes may directly or indirectly interfere with the activity of ribonucleotide reductase and affect dNTP synthesis. To estimate the impact of dNTP concentration on the CanR mutation rates in the wild-type, trr1Δ and trx1Δ trx2Δ contexts, we increased ribonucleotide reductase activity by deleting SML1 gene whose product is an ribonucleotide reductase inhibitor [56], and by placing RNR1 gene under the control of the strong TEF1 promoter. As shown in Figure 5 and Table S5, the dNTP levels of trr1Δ and trx1Δ trx2Δ were approximately 2-fold lower than that of wild-type cells grown asynchronously in YPD medium. SML1 deletion and RNR1 overexpression increased dNTP level from 1 to 5.22 in the wild-type cells (basal dNTP level in the wild type was set as 1), from 0.50 to 5.52 in the trr1Δ and from 0.52 to 3.93 in the trx1Δ trx2Δ context. In the trr1Δ and trx1Δ trx2Δ contexts, large increases of dNTP concentration produced 2.8-fold and 5.5-fold increases in the CanR mutation rate, respectively (Table 8). In contrast, the increase of dNTP concentration had almost no effect in wild-type cells. In E. coli, lower dNTP levels correlate with a decreased spontaneous point mutation rate while higher dNTP levels correlate with an increased rate [57], [58]. In S. cerevisiae, such correlations were also observed in mutants affecting DNA damage checkpoints, RNR, replication factors or DNA repair components [14], [59], [60]. Taken together, dNTP fluctuations have more pronounced impact on the mutation formation in mutant cells than in wild-type cells.
Figure 5. dNTP concentrations in yeast strains.

dNTP levels were measured in wild-type, trr1Δ trx1Δ trx2Δ, sml1Δ RNR1TEF1, trr1Δ sml1Δ RNR1TEF1 and trx1Δ trx2Δ sml1Δ RNR1TEF1 strains. Values are the average of two experiments and error bars represent the standard error of the mean.
Table 8. Impact of dNTP levels on the CanR mutation rates.
| Strain | Genotype | Yap1 activation | dNTP concentration* | CanR rate (× 10−7) |
| GF4729 | wild-type | no | 1 | 4.20 (3.98–4.82) |
| GF5270 | tsa1Δ | no | 2.5** | 70.70 (61.60–101.50) |
| GF5505 | trr1Δ | strong | 0.50 | 0.84 (0.55–1.05) |
| GF5499 | trr1Δ tsa1Δ | strong | ND | 2.15 (1.88–2.78) |
| GF5668 | trx1Δ trx2Δ | moderate | 0.52 | 2.11 (1.74–2.42) |
| GF5674 | trx1Δ trx2Δ tsa1Δ | moderate | ND | 5.75 (5.21–7.05) |
| GF6080 | sml1Δ RNR1TEF1 | ND | 5.22 | 5.04 (4.34–6.13) |
| GF6084 | trr1Δ sml1Δ RNR1TEF1 | ND | 5.52 | 2.35 (2.08–2.54) |
| GF6129 | trx1Δ trx2Δ sml1Δ RNR1TEF1 | ND | 3.93 | 11.53 (9.55–12.25) |
*dNTP concentration in mutant strains relative to wild-type cells (set to be 1).
** Davidson et al. 2012; ND, not determined.
Low levels of dNTP in trr1Δ and trx1Δtrx2Δ mutants compared to wild-type strains suggest that the decrease of CanR mutation rate in these mutants may be due to the combined effects of Yap1 activation and dNTP pool reduction. To estimate their relative contributions, we propose a tentative model which links the quantity of endogenous DNA lesion and the cellular dNTP concentration to the production of CanR mutations (Text S1). This model predicts that ROS level in trr1Δ is about 21 times lower than that in trx1Δ trx2Δ cells. These estimations are consistent with the observation above that the oxido-reductasesinvolved in the reduction of ROS are more actively synthesized in trr1Δ than in trx1Δ trx2Δ (Table 5, Table S6). On the other hand, the model predicts that the impact of dNTP concentration on the CanR mutation rate is dependent on the level of lesions produced by ROS and processed by translesion polymerases. As more lesions are present in trx1Δ trx2Δ cells than in trr1Δ cells, changes in dNTP concentration will have more impact in the generation of CanR mutations in trx1Δ trx2Δ than in trr1Δ cells. We note that these proposals seem to contradict the fact that trr1Δ cells are more sensitive to hydrogen peroxide than wild-type cells [45], [49]. We compared the growth of wild-type, trr1Δ and trr1Δ trx1Δ trx2Δ strains on SC plates containing 0.10 and 0.25 mM H2O2 (Figure 6). In these H2O2 concentration ranges, trr1Δ trx1Δ trx2Δ strain was almost as resistant as the wild-type strain, pointing to the possibility that the sensitivity of the trr1Δ cells is primarily due to the accumulation of toxic oxidized thioredoxins which disturb the cellular redox homeostasis, as previously suggested [4].
Figure 6. Sensitivity of wild-type, trr1Δ and trr1Δ trx1Δ trx2Δ strains to H2O2.
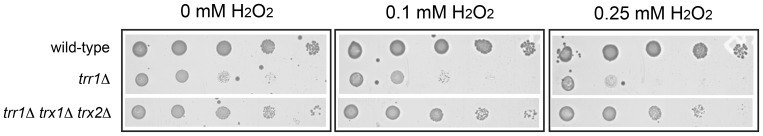
Equal numbers of cells were serially diluted (10-fold dilution) and spotted onto SC medium containing indicated concentrations of H2O2. Yeasts growth was assessed after 4 days at 30°C.
Discussion
Among the five S. cerevisiae genes encoding peroxiredoxins, deletion of TSA1 causes the accumulation of a broad spectrum of mutations. The mutator phenotype of tsa1Δ cells might be primarily due to their inability to reduce H2O2. The endogenous concentrations of H2O2 and alkyl hydroperoxides in wild-type and tsa1Δ strains have so far not been precisely determined because of the lack of the selective, quantitative and sensitive ROS probes [19]. Nevertheless, several laboratories report an increased intracellular ROS in tsa1Δ strains [16], [20]–[23]. Importantly, CanR mutation rate of a tsa1Δ strain decreases substantially under anaerobiosis, further suggesting that the increase of intracellular H2O2 concentration in tsa1Δ cells is the primary cause of the genome instability of these cells under aerobiosis. In such conditions, mitochondria and other sources of ROS (peroxisome and endoplasmic reticulum) generate less ROS, due to the restricted availability of O2 which is a substrate for electrons [61], [62]. Consistently, loss of Trr1 function reduces substantially the CanR mutation rate of tsa1Δ strain, most probably through over-expression of Yap1-regulated oxido-reductases that reduce substantially ROS-associated DNA damages. On the other hand, the associated expansion of the dNTP pool in tsa1Δ cells also contributes to its mutator phenotype [16], [60]. The excess of ROS in tsa1Δ cells increases the level of DNA lesions that in turn activates the DNA damage checkpoint pathway leading to the induction of RNR and the consequent overproduction of dNTPs. This double effect of increased ROS accumulation: elevation of DNA lesions level and stimulation of ribonucleotide reductase activity, may explain why the tsa1Δ mutant deviates from a correlation linking the increased dNTP level with increased spontaneous mutagenesis [60]. All these evidences support the view that the increase of intracellular H2O2 concentration in tsa1Δ cells is the primary cause of the genome instability of these cells under aerobiosis.
The transcription factor Yap1 is a central regulator of the response to oxidative stress in S. cerevisiae. In yeast cells with functional Gpx3 and Ybp1 proteins, Gpx3 senses exogenous added H2O2 and mediates H2O2 signal into a cysteine-based redox cascade that culminates in the oxidation of Yap1 [38], [39]. In strains harboring the W303 genetic background, the YBP1 gene is mutated and nonfunctional, and Yap1 activation appears to be Gpx3-independent but requires Tsa1 [35]. Thioredoxins are physiological reducing agents and are responsible for the negative regulation of the Yap1 transcription factor activity [42]–[44]. Indeed, thioredoxins should be able to reduce the activated disulfide form of Yap1, preventing its translocation to nucleus or facilitating its exit to cytoplasm. In the present study, we have shown that the loss of Trr1 induces Yap1 accumulation in nucleus and over-expression of a set of Yap1-regulated oxido-reductases. Several evidence strongly suggest that oxidized thioredoxins are a key activator of Yap1 in trr1Δ mutants. First, as thioredoxins reduce the activated disulfide form of Yap1, highly oxidation of thioredoxins in trr1Δ cells should prevent their capacity to reduce oxidized Yap1 and facilitate its accumulation in nucleus. Secondly, we observed that a set of Yap1-regulated genes is significantly more over-expressed in a trr1Δ, trr1Δ trx1Δ, or trr1Δ trx2Δ strain than in trr1Δ trx1Δ trx2Δ strain, suggesting that the presence of Trx1 and/or Trx2 largely contributes to the strong activation of the tested genes in trr1Δ cells. Furthermore, as Yap1 activity in trx1Δ trx2Δ mutants is lower than in trr1Δ mutants, this suggests that Yap1 activation in trr1Δ cells is due to a high concentration of oxidized thioredoxins but not the decreased level of reduced thioredoxins. In addition to the thioredoxins, Tsa1 contributes also to the Yap1 activation, as expression levels of Yap1-regulated genes are significantly lower in trr1Δ tsa1Δ than in trr1Δ mutants. Finally, a moderate but significant Yap1 activation is noteworthy in trx1Δ trx2Δ cells suggesting the existence of some Trx1/Trx2-independent mechanisms. The precise mechanisms in which oxidized thioredoxins and Tsa1 or other factors are involved in Yap1 oxidation and activation remain to be determined in the contexts of present mutant strains.
The tsa1Δ rad51Δ double mutant is not viable under aerobic conditions but viable under anaerobic conditions [18]. This observation suggests that a large excess of DNA lesions produced by ROS are the cause of lethality in the absence of efficient recombination repair. In searching for spontaneous suppressors of synthetic lethality of the tsa1Δ rad51Δ double mutant, we identified that loss of thioredoxin reductase Trr1 rescues their viability. This unexpected effect seems to result from the strong impact of TRR1 deletion on mutation formation. Additional deletion of TRR1 in tsa1Δ mutants reduces substantially the CanR mutation rate of tsa1Δ strain (33-fold), and to a less extend, of rad51Δ strain (4-fold). We reason that the synthetic lethality of tsa1Δ rad51Δ double mutant may result from excessive DNA damage. Substantial reduction of tsa1Δ and rad51Δ mutation rates in the trr1Δ context rescues the viability of the double mutant.
In conclusion, we propose that the trr1Δ -induced mutation suppression effect results from at least two distinct mechanisms both mediated by oxidized thioredoxins (Figure 7). Constitutive oxidation of thioredoxins strongly activates Yap1, induces over-expression of a set of Yap1-regulated oxido-reductases with antioxidant properties that ultimately reduces substantially intracellular ROS concentration and consequently ROS associated DNA damages. Besides, the oxidized thioredoxins may directly interfere with the activity of ribonucleotide reductase, leading to a reduction in the dNTPs synthesis and the translesion DNA synthesis (TLS), further reducing the mutagenesis. Although Yap1 could regulate the expression of several chaperones and components involved in post-replication repair (http://www.yeastract.com), it remains to determine whether these elements could contribute significantly to suppress the mutator phenotype of tsa1Δ mutants. In combination with the model described in Text S1, we strengthen the view that in yeast cells, the large majority of spontaneous mutations originates from ROS-induced lesions.
Figure 7. Schematics for trr1Δ-mediated decrease of CanR mutation.
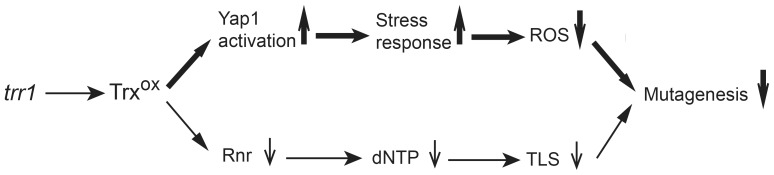
Loss-of-function of thioredoxin reductase Trr1 results in constitutive oxidation of thioredoxins, which leads to suppression of mutation formation through two distinct mechanisms. Yap1 activation and consequent over-expression of Yap1-regulated oxido-reductases play a major role in reducing mutagenesis. TLS, translesion DNA synthesis.
Materials and Methods
Media
Complete medium (YPD) contained 1% (w/v) yeast extract, 1% (w/v) bactopeptone, 2% (w/v) glucose. Synthetic complete medium (SC) with 2% glucose was prepared as described by Sherman [63]. SPO2 sporulation medium contained 0.5% yeast extract, 0.5% bactopeptone, 2% potassium acetate. Canavanine-resistant mutants (CanR) caused by inactivation of the CAN1 gene were selected on SC-arginine dropout plates containing 60 mg/liter canavanine. 5-fluoroorotic acid (5-FOA) was used at 1 g/liter in SC medium.
Plasmid and strain constructions
The constructions of plasmids and strains used in this work are described in Text S2 and Text S3. All yeast strains used in this study are listed in Table S1. All oligonucleotides used for plasmid and strain constructions are listed in Table S2.
Fluctuation analysis
The rate of accumulation of CanR mutations in cell populations was determined by fluctuation analysis [26]. 2-ml cell cultures were incubated under aerobic conditions at 30°C for 3 days with agitation. The number of mutant cells per culture among 19–57 parallel cultures was calculated and the median of the distribution was used to determine the mutation rate of a given strain. The 95% confidence intervals for a median rate were calculated according to Huang and Kolodner [17].
Anaerobic growth conditions
YPD medium was supplemented with Tween 80 and ergosterol to a final concentration of 1.32 g/liter and 6.75 mg/liter, respectively. For determining mutations rates, 2-ml cell cultures were placed in an 8-litre airtight jar containing 3 disposable hydrogen- and carbon dioxide-generating envelopes (BD BBL GasPak Plus, Becton Dickinson) and grown anaerobically at 30°C for 5 days without agitation. Anaerobic conditions were monitored with the redox indicators Anaerotest (Merck, Darmstadt) placed inside the airtight jar. As controls, cell cultures were grown under the same conditions except aerobically.
Quantitative RT-PCR
Wild-type and mutated strains were grown to mid-exponential phase (OD 0.5, 600 nm) in YPD medium. Total RNA was extracted according to a protocol previously described [64]. To ensure the absence of trace DNA in the samples, extracted RNA were treated with DNase and verified by conventional PCR, using primers DAN2UP and DAN2DW. cDNA were generated using an iScript cDNA synthesis Kit (Bio-Rad). Appropriately diluted cDNA from each strain were amplified using iQ SYBR Green Supermix (Bio-Rad) and a set of 13 primer pairs (Table S3) to follow the expression of 13 oxido-reductases. Expression of ACT1, CDC11 and LRE1 were used as internal controls. Quantitative RT-PCR reactions were carried out in the CFX96 Bio-Rad Thermal cycler. mRNA relative levels were calculated with Bio-Rad CFX Manager software. The oxido-reductase mRNA levels were normalized with respect to the ACT1, CDC11 and LRE1 mRNA levels. mRNA levels of oxido-reductase genes were calculated as ratios relative to that of a wild-type strain (set as 1).
Trx2 polyclonal antibody
The BL21(DE3) E. coli strain (New England Biolabs) was transformed with plasmid p1672 (TRX2). Transformed cells were grown with shaking at 37°C in LB in the presence of kanamycin (30 mg/liter). When the OD (580 nm) of the culture reached 0.6, isopropyl-ß-D-thiogalactoside (IPTG) was added to a final concentration of 0.8 mM and growth was continued overnight at 18°C. Cell lysates were prepared using a French Press. Trx2-6His was purified by affinity chromatography on a Ni-column (Qiagen). Then, the 6-His tag was cleaved off with tobacco etch virus protease (TEV) and the uncleaved material was purified by passage through a Ni-column. The Trx2 lacking the His tag was used for antibody production in rabbits by a commercial facility (Agro-Bio).
Western blotting
5 ml of cell cultures at logarithmic phase in complete medium were acid-quenched with 20% trichloroacetic acid (TCA, Sigma) at 4°C. Cells were harvested, resuspended in 1 ml of 20% TCA and pelleted by centrifugation. For protein extraction, the cell pellet was resuspended in 400 µl of 20% TCA and lysed with glass beads (200 µl) by vortexing for 5 min at 4°C. The lysed cells were centrifuged and the pellet was washed with cold acetone. The dry pellet was resuspended in 50 µl of TES buffer (100 mM Tris-Cl pH 8.8, 10 mM EDTA, 1% SDS) containing 30 mM 4-acetamido-4’-maleimidylstilbene-2,2’ -disulfonic acid, disodium salt (AMS, Molecular Probe) [38]. Following a 2 h incubation at 37°C with agitation in the dark, insoluble protein was removed by centrifugation. Each sample containing about 30 µg of protein was prepared with NuPAGE LDS sample buffer and was separated on a 12% NuPAGE Novex Bis-Tris gel with MES SDS buffer (Life Technologies). Upon transferring to a nitrocellulose membrane, Trx1 or Trx2 were probed with a rabbit anti-Trx1 antibody (a generous gift of C. Grant) or rabbit anti-Trx2 antibody, respectively and a secondary anti-rabbit IgG (IRDye, LI-COR) and then analyzed by quantitative immunoblot using an Odyssey Infrared Imaging System (LI-COR).
Isolation of suppressors
Strains GF5297 and GF5305 (Table S1) were streaked out on YPD plates supplemented with Tween 80 and ergosterol and incubated under anaerobic conditions at 30°C. Ten single colonies from each strain were inoculated into YPD-Tween 80-ergosterol and grown anaerobically for 4 days. 100 µl of each culture were then plated onto YPD and incubated aerobically. About 3 to 40 colonies appeared on each plate after 3 days at 30°C. One colony from each plate was retained and these were named GF5297-1 to GF5297-10 and GF5305-1 to GF5305-10. To determine whether suppression was due to single mutations, the suppressor strains were crossed with strains GF5265 or GF5269 and the meiotic products of the diploids obtained were analyzed. The proportions of the three types of tetrads (tetratypes, parental ditypes and non-parental ditypes) suggested that the putative suppressor mutations were single mutations that were not linked to the RAD51 locus. For determining whether the 20 suppressor mutations were recessive or dominant, the suppressor strains were crossed individually with strains GF5335 or GF5336. All diploids obtained were unable to grow on 5-FOA plates, suggesting that the suppressor mutations were recessive.
Identification of the suppressor mutation of strain GF5305–6
To clone the sup gene from the suppressor-containing strain GF5305-6, we used the red/white colony assay described by Zhao et al [47]. We first disrupted genes ADE2 and ADE3 of strain GF5305-6, yielding strain GF5374. Plasmid p1591 was introduced by transformation, yielding strain GF5377. GF5377 colonies were red on SC-uracil, 8% glucose medium but became white or sectored on YPD, 8% glucose plates because plasmid p1591 (TSA1, ADE3) was easily lost without selection. Strain GF5377 was transformed with a CEN-LEU2 plasmid-based genomic library (ATCC number 77162). Approximately 40,000 transformants were selected on SC-leucine, 8% glucose plates, of which 137 appeared red or pink. These 137 strains were restreaked on SC-leucine, 8% glucose plates and 21 strains continued to appear red or pink. These 21 strains were then tested for growth on SC-leucine, 5-FOA plates. Plasmids from the seven strains that failed to grow on SC-leucine, 5-FOA plates were recovered into E. coli using standard methods for further analysis.
Fluorescence microscopy
Exponentially growing cells expressing Yap1-GFP under the control of native Yap1 promoter were washed with PBS, resuspended in mounting solution (75% glycerol in PBS) containing 4′,6-diamidino-2-phenylindole (DAPI) at concentration of 50 µg/ml, and mounted on a glass slide covered with polylysine (Sigma). Fluorescent images were captured with a Leica microscope (DMRXA) equipped with a cooled CCD camera MicroMAX (Princeton Instruments) under control of the MetaMorph software (Molecular Devices). Images obtained were processed using ImageJ software.
Determination of dNTP levels
Approximately 3.7×108 (as determined by the optical density at 600 nm [OD600]) cells were harvested by filtration through 25-mm white AAWP nitrocellulose filters (0.8 mm; Millipore AB). The filters were immersed in 700 µl of ice-cold extraction solution (12% TCA, 15 mM MgCl2) in Eppendorf tubes and frozen in liquid nitrogen. The following steps were carried out at 4°C. The tubes were vortexed for 30 s, incubated for 15 min, and vortexed again for 30 s. The filters were removed, and 700 µl supernatants were collected after centrifugation at 20,000×g for 1 min and added to 800 µl of ice-cold Freon-trioctylamine mixture (10 ml of 99% pure Freon [1,1,2-trichlorotrifluoroethane; Sigma-Aldrich] and 2.8 ml of 98% pure trioctylamine [Sigma-Aldrich]). The samples were vortexed and centrifuged for 1 min at 20,000×g. The aqueous phase was collected and added to 700 µl of ice-cold Freon-trioctylamine mixture. Volumes of 475 and 47.5 µl of the aqueous phase were collected. The 475-μl aliquots of the aqueous phase were pH adjusted with 1 M NH4HCO3 (pH 8.9), loaded on boronate columns (Affi-Gel 601; Bio-Rad), and eluted with 50 mM NH4HCO3, pH 8.9, 15 mM MgCl2 to separate dNTPs and NTPs. The eluates with purified dNTPs were adjusted to pH 3.4 with 6 M HCl, separated on a Partisphere SAX HPLC column (125 mm×4.6 mm, Hichrome), under isocratic elution with 0.35 M potassium phosphate buffer (pH 3.4; containing 2.5% [vol/vol] acetonitrile) and quantified using a LaChrom Elite HPLC system (VWR International). The 47.5-μl aliquots of the aqueous phase were adjusted to pH 3.4 and used to quantify NTPs by HPLC in the same way.
Supporting Information
Cytoplasmic and nuclear GSH/GSSG redox status measured by rxYFP.
(TIF)
Yeast strains used in this study.
(DOC)
Oligonucleotides used for plasmid and strain constructions.
(DOC)
Primer pairs used for quantitative RT-PCR.
(DOC)
Characterization of mutations affecting gene TRR1 in the 20 suppressors.
(DOC)
dNTP levels in wild-type, trr1Δ trx1Δ trx2Δ, sml1Δ RNR1TEF1, trr1Δ sml1Δ RNR1TEF1 and trx1Δ trx2Δ sml1Δ RNR1TEF1 strains.
(DOC)
Average expression of TSA2 , AHP1 , CCP1 and YKL071w in Set A, B and C.
(DOC)
A tentative model.
(DOC)
Plasmid constructions.
(DOC)
Strain constructions.
(DOC)
Acknowledgments
We thank Andrei Chabes for his generous help in the determination of dNTP levels, Chris Grant for his kind gift of anti-Trx1 antibody, Judith Souphron for providing TEV protease. We thank Chitranshu Kumar, Marie Dutreix, Dorothée Baïlle, Pierre-Marie Girard for helpful discussions.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.
Funding Statement
This study was funded by the Centre National de la Recherche Scientifique and the Institut Curie. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Fu Y, Pastushok L, Xiao W (2008) DNA damage-induced gene expression in Saccharomyces cerevisiae. FEMS Microbiol Rev 32: 908–926. [DOI] [PubMed] [Google Scholar]
- 2. Kemp M, Go YM, Jones DP (2008) Nonequilibrium thermodynamics of thiol/disulfide redox systems: a perspective on redox systems biology. Free Radic Biol Med 44: 921–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lopez-Mirabal HR, Winther JR (2008) Redox characteristics of the eukaryotic cytosol. Biochim Biophys Acta 1783: 629–640. [DOI] [PubMed] [Google Scholar]
- 4. Toledano MB, Kumar C, Le Moan N, Spector D, Tacnet F (2007) The system biology of thiol redox system in Escherichia coli and yeast: differential functions in oxidative stress, iron metabolism and DNA synthesis. FEBS Lett 581: 3598–3607. [DOI] [PubMed] [Google Scholar]
- 5. Camier S, Ma E, Leroy C, Pruvost A, Toledano M, et al. (2007) Visualization of ribonucleotide reductase catalytic oxidation establishes thioredoxins as its major reductants in yeast. Free Radic Biol Med 42: 1008–1016. [DOI] [PubMed] [Google Scholar]
- 6. Muller EG (1991) Thioredoxin deficiency in yeast prolongs S phase and shortens the G1 interval of the cell cycle. J Biol Chem 266: 9194–9202. [PubMed] [Google Scholar]
- 7. Herrero E, Ros J, Belli G, Cabiscol E (2008) Redox control and oxidative stress in yeast cells. Biochim Biophys Acta 1780: 1217–1235. [DOI] [PubMed] [Google Scholar]
- 8. Lee J, Godon C, Lagniel G, Spector D, Garin J, et al. (1999) Yap1 and Skn7 control two specialized oxidative stress response regulons in yeast. J Biol Chem 274: 16040–16046. [DOI] [PubMed] [Google Scholar]
- 9. Godon C, Lagniel G, Lee J, Buhler JM, Kieffer S, et al. (1998) The H2O2 stimulon in Saccharomyces cerevisiae. J Biol Chem 273: 22480–22489. [DOI] [PubMed] [Google Scholar]
- 10. Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, et al. (2000) Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell 11: 4241–4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Park SG, Cha MK, Jeong W, Kim IH (2000) Distinct physiological functions of thiol peroxidase isoenzymes in Saccharomyces cerevisiae. J Biol Chem 275: 5723–5732. [DOI] [PubMed] [Google Scholar]
- 12. Jang HH, Lee KO, Chi YH, Jung BG, Park SK, et al. (2004) Two enzymes in one; two yeast peroxiredoxins display oxidative stress-dependent switching from a peroxidase to a molecular chaperone function. Cell 117: 625–635. [DOI] [PubMed] [Google Scholar]
- 13. Huang ME, Rio AG, Nicolas A, Kolodner RD (2003) A genomewide screen in Saccharomyces cerevisiae for genes that suppress the accumulation of mutations. Proc Natl Acad Sci U S A 100: 11529–11534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chabes A, Georgieva B, Domkin V, Zhao X, Rothstein R, et al. (2003) Survival of DNA damage in yeast directly depends on increased dNTP levels allowed by relaxed feedback inhibition of ribonucleotide reductase. Cell 112: 391–401. [DOI] [PubMed] [Google Scholar]
- 15. Sabouri N, Viberg J, Goyal DK, Johansson E, Chabes A (2008) Evidence for lesion bypass by yeast replicative DNA polymerases during DNA damage. Nucleic Acids Res 36: 5660–5667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tang HM, Siu KL, Wong CM, Jin DY (2009) Loss of yeast peroxiredoxin Tsa1p induces genome instability through activation of the DNA damage checkpoint and elevation of dNTP levels. PLoS Genet 5: e1000697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huang ME, Kolodner RD (2005) A biological network in Saccharomyces cerevisiae prevents the deleterious effects of endogenous oxidative DNA damage. Mol Cell 17: 709–720. [DOI] [PubMed] [Google Scholar]
- 18. Ragu S, Faye G, Iraqui I, Masurel-Heneman A, Kolodner RD, et al. (2007) Oxygen metabolism and reactive oxygen species cause chromosomal rearrangements and cell death. Proc Natl Acad Sci U S A 104: 9747–9752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wardman P (2007) Fluorescent and luminescent probes for measurement of oxidative and nitrosative species in cells and tissues: progress, pitfalls, and prospects. Free Radic Biol Med 43: 995–1022. [DOI] [PubMed] [Google Scholar]
- 20. Wong CM, Zhou Y, Ng RW, Kung Hf HF, Jin DY (2002) Cooperation of yeast peroxiredoxins Tsa1p and Tsa2p in the cellular defense against oxidative and nitrosative stress. J Biol Chem 277: 5385–5394. [DOI] [PubMed] [Google Scholar]
- 21. Wong CM, Siu KL, Jin DY (2004) Peroxiredoxin-null yeast cells are hypersensitive to oxidative stress and are genomically unstable. J Biol Chem 279: 23207–23213. [DOI] [PubMed] [Google Scholar]
- 22. Watanabe T, Irokawa H, Ogasawara A, Iwai K, Kuge S (2014) Requirement of peroxiredoxin on the stationary phase of yeast cell growth. J Toxicol Sci 39: 51–58. [DOI] [PubMed] [Google Scholar]
- 23. Ogusucu R, Rettori D, Netto LE, Augusto O (2009) Superoxide dismutase 1-mediated production of ethanol- and DNA-derived radicals in yeasts challenged with hydrogen peroxide: molecular insights into the genome instability of peroxiredoxin-null strains. J Biol Chem 284: 5546–5556. [DOI] [PubMed] [Google Scholar]
- 24. Shackelford RE, Kaufmann WK, Paules RS (2000) Oxidative stress and cell cycle checkpoint function. Free Radic Biol Med 28: 1387–1404. [DOI] [PubMed] [Google Scholar]
- 25. Dardalhon M, Kumar C, Iraqui I, Vernis L, Kienda G, et al. (2012) Redox-sensitive YFP sensors monitor dynamic nuclear and cytosolic glutathione redox changes. Free Radic Biol Med 52: 2254–2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lea D, Coulson C (1948) The distribution of the numbers of mutants in bacterial population. J Genet 49: 264–285. [DOI] [PubMed] [Google Scholar]
- 27. Northam MR, Robinson HA, Kochenova OV, Shcherbakova PV (2010) Participation of DNA polymerase zeta in replication of undamaged DNA in Saccharomyces cerevisiae. Genetics 184: 27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lai LC, Kosorukoff AL, Burke PV, Kwast KE (2005) Dynamical remodeling of the transcriptome during short-term anaerobiosis in Saccharomyces cerevisiae: differential response and role of Msn2 and/or Msn4 and other factors in galactose and glucose media. Mol Cell Biol 25: 4075–4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lai LC, Kosorukoff AL, Burke PV, Kwast KE (2006) Metabolic-state-dependent remodeling of the transcriptome in response to anoxia and subsequent reoxygenation in Saccharomyces cerevisiae. Eukaryot Cell 5: 1468–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Parker KR, von Borstel RC (1987) Base-substitution and frameshift mutagenesis by sodium chloride and potassium chloride in Saccharomyces cerevisiae. Mutat Res 189: 11–14. [DOI] [PubMed] [Google Scholar]
- 31. Shor E, Fox CA, Broach JR (2013) The yeast environmental stress response regulates mutagenesis induced by proteotoxic stress. PLoS Genet 9: e1003680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mihaylova VT, Bindra RS, Yuan J, Campisi D, Narayanan L, et al. (2003) Decreased expression of the DNA mismatch repair gene Mlh1 under hypoxic stress in mammalian cells. Mol Cell Biol 23: 3265–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Salin H, Fardeau V, Piccini E, Lelandais G, Tanty V, et al. (2008) Structure and properties of transcriptional networks driving selenite stress response in yeasts. BMC Genomics 9: 333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yan C, Lee LH, Davis LI (1998) Crm1p mediates regulated nuclear export of a yeast AP-1-like transcription factor. EMBO J 17: 7416–7429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Okazaki S, Naganuma A, Kuge S (2005) Peroxiredoxin-mediated redox regulation of the nuclear localization of Yap1, a transcription factor in budding yeast. Antioxid Redox Signal 7: 327–334. [DOI] [PubMed] [Google Scholar]
- 36. Tachibana T, Okazaki S, Murayama A, Naganuma A, Nomoto A, et al. (2009) A major peroxiredoxin-induced activation of Yap1 transcription factor is mediated by reduction-sensitive disulfide bonds and reveals a low level of transcriptional activation. J Biol Chem 284: 4464–4472. [DOI] [PubMed] [Google Scholar]
- 37. Monje-Casas F, Michan C, Pueyo C (2004) Absolute transcript levels of thioredoxin- and glutathione-dependent redox systems in Saccharomyces cerevisiae: response to stress and modulation with growth. Biochem J 383: 139–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Delaunay A, Pflieger D, Barrault MB, Vinh J, Toledano MB (2002) A thiol peroxidase is an H2O2 receptor and redox-transducer in gene activation. Cell 111: 471–481. [DOI] [PubMed] [Google Scholar]
- 39. Toledano MB, Delaunay A, Monceau L, Tacnet F (2004) Microbial H2O2 sensors as archetypical redox signaling modules. Trends Biochem Sci 29: 351–357. [DOI] [PubMed] [Google Scholar]
- 40. Veal EA, Ross SJ, Malakasi P, Peacock E, Morgan BA (2003) Ybp1 is required for the hydrogen peroxide-induced oxidation of the Yap1 transcription factor. J Biol Chem 278: 30896–30904. [DOI] [PubMed] [Google Scholar]
- 41. Gulshan K, Lee SS, Moye-Rowley WS (2011) Differential oxidant tolerance determined by the key transcription factor Yap1 is controlled by levels of the Yap1-binding protein, Ybp1. J Biol Chem 286: 34071–34081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Delaunay A, Isnard AD, Toledano MB (2000) H2O2 sensing through oxidation of the Yap1 transcription factor. EMBO J 19: 5157–5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mason JT, Kim SK, Knaff DB, Wood MJ (2006) Thermodynamic basis for redox regulation of the Yap1 signal transduction pathway. Biochemistry 45: 13409–13417. [DOI] [PubMed] [Google Scholar]
- 44. Izawa S, Maeda K, Sugiyama K, Mano J, Inoue Y, et al. (1999) Thioredoxin deficiency causes the constitutive activation of Yap1, an AP-1-like transcription factor in Saccharomyces cerevisiae. J Biol Chem 274: 28459–28465. [DOI] [PubMed] [Google Scholar]
- 45. Trotter EW, Grant CM (2005) Overlapping roles of the cytoplasmic and mitochondrial redox regulatory systems in the yeast Saccharomyces cerevisiae. Eukaryot Cell 4: 392–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Okazaki S, Tachibana T, Naganuma A, Mano N, Kuge S (2007) Multistep disulfide bond formation in Yap1 is required for sensing and transduction of H2O2 stress signal. Mol Cell 27: 675–688. [DOI] [PubMed] [Google Scholar]
- 47. Zhao X, Muller EG, Rothstein R (1998) A suppressor of two essential checkpoint genes identifies a novel protein that negatively affects dNTP pools. Mol Cell 2: 329–340. [DOI] [PubMed] [Google Scholar]
- 48. Trotter EW, Grant CM (2002) Thioredoxins are required for protection against a reductive stress in the yeast Saccharomyces cerevisiae. Mol Microbiol 46: 869–878. [DOI] [PubMed] [Google Scholar]
- 49. Carmel-Harel O, Stearman R, Gasch AP, Botstein D, Brown PO, et al. (2001) Role of thioredoxin reductase in the Yap1p-dependent response to oxidative stress in Saccharomyces cerevisiae. Mol Microbiol 39: 595–605. [DOI] [PubMed] [Google Scholar]
- 50. Boisnard S, Lagniel G, Garmendia-Torres C, Molin M, Boy-Marcotte E, et al. (2009) H2O2 activates the nuclear localization of Msn2 and Maf1 through thioredoxins in Saccharomyces cerevisiae. Eukaryot Cell 8: 1429–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sha W, Martins AM, Laubenbacher R, Mendes P, Shulaev V (2013) The genome-wide early temporal response of Saccharomyces cerevisiae to oxidative stress induced by cumene hydroperoxide. PLoS One 8: e74939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Heer D, Heine D, Sauer U (2009) Resistance of Saccharomyces cerevisiae to high concentrations of furfural is based on NADPH-dependent reduction by at least two oxireductases. Appl Environ Microbiol 75: 7631–7638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Trotter EW, Grant CM (2003) Non-reciprocal regulation of the redox state of the glutathione-glutaredoxin and thioredoxin systems. EMBO Rep 4: 184–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kumar C, Igbaria A, D'Autreaux B, Planson AG, Junot C, et al. (2011) Glutathione revisited: a vital function in iron metabolism and ancillary role in thiol-redox control. EMBO J 30: 2044–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Garcia-Santamarina S, Boronat S, Calvo IA, Rodriguez-Gabriel M, Ayte J, et al. (2013) Is oxidized thioredoxin a major trigger for cysteine oxidation? Clues from a redox proteomics approach. Antioxid Redox Signal 18: 1549–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tsaponina O, Barsoum E, Astrom SU, Chabes A (2011) Ixr1 is required for the expression of the ribonucleotide reductase Rnr1 and maintenance of dNTP pools. PLoS Genet 7: e1002061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Laureti L, Selva M, Dairou J, Matic I (2013) Reduction of dNTP levels enhances DNA replication fidelity in vivo. DNA Repair (Amst) 12: 300–305. [DOI] [PubMed] [Google Scholar]
- 58. Gon S, Napolitano R, Rocha W, Coulon S, Fuchs RP (2011) Increase in dNTP pool size during the DNA damage response plays a key role in spontaneous and induced-mutagenesis in Escherichia coli. Proc Natl Acad Sci U S A 108: 19311–19316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Datta A, Schmeits JL, Amin NS, Lau PJ, Myung K, et al. (2000) Checkpoint-dependent activation of mutagenic repair in Saccharomyces cerevisiae pol3-01 mutants. Mol Cell 6: 593–603. [DOI] [PubMed] [Google Scholar]
- 60. Davidson MB, Katou Y, Keszthelyi A, Sing TL, Xia T, et al. (2012) Endogenous DNA replication stress results in expansion of dNTP pools and a mutator phenotype. EMBO J 31: 895–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. de Groot MJ, Daran-Lapujade P, van Breukelen B, Knijnenburg TA, de Hulster EA, et al. (2007) Quantitative proteomics and transcriptomics of anaerobic and aerobic yeast cultures reveals post-transcriptional regulation of key cellular processes. Microbiology 153: 3864–3878. [DOI] [PubMed] [Google Scholar]
- 62. van den Brink J, Daran-Lapujade P, Pronk JT, de Winde JH (2008) New insights into the Saccharomyces cerevisiae fermentation switch: dynamic transcriptional response to anaerobicity and glucose-excess. BMC Genomics 9: 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sherman F (1991) Getting started with yeast. Methods Enzymol 194: 3–21. [DOI] [PubMed] [Google Scholar]
- 64.Collart M, Oliviero S (1993) Preparation of yeast RNA. In: Current protocols in molecular biology. supplement 23. John Wiley & Sons. unit 13.12. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cytoplasmic and nuclear GSH/GSSG redox status measured by rxYFP.
(TIF)
Yeast strains used in this study.
(DOC)
Oligonucleotides used for plasmid and strain constructions.
(DOC)
Primer pairs used for quantitative RT-PCR.
(DOC)
Characterization of mutations affecting gene TRR1 in the 20 suppressors.
(DOC)
dNTP levels in wild-type, trr1Δ trx1Δ trx2Δ, sml1Δ RNR1TEF1, trr1Δ sml1Δ RNR1TEF1 and trx1Δ trx2Δ sml1Δ RNR1TEF1 strains.
(DOC)
Average expression of TSA2 , AHP1 , CCP1 and YKL071w in Set A, B and C.
(DOC)
A tentative model.
(DOC)
Plasmid constructions.
(DOC)
Strain constructions.
(DOC)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.